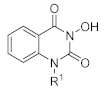
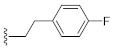
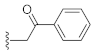
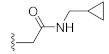
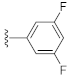
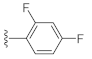
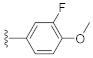
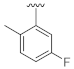
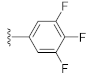
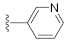
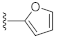
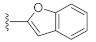
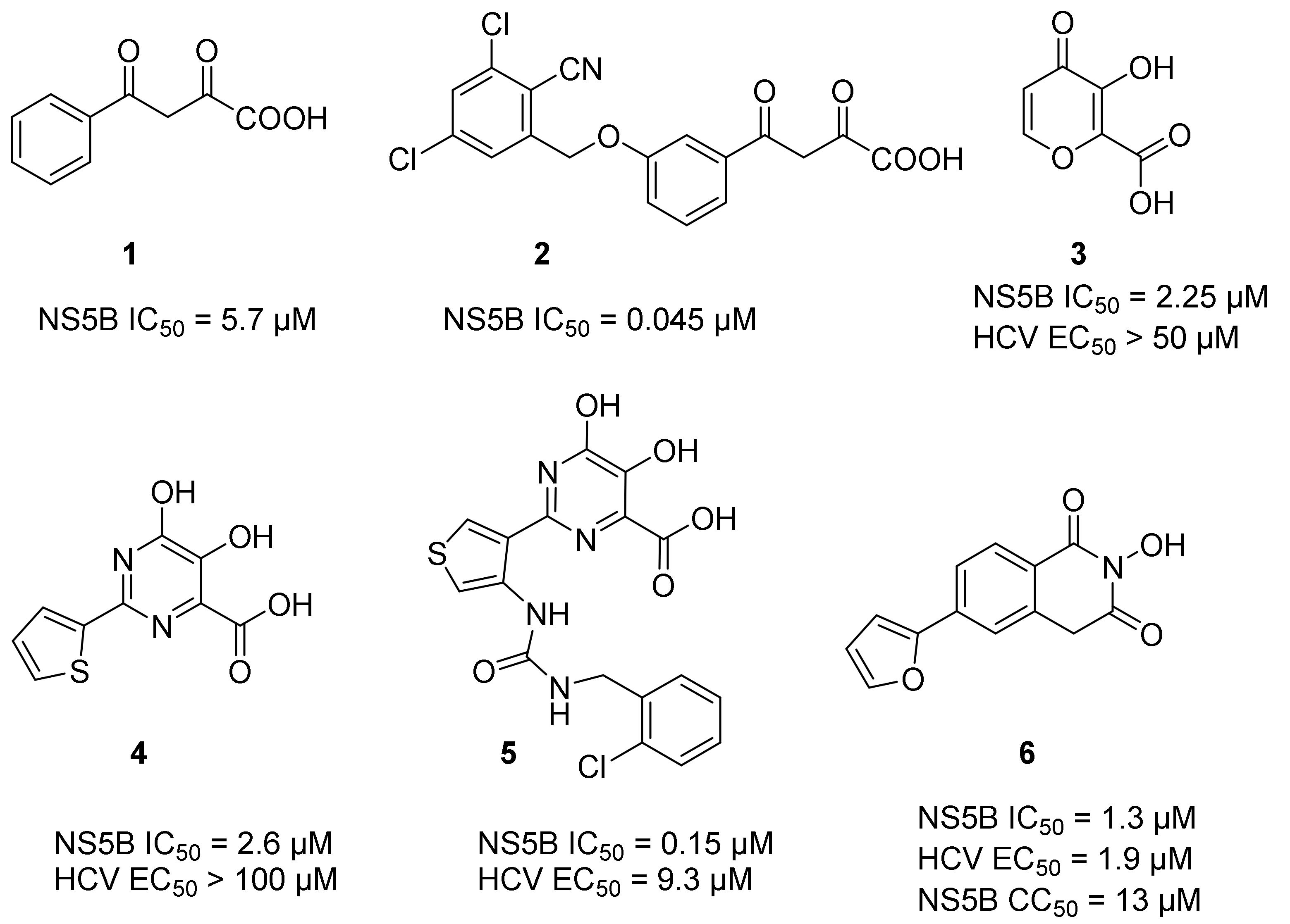
3.1. Chemistry
The reagents used in the chemical experiments were purchased from qualified chemical sellers, with purity greater than 95%. Most of the solvents used in the synthesis experiments were obtained from the China National Pharmaceutical Group Corporation. Unless otherwise specified, all solvents used in the reactions are of analytical grade and have not been further processed. Anhydrous solvents such as DMF, DCM, THF, etc., were purchased from the Innovative Technology Solvent Purification System (Innovative Technology Ltd., Hong Kong, China). Some reaction products were purified by flash column chromatography (CombiFlash® EZ Prep, Teledyne ISCO, Lincoln, NE, USA) with 200–300 mesh silica gel (Qingdao Haiyang Chemical Co., Ltd., Qingdao, China).
The nuclear magnetic resonance spectra (1H NMR, 13C NMR) of the synthesized compounds were recorded by Varian Mercury Plus 400 MHz and Bruker AscendTM 600 MHz spectrometers with TMS as an internal standard to calibrate the chemical shifts (δ). Deuterated DMSO or Deuterated trifluoroacetic acid purchased from J&K Scientific was used as the solvent for NMR spectroscopy. The purities and molecular weights of the compounds were determined using an Agilent 1100s mass spectrometer and an Agilent 1260 LC-Agilent 6120 MS liquid chromatography-mass spectrometer with an ESI ion source. The mobile phase was a chromatographically pure water/methanol mixture.
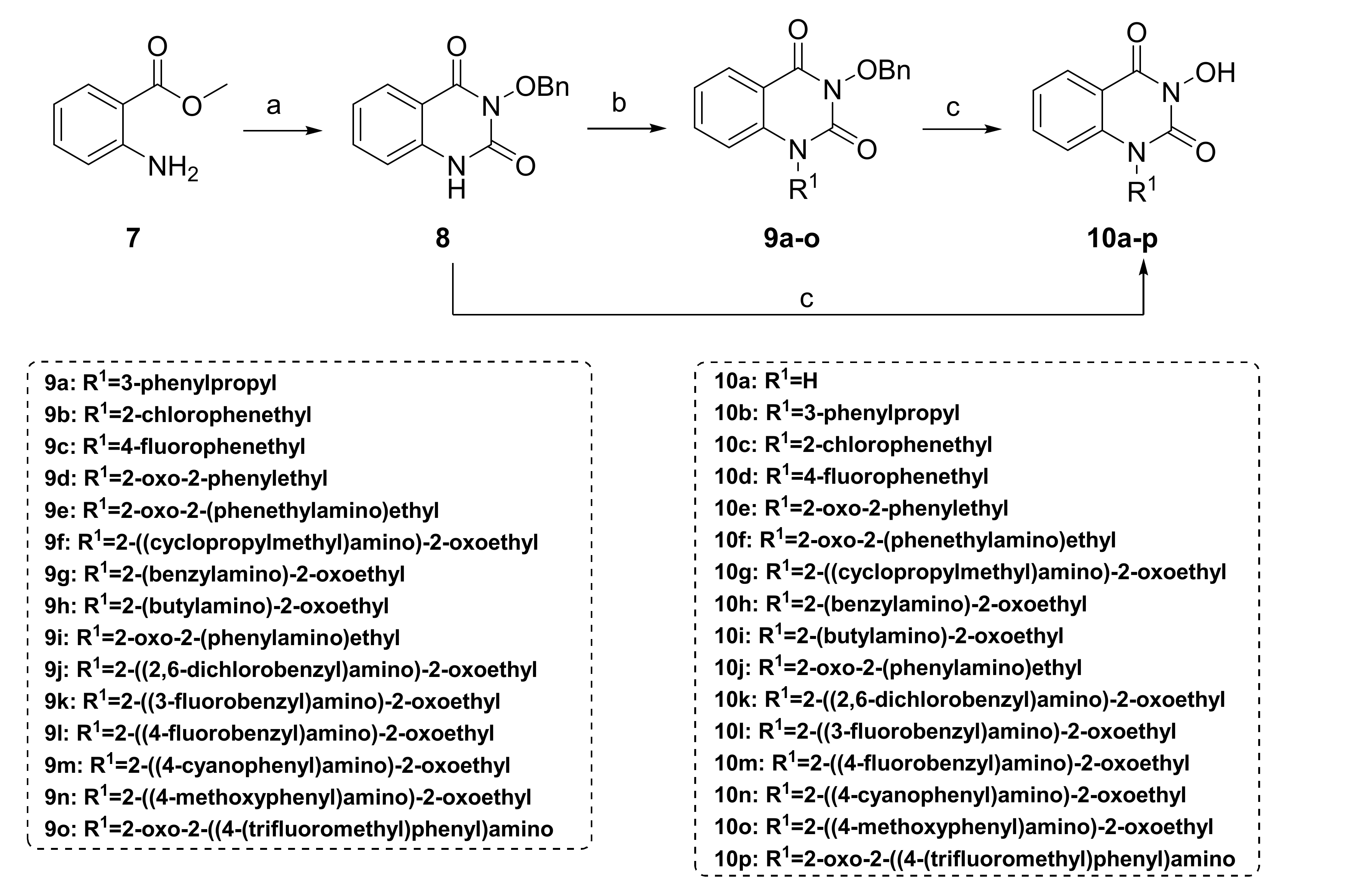
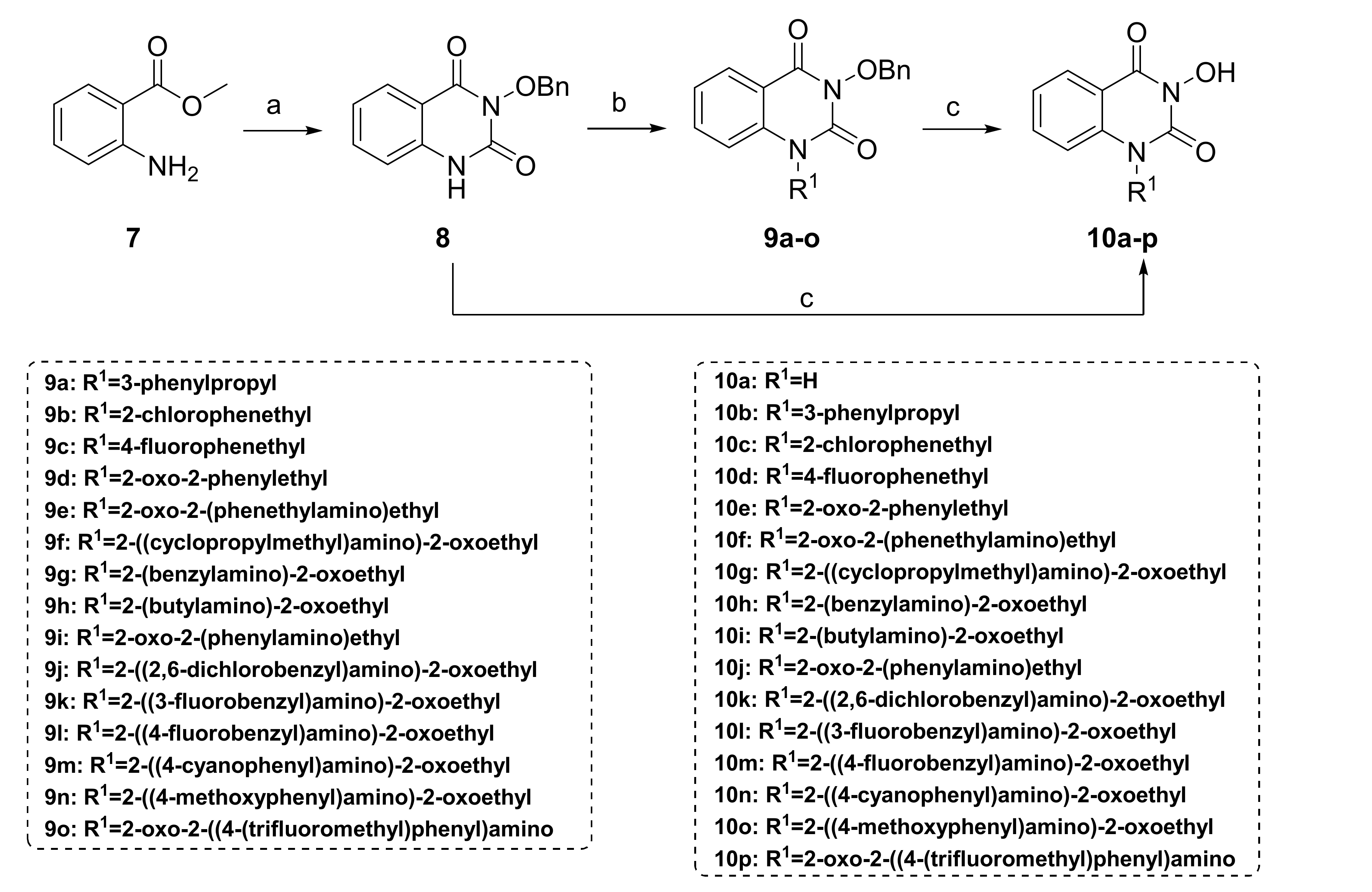
3.1.1. Synthesis of Compounds 8 and 10a
3-(Benzyloxy)quinazoline-2,4(1H,3H)-dione (8). To O-benzylhydroxylamine hydrochloride (6.25 g, 39.1 mmol, 1.5 equiv.) was added 5% NaOH aqueous solution (80 mL) and ether (230 mL), and the mixture was stirred at room temperature for 2 h. The layers were left to stand, and the ether layer was washed three times with saturated brine. The ether layer was dried by Na2SO4 and concentrated to give O-benzylhydroxylamine (4.8 g, 100%), which was stored at 4 °C for later use. A suspension of methyl anthranilate (3.95 g, 26.1 mmol, 1.0 equiv.) and CDI (5.3 g, 32.6 mmol, 1.25 equiv.) in toluene (240 mL) was heated under reflux for 2 h. After the reaction mixture was cooled, the above prepared O-benzylhydroxylamine (4.8 g, 39.1 mmol, 1.5 equiv.) was added, and the suspension was heated under reflux for 4 h and then evaporated. Subsequently, ethanol (70 mL) and 2 mol/L NaOH aqueous solution (18 mL) were added to the flask, and the mixture was refluxed for 2 h. After cooling, 15% volume fraction of acetic acid aqueous solution (240 mL) was slowly added to the reaction mixture. The white precipitate was filtrated and recrystallized with methanol (40 mL) to obtain 8 as a white solid (4.2 g, 60% in all steps). MS (ESI) m/z: 269.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (dd, J = 8.7, 1.5 Hz, 1H), 7.54 (td, J = 7.9, 1.5 Hz, 1H), 7.38–7.31 (m, 4H), 7.35–7.17 (m, 4H), 5.01 (s, 2H).
3-Hydroxyquinazoline-2,4(1H,3H)-dione (10a). To compound 8 (100 mg, 0.373 mmol, 1.0 equiv.) was added 10 wt.% loading palladium–carbon catalyst (10% of the mass of 8), THF (4 mL), and methanol (1 mL). The solution was stirred under hydrogen (1 atm) at room temperature for 8 h until the raw material 8 was completely converted. The palladium–carbon catalyst was removed by filtration. The filtrate was evaporated to dryness, recrystallized with methanol to give 10a as a white solid (38 mg, 57%). MS (ESI) m/z: 177.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 10.56 (s, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.66 (t, J = 7.8 Hz, 1H), 7.27–7.12 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 159.23, 148.58, 138.18, 134.53, 126.90, 122.36, 115.10, 113.97.
3.1.2. Synthesis of Compounds 10b–p
A suspension of compound 8 (300 mg, 1.12 mmol, 1.0 equiv.), K2CO3 (309.6 mg, 2.24 mmol, 2.0 equiv.), the corresponding halide (1.34 mmol, 1.2 equiv.) was stirred in DMF (3 mL) at 80 °C for 2 h. After cooling, the reaction mixture was poured into water. The precipitate was washed with water and ether and dried to give the crude product 9a–o, respectively. Debenzylation reactions of 9a–o were subsequently conducted using two different conditions to obtain 10b–p, respectively.
For 10b–e: to compound 9a–d (0.388 mmol, 1.0 equiv.) was added 48% hydrobromic acid (1.5 mL) and glacial acetic acid (1.5 mL). The solution was refluxed for 2 h until the complete conversion of 9a–d. The reaction mixture was cooled to 0 °C under an ice bath and neutralized with 1 mol/L sodium hydroxide aqueous solution. The precipitate was collected by filtration, washed with water and diethyl ether, dried, and re-slurried or recrystallized with a mixture of ethyl acetate/methanol to obtain 10b–e as a solid, respectively.
For 10f–p: A suspension of compound 9e–o (0.233 mmol, 1.0 equiv.), 10 wt.% loading palladium–carbon catalyst (10% of the mass of 9e–o), THF (4 mL), and methanol (1 mL) was stirred under hydrogen (1 atm) at room temperature for 4–12 h until the raw material 9e–o was completely consumed. The catalyst was removed by filtration. The filtrate was evaporated to dryness, recrystallized or re-slurried with methanol, and dried to give 10f–p as a solid, respectively.
3-Hydroxy-1-(3-phenylpropyl)quinazoline-2,4(1H,3H)-dione (10b). According to the above general procedure with 1-bromo-3-phenylpropane as the halide in the reactants, the crude 9a was obtained as a white solid. MS (ESI) m/z: 387.0 [M + H]+. Subsequent debenzylation in the mixture of hydrobromic acid/glacial acetic acid gave 10b as a pale yellow solid (82.2 mg, 55% over two steps). MS (ESI) m/z: 294.9 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.31–7.07 (m, 6H), 4.09 (t, J = 7.7 Hz, 2H), 2.67 (d, J = 8.2 Hz, 2H), 1.89 (t, J = 8.6 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 158.71, 149.74, 141.08, 137.98, 134.21, 128.51, 128.25, 128.17, 128.09, 127.36, 125.74, 122.30, 115.13, 114.14, 42.74, 31.98, 28.40.
1-(2-Chlorophenethyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (10c). According to the above general procedure with 1-(2-bromoethyl)-2-chlorobenzene as the halide in the reactants, the crude 9b was obtained as a white solid. MS (ESI) m/z: 406.9 [M + H]+. Subsequent debenzylation in the mixture of hydrobromic acid/glacial acetic acid gave 10c as a pale yellow solid (43 mg, 18% over two steps). MS (ESI) m/z: 315.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.07 (d, J = 7.5 Hz, 1H), 7.74 (t, J = 7.6 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.40 (d, J = 7.2 Hz, 2H), 7.29 (dt, J = 14.7, 6.9 Hz, 3H), 4.37 (t, J = 7.5 Hz, 2H), 3.09 (t, J = 7.3 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 158.28, 148.91, 138.39, 135.50, 134.87, 133.10, 131.35, 129.13, 128.55, 127.67, 127.36, 122.73, 114.89, 114.25, 42.87, 30.58.
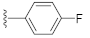
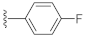
1-(4-Fluorophenethyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (10d). According to the above general procedure with 1-(2-bromoethyl)-4-fluorobenzene as the halide in the reactants, the crude 9c was obtained as a white solid. MS (ESI) m/z: 391.1 [M + H]+. Subsequent debenzylation in the mixture of hydrobromic acid/glacial acetic acid gave 10d as a pale yellow solid (60 mg, 65% over two steps). MS (ESI) m/z: 299.0 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.92–7.71 (m, 1H), 7.56 (d, J = 8.7 Hz, 1H), 7.49–7.27 (m, 3H), 7.23–7.07 (m, 2H), 4.33 (d, J = 7.7 Hz, 2H), 2.95 (d, J = 7.4 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 161.74, 160.14, 158.29, 148.90, 138.32, 134.98, 134.03, 130.66, 130.61, 127.64, 122.78, 115.12, 114.98, 114.86, 114.67, 44.34, 31.94.
3-Hydroxy-1-(2-oxo-2-phenylethyl)quinazoline-2,4(1H,3H)-dione (10e). According to the above general procedure with 2-bromo-1-phenylethan-1-one as the halide in the reactants, the crude 9d was obtained as a white solid. MS (ESI) m/z: 386.9 [M + H]+. Subsequent debenzylation in the mixture of hydrobromic acid/glacial acetic acid gave 10e as a pale yellow solid (30 mg, 27% over two steps). MS (ESI) m/z: 295.0 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 8.14 (d, J = 7.2 Hz, 3H), 7.85–7.57 (m, 4H), 7.43–7.26 (m, 2H), 5.82 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 192.91, 158.44, 149.36, 139.06, 135.01, 134.15, 128.85 (2C), 128.17 (2C), 127.58, 123.02, 114.75, 114.61, 50.12.
2-(3-Hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)-N-phenethylacetamide (10f). According to the above general procedure with 2-bromo-N-phenethylacetamide as the halide in the reactants, the crude 9e was obtained as a white solid. MS (ESI) m/z: 430.2 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10f as a white solid (36.3 mg, 35% over two steps). MS (ESI) m/z: 338.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 8.36 (t, J = 5.9 Hz, 1H), 8.05 (d, J = 7.5 Hz, 1H), 7.70 (t, J = 7.7 Hz, 1H), 7.28 (t, J = 8.0 Hz, 3H), 7.20 (t, J = 8.6 Hz, 3H), 7.11 (d, J = 7.9 Hz, 1H), 4.72 (d, J = 4.9 Hz, 2H), 3.32 (d, J = 7.3 Hz, 2H), 2.71 (dd, J = 10.0, 4.6 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.20, 158.55, 149.50, 139.12, 138.95, 134.68, 128.55 (2C), 128.20 (2C), 127.38, 125.99, 122.79, 114.92, 114.30, 45.89, 40.21, 34.89.
N-(Cyclopropylmethyl)-2-(3-hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (10g). According to the above general procedure with 2-bromo-N-(cyclopropylmethyl)acetamide as the halide in the reactants, the crude 9f was obtained as a white solid. MS (ESI) m/z: 380.2 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10g as a white solid (85 mg, 79% over two steps). MS (ESI) m/z: 288.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 8.33 (s, 1H), 8.00 (d, J = 6.8 Hz, 1H), 7.69 (s, 1H), 7.26 (d, J = 7.5 Hz, 1H), 7.17 (d, J = 8.1 Hz, 1H), 4.73 (s, 2H), 2.97 (s, 2H), 2.50 (s, 2H), 0.88 (s, 1H), 0.38 (d, J = 5.7 Hz, 2H), 0.13 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.23, 158.80, 149.91, 138.78, 134.23, 127.21, 122.53, 115.06, 114.16, 45.84, 42.81, 10.58, 3.06 (2C).
N-Benzyl-2-(3-hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (10h). According to the above general procedure with N-benzyl-2-bromoacetamide as the halide in the reactants, the crude 9g was obtained as a white solid. MS (ESI) m/z: 416.1 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10h as a pale yellow solid (90 mg, 81% over two steps). MS (ESI) m/z: 324.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 8.78 (s, 1H), 8.06 (d, J = 7.6 Hz, 1H), 7.73 (s, 1H), 7.31 (t, J = 8.0 Hz, 3H), 7.27–7.19 (m, 4H), 4.84 (s, 2H), 4.30 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.49, 158.61, 149.57, 138.90, 134.63, 128.14, 127.38 (2C), 126.99 (2C), 126.71, 122.84, 114.98, 114.41, 46.03, 42.02.
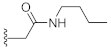
N-Butyl-2-(3-hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (10i). According to the above general procedure with 2-bromo-N-butylacetamide as the halide in the reactants, the crude 9h was obtained as a white solid. MS (ESI) m/z: 382.2 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10i as a white solid (19 mg, 11% over two steps). MS (ESI) m/z: 290.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H), 8.20 (d, J = 5.7 Hz, 1H), 8.07 (d, J = 7.9 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.31 (t, J = 7.7 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 4.73 (s, 2H), 3.06 (p, J = 5.6, 5.0 Hz, 2H), 1.37 (t, J = 8.0 Hz, 2H), 1.24 (q, J = 7.5 Hz, 2H), 0.90–0.78 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 166.03, 158.51, 149.41, 139.05, 134.74, 127.43, 122.84, 114.90, 114.37, 45.92, 38.18, 30.97, 19.33, 13.52.
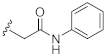
2-(3-Hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)-N-phenylacetamide (10j). According to the above general procedure with 2-bromo-N-phenylacetamide as the halide in the reactants, the crude 9i was obtained as a white solid. MS (ESI) m/z: 402.1 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10j as a white solid (13 mg, 7% over two steps). MS (ESI) m/z: 310.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 10.38 (s, 1H), 8.10 (d, J = 7.0 Hz, 1H), 7.76 (t, J = 8.0 Hz, 1H), 7.57 (d, J = 6.8 Hz, 2H), 7.40 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 6.7 Hz, 3H), 7.08 (d, J = 7.1 Hz, 1H), 5.00 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 165.13, 158.41, 149.42, 139.15, 138.39, 134.89, 128.67 (2C), 127.45, 123.42, 122.90, 119.01 (2C), 114.62, 114.57, 46.30.
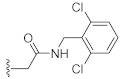
N-(2,6-Dichlorobenzyl)-2-(3-hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (10k). According to the above general procedure with 2-bromo-N-(2,6-dichlorobenzyl)acetamide as the halide in the reactants, the crude 9j was obtained as a white solid. MS (ESI) m/z: 484.1 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10k as a grey solid (28.4 mg, 25% over two steps). MS (ESI) m/z: 393.0 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 1H), 8.56 (s, 1H), 8.07 (d, J = 8.5 Hz, 1H), 7.71 (t, J = 8.3 Hz, 1H), 7.50 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 6.8 Hz, 1H), 7.34–7.24 (m, 1H), 7.18 (d, J = 8.8 Hz, 1H), 4.79 (s, 2H), 4.54 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.11, 158.44, 149.40, 139.02, 135.46 (2C), 134.74, 132.83, 130.27, 128.50 (2C), 127.44, 122.88, 114.78, 114.41, 45.55, 38.80.
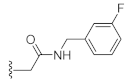
N-(3-Fluorobenzyl)-2-(3-hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (10l). According to the above general procedure with 2-bromo-N-(3-fluorobenzyl)acetamide as the halide in the reactants, the crude 9k was obtained as a white solid. MS (ESI) m/z: 434.1 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10l as a white solid (40 mg, 30% over two steps). MS (ESI) m/z: 342.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.80 (d, J = 6.7 Hz, 1H), 8.08 (d, J = 7.8 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.39–7.24 (m, 3H), 7.05 (dd, J = 17.2, 9.0 Hz, 3H), 4.85 (d, J = 2.9 Hz, 2H), 4.32 (d, J = 6.0 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.70, 162.11 (d, J = 243.3 Hz), 158.55, 149.49, 142.02 (d, J = 7.2 Hz), 139.03, 134.76, 130.10 (d, J = 8.4 Hz), 127.49, 122.94, 115.01, 114.46, 113.52.
N-(4-Fluorobenzyl)-2-(3-hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (10m). According to the above general procedure with 2-bromo-N-(4-fluorobenzyl)acetamide as the halide in the reactants, the crude 9l was obtained as a white solid. MS (ESI) m/z: 434.1 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10m as a white solid (49.4 mg, 36% over two steps). MS (ESI) m/z: 342.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.79 (d, J = 6.0 Hz, 1H), 8.08 (d, J = 7.7 Hz, 1H), 7.74 (t, J = 7.9 Hz, 1H), 7.32 (t, J = 8.0 Hz, 1H), 7.26 (d, J = 7.7 Hz, 3H), 7.19–7.09 (m, 2H), 4.83 (s, 2H), 4.28 (d, J = 5.7 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.53, 161.08 (d, J = 242.2 Hz), 158.54, 149.47, 139.03, 135.15, 134.79, 128.99 (d, J = 8.3 Hz), 127.47, 122.93, 114.97 (d, J = 4.4 Hz), 114.81, 114.44, 46.05, 41.33.
N-(4-Cyanophenyl)-2-(3-hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (10n). According to the above general procedure with 2-bromo-N-(4-cyanophenyl)acetamide as the halide in the reactants, the crude 9m was obtained as a white solid. MS (ESI) m/z: 427.1 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10n as a white solid (20 mg, 19% over two steps). MS (ESI) m/z: 335.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 10.85 (s, 1H), 8.11 (dd, J = 7.5, 3.6 Hz, 1H), 7.78 (d, J = 11.4 Hz, 5H), 7.47–7.40 (m, 1H), 7.34 (td, J = 7.4, 3.6 Hz, 1H), 5.05 (d, J = 3.7 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 166.14, 158.40, 149.41, 142.57, 139.10, 134.96, 133.27, 123.00, 119.07, 118.79, 114.61, 105.23, 46.50.
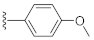
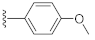
2-(3-Hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)-N-(4-methoxyphenyl)acetamide (10o). According to the above general procedure with 2-bromo-N-(4-methoxyphenyl)acetamide as the halide in the reactants, the crude 9n was obtained as a white solid. MS (ESI) m/z: 432.1 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10o as a white solid (23.8 mg, 18% over two steps). MS (ESI) m/z: 340.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 10.22 (s, 1H), 8.09 (d, J = 7.9 Hz, 1H), 7.74 (t, J = 8.2 Hz, 1H), 7.47 (d, J = 8.6 Hz, 2H), 7.35 (dd, J = 20.2, 8.4 Hz, 2H), 6.88 (d, J = 8.4 Hz, 2H), 4.96 (s, 2H), 3.71 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 164.60, 158.41, 155.22, 149.41, 139.14, 134.85, 131.47, 127.41, 122.85, 120.59, 114.63, 114.54, 113.73, 54.96, 46.17.
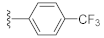
2-(3-Hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)-N-(4-(trifluoromethyl)phenyl)acetamide (10p). According to the above general procedure with 2-bromo-N-(4-(trifluoromethyl)phenyl)acetamide as the halide in the reactants, the crude 9o was obtained as a white solid. MS (ESI) m/z: 470.1 [M + H]+. Subsequent debenzylation by catalytic hydrogenation gave 10p as a white solid (30 mg, 18% over two steps). MS (ESI) m/z: 378.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 10.77 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 9.0 Hz, 2H), 7.74 (d, J = 7.3 Hz, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 7.9 Hz, 1H), 7.33 (d, J = 7.3 Hz, 1H), 5.05 (s, 2H). 13C NMR (151 MHz, Chloroform-d) δ 165.95, 158.42, 149.44, 141.95, 139.12, 134.92, 127.48, 126.04, 125.04, 123.54, 123.28 (d, J = 12.2 Hz), 122.96, 118.96, 114.61, 46.44.
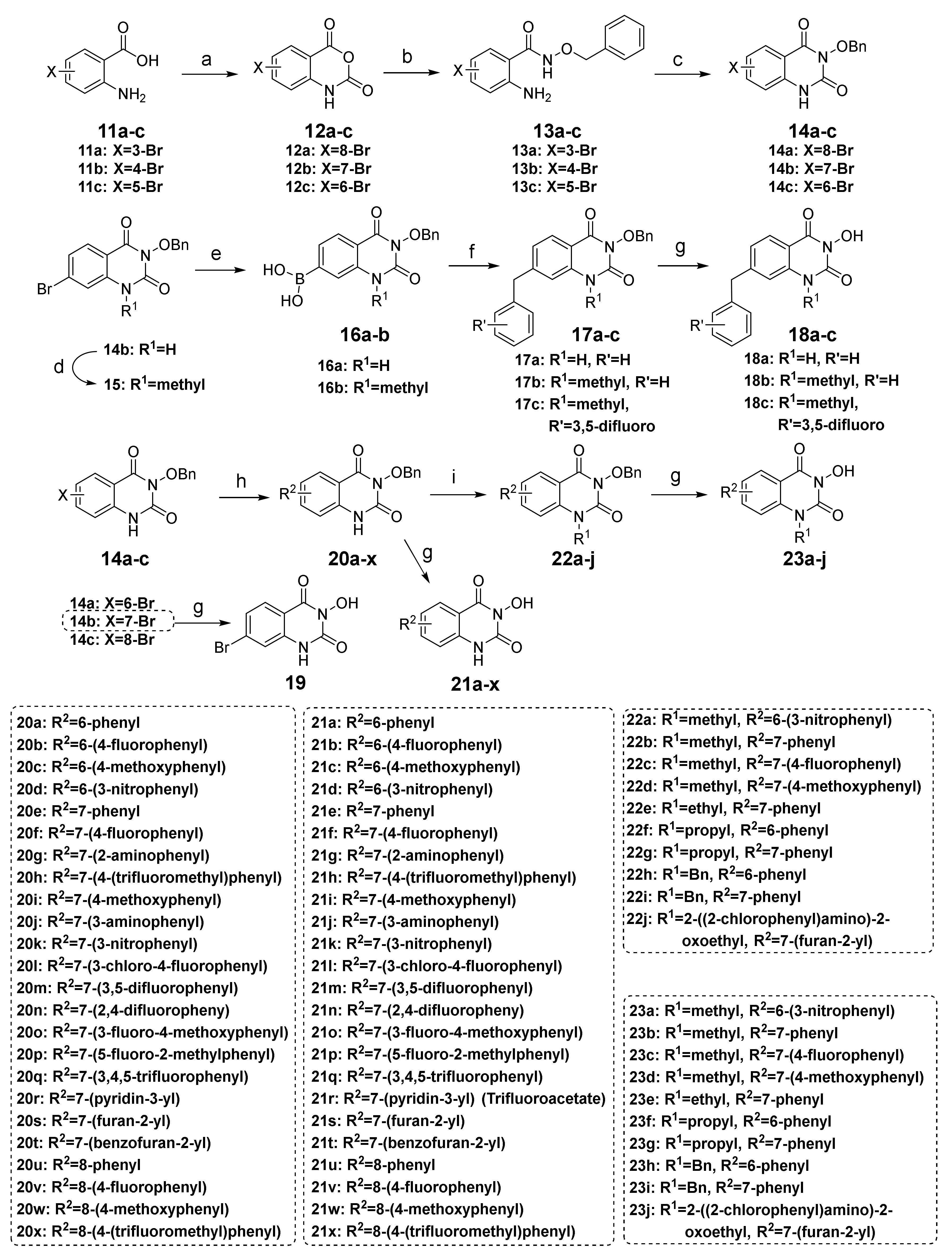
3.1.3. Synthesis of Compounds 14a–c
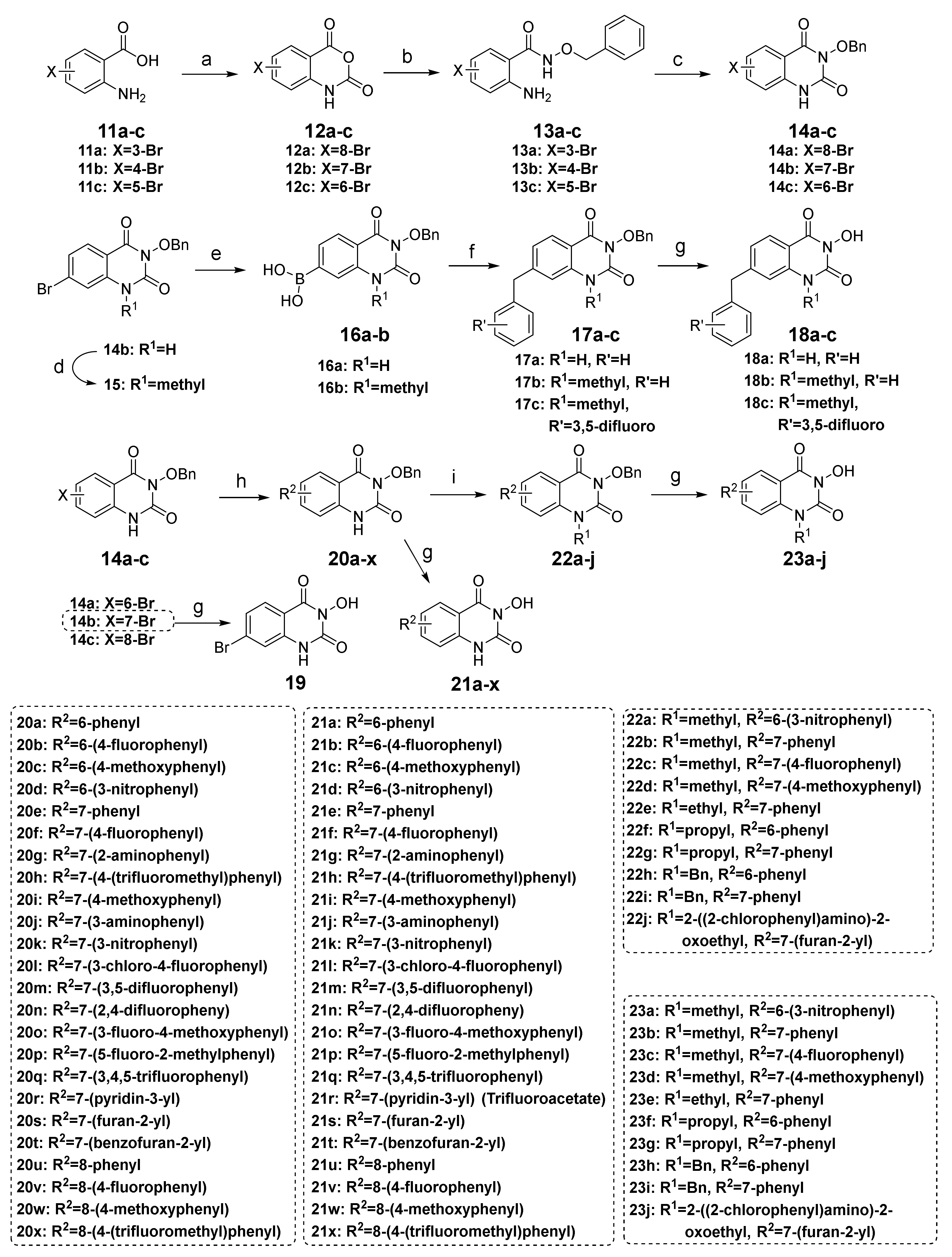
To compound 11a–c (37.0 mmol, 1.0 equiv.) was added triphosgene (3.84 g, 12.9 mmol, 0.35 equiv.), and 1,4-dioxane (40 mL). The suspension was refluxed at 110 °C for 6 h and then evaporated to 1/3 of the volume. The precipitate was collected by filtration, washed with petroleum ether (50 mL) and ethyl acetate (10 mL) successively, and dried to give the crude product 12a–c. A suspension of O-benzylhydroxylamine hydrochloride (3.72 g, 23.3 mmol, 1.02 equiv.) and triethylamine (2.36 g, 23.3 mmol, 1.02 equiv.) in ethanol (100 mL) was stirred at room temperature for 1 h, and then crude 12a–c (22.9 mmol, 1.0 equiv.) was added. The reaction mixture was refluxed for 3 h, then poured into water (350 mL). The precipitate was collected by filtration, washed with water, and dried to obtain crude 13a–c. Without further purification, crude 13a–c (16.7 mmol, 1.0 equiv.) was stirred with triphosgene (1.98 g, 6.63 mmol, 0.4 equiv.) and triethylamine (4.05 g, 40.0 mmol, 2.4 equiv.) in THF (200 mL) at room temperature for 2 h, then quenched with water (600 mL). The precipitate was filtrated, washed with water, dried, and re-slurried with petroleum ether/ethyl acetate to give compound 14a–c, respectively.
3-(Benzyloxy)-8-bromoquinazoline-2,4(1H,3H)-dione (14a). According to the above general procedure, cyclization of 11a with triphosgene afforded crude 12a. MS (ESI) m/z: 242.1 [M + H]+. Reaction of 12a with O-benzylhydroxylamine gave crude 13a. MS (ESI) m/z: 321.0 [M + H]+. Compound 14a was finally obtained from crude 13a as a white solid (4.52 g, 36% over three steps). MS (ESI) m/z: 347.1 [M + H]+.
3-(Benzyloxy)-7-bromoquinazoline-2,4(1H,3H)-dione (14b). According to the above general procedure, cyclization of 11b with triphosgene afforded crude 12b. MS (ESI) m/z: 242.1 [M + H]+. Reaction of 12b with O-benzylhydroxylamine gave crude 13b. MS (ESI) m/z: 321.0 [M + H]+. Compound 14b was finally obtained from crude 13b as a white solid (5.28 g, 42% over three steps). MS (ESI) m/z: 347.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.78 (s, 1H), 7.88 (d, J = 8.5 Hz, 1H), 7.62–7.54 (m, 2H), 7.42 (d, J = 7.3 Hz, 4H), 7.37 (d, J = 1.8 Hz, 1H), 5.09 (s, 2H).
3-(Benzyloxy)-6-bromoquinazoline-2,4(1H,3H)-dione (14c). According to the above general procedure, cyclization of 11c with triphosgene afforded crude 12c. MS (ESI) m/z: 242.1 [M + H]+. Reaction of 12c with O-benzylhydroxylamine gave crude 13c. MS (ESI) m/z: 321.0 [M + H]+. Compound 14c was finally obtained from crude 13c as a white solid (1.1 g, 18% over three steps). MS (ESI) m/z: 347.1 [M + H]+.
3.1.4. Synthesis of Compounds 18a–c and 19
A mixture of 14b or 15 (2.88 mmol, 1.0 equiv.), bis(pinacolato)diboron (3.47 mmol, 1.2 equiv.), Pd(dppf)Cl2 (0.145 mmol, 0.05 equiv.), and KOAc (8.66 mmol, 3.0 equiv.) were heated at 100 °C under nitrogen atmosphere for 8 h. The suspension was filtered while hot to remove the catalyst and KOAc, and the filtrate was poured into water (175 mL). The precipitate was collected by filtration, washed with water and petroleum ether, and dried to give crude boron 16a or 16b. A portion of crude 16a or 16b (0.769 mmol, 1.0 equiv.) was added to benzyl bromide or substituted benzyl bromide (0.641 mmol, 1.2 equiv.), Pd(dppf)Cl2 (0.032 mmol, 0.05 equiv.), KOAc (1.28 mmol, 2.0 equiv.), and water (2 mL) in 1,4-dioxane (20 mL). The suspension was heated at 80 °C for 3 h and then filtered while hot. The filtrate was concentrated, and the residue was purified by chromatography on silica gel with 20–30% ethyl acetate in petroleum ether to afford 17a–c, which was subsequently subjected to catalytic hydrogenation following the same procedure as described in the preparation of 10a to obtain 18a–c.
7-Benzyl-3-hydroxyquinazoline-2,4(1H,3H)-dione (18a). According to the above general procedure, 14b was first converted to the boron intermediate 16a. MS (ESI) m/z: 310.8 [M−H]−. The crude 16a was then subjected to the Suzuki coupling reaction with benzyl bromide to afford crude 17a. MS (ESI) m/z: 359.1 [M + H]+. Subsequent debenzylation of 17a by catalytic hydrogenation gave 18a as a white solid (11 mg, 10% over three steps). MS (ESI) m/z: 266.9 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H), 10.50 (s, 1H), 7.86 (dd, J = 8.1, 2.3 Hz, 1H), 7.32 (dd, J = 10.9, 4.6 Hz, 2H), 7.23 (d, J = 6.3 Hz, 3H), 7.09 (d, J = 8.1 Hz, 1H), 7.00 (s, 1H), 4.01 (d, J = 4.1 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 159.09, 148.64, 148.55, 139.79, 138.36, 128.73 (2C), 128.45 (2C), 127.16, 126.18, 123.33, 114.66, 112.13, 40.83.
7-Benzyl-3-hydroxy-1-methylquinazoline-2,4(1H,3H)-dione (18b). According to the above general procedure, 15 was first converted to the boron intermediate 16b. MS (ESI) m/z: 324.8 [M−H]−. The crude 16b was then subjected to the Suzuki coupling reaction with benzyl bromide to afford crude 17b. MS (ESI) m/z: 373.1 [M + H]+. Subsequent debenzylation of 17b by catalytic hydrogenation gave 18b as a white solid (30 mg, 18% over three steps). MS (ESI) m/z: 280.9 [M−H]−. 1H NMR (400 MHz, DMSO-d6) 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.41 (s, 1H), 7.30 (d, J = 3.5 Hz, 4H), 7.21 (p, J = 4.2 Hz, 1H), 7.15 (d, J = 7.9 Hz, 1H), 4.09 (s, 2H), 3.52 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 158.21, 149.24, 148.95, 140.01, 139.42, 128.66 (2C), 128.40 (2C), 127.57, 126.14, 123.38, 114.51, 112.77, 41.11, 30.59.
7-(3,5-Difluorobenzyl)-3-hydroxy-1-methylquinazoline-2,4(1H,3H)-dione (18c). According to the above general procedure, 14b was first converted to the boron intermediate 16a. MS (ESI) m/z: 310.8 [M−H]−. The crude 16a was then subjected to the Suzuki coupling reaction with 1-(bromomethyl)-3,5-difluorobenzene to afford crude 17c. MS (ESI) m/z: 373.1 [M + H]+. Subsequent debenzylation of 17c by catalytic hydrogenation gave 18c as a white solid (30 mg, 18% over three steps). MS (ESI) m/z: 316.8 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.46 (s, 1H), 7.21 (d, J = 7.9 Hz, 1H), 7.14–7.00 (m, 3H), 4.11 (s, 2H), 3.54 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 162.22 (dd, J = 246.0, 13.3 Hz, 2C), 158.18, 149.23, 147.52, 144.63, 139.51, 127.71, 123.32, 114.75, 113.07, 111.88 (d, J = 5.0 Hz), 111.75 (d, J = 4.9 Hz), 101.69 (t, J = 25.7 Hz), 40.41, 30.65.
7-Bromo-3-hydroxyquinazoline-2,4(1H,3H)-dione (19). To compound 14b (200 mg, 0.576 mmol, 1.0 equiv.) was added 48% hydrobromic acid (1.5 mL) and glacial acetic acid (1.5 mL). The solution was refluxed for 2 h. The reaction mixture was cooled to 0 °C under an ice bath and neutralized with 1 mol/L sodium hydroxide aqueous solution. The precipitate was collected by filtration, washed with water, dried, and recrystallized with methanol to obtain 19 as a pale yellow solid (120 mg, 81%). MS (ESI) m/z: 255.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.65 (s, 1H), 7.86 (s, 1H), 7.39 (d, J = 13.7 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 158.78, 148.45, 139.33, 129.04, 127.84, 125.40, 117.51, 113.36.
3.1.5. Synthesis of Compounds 21a–x
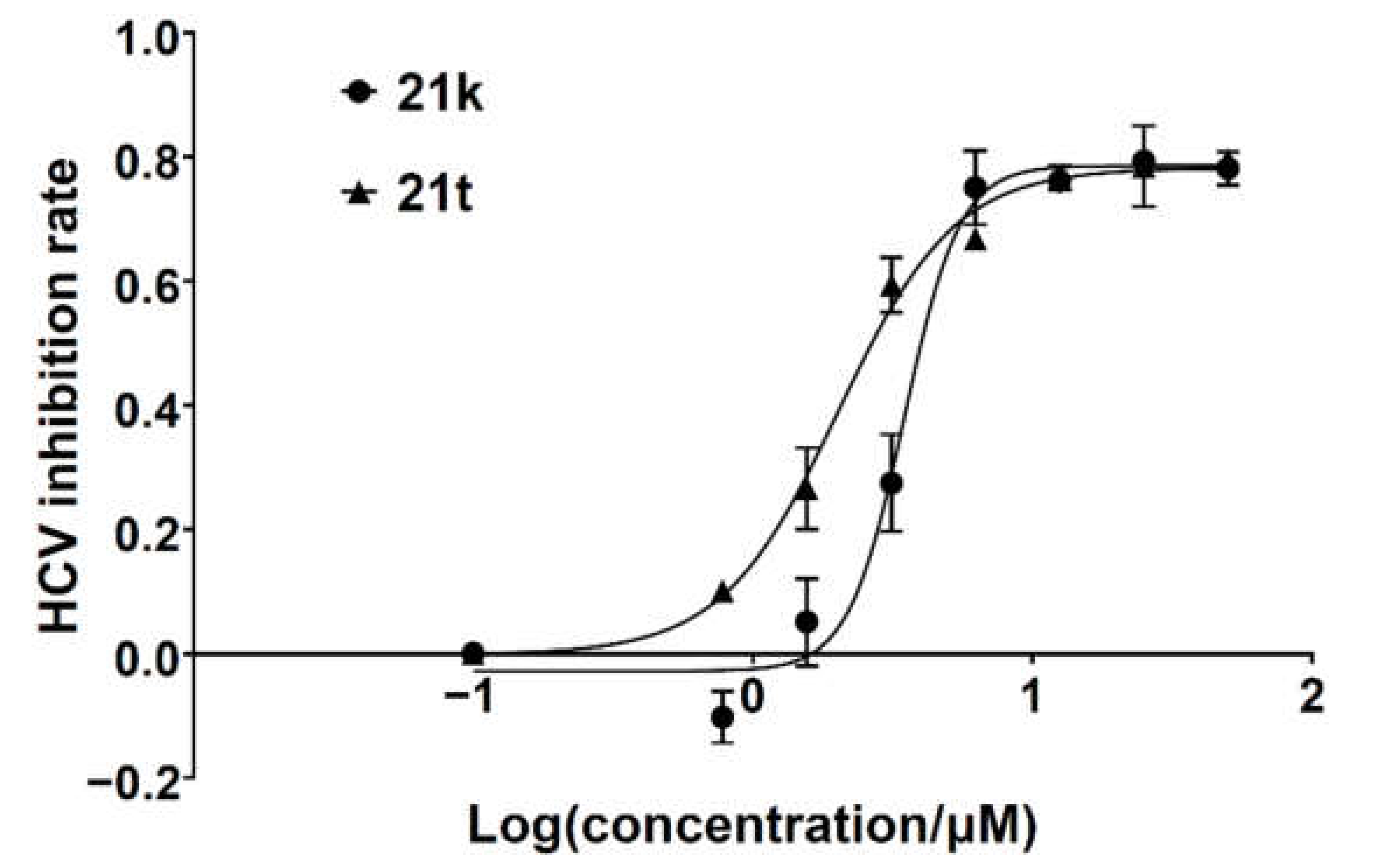
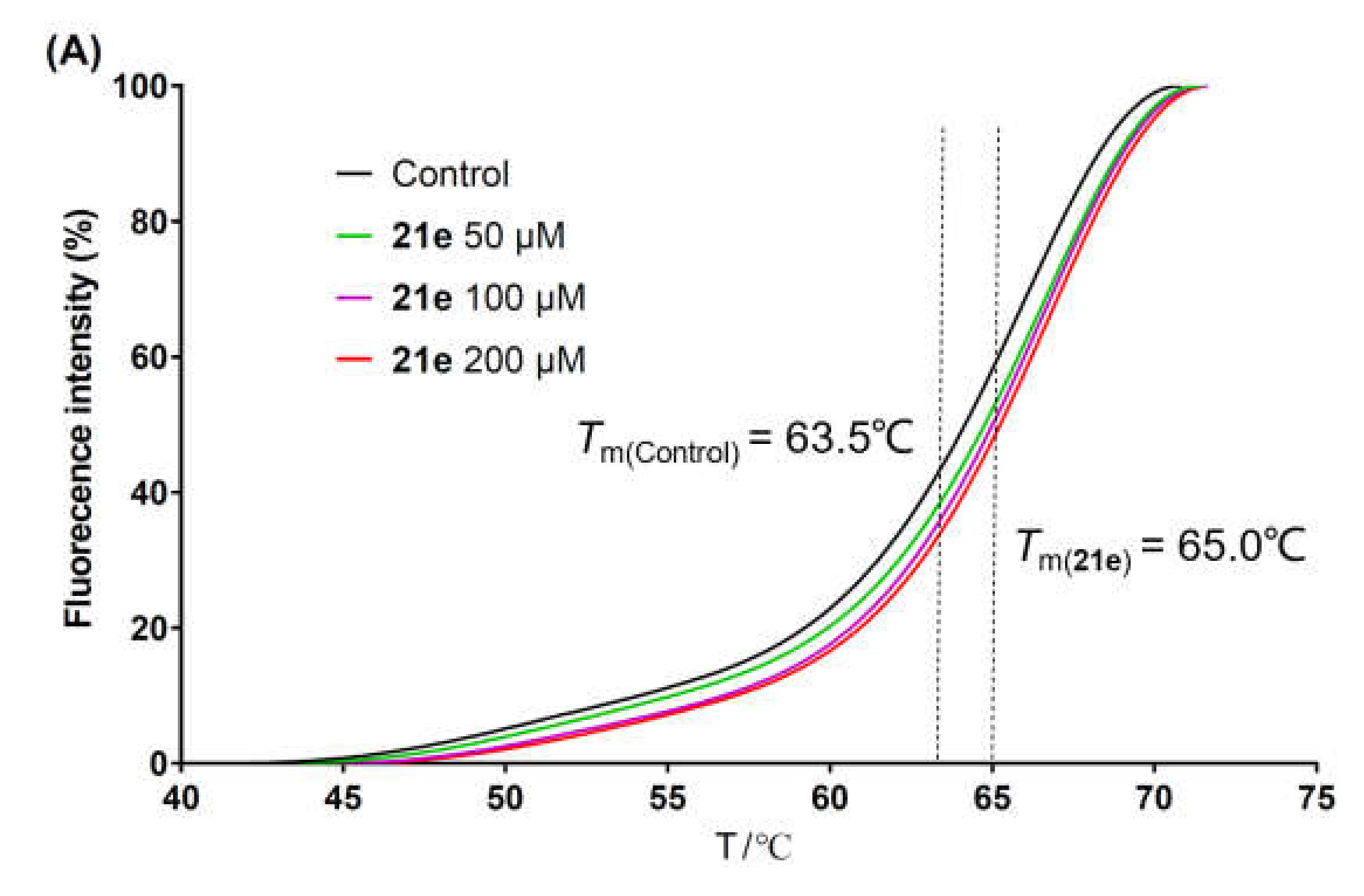
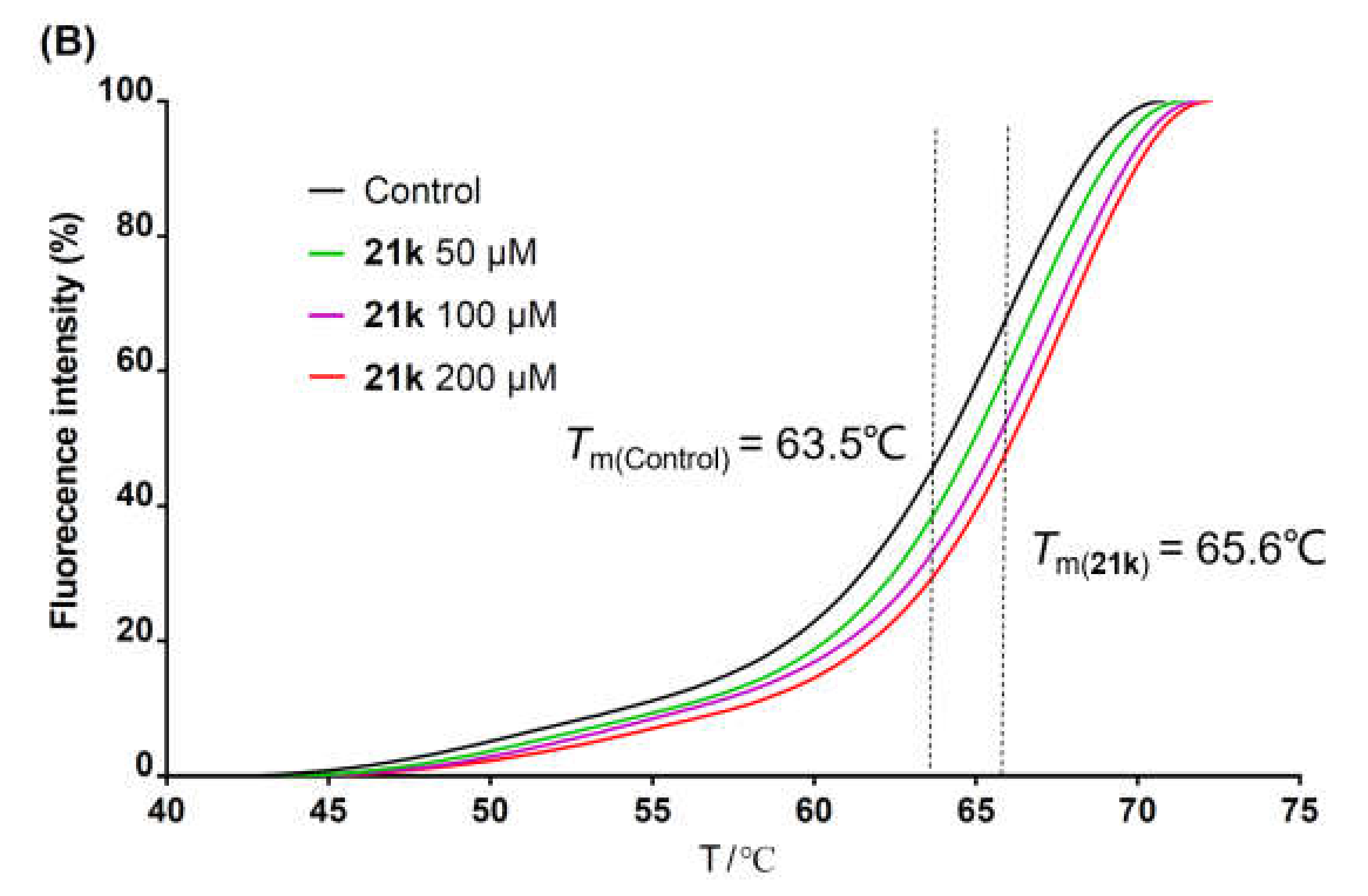
The brominated compound 14a, 14b, or 14c (0.576 mmol, 1.0 equiv.), the corresponding arylboronic acid (0.864 mmol, 1.5 equiv.), Pd(PPh3)4 (0.029 mmol, 0.05 equiv.), K2CO3 (2.88 mmol, 5.0 equiv.), and water (1.5 mL) were suspended in 1,4-dioxane (15 mL). The mixture was heated at 100 °C under nitrogen for 12 h until the complete conversion of 14a–c and then filtrated while hot. The filtrate was poured into water (15 mL), and the precipitate was collected by filtration, washed with water, dried, and re-slurried with methanol or ethyl acetate (5 mL) to give the crude solid 20a–x. Debenzylation reactions of 20a–x were subsequently performed using four different conditions to obtain 21a–x, respectively. For 20a–i, 20m–q, and 20t–x, the same method of catalytic hydrogenation as described in the preparation of 10f–q was used to give 21a–i, 21m–q, and 20t–x, respectively. For 20j–l, the same procedure of debenzylation in the mixture of hydrobromic acid/glacial acetic acid as described in the preparation of 10b–e was conducted to afford 21j–l, respectively. 21r or 21s was obtained from the debenzylation of 20r or 20s using trifluoroacetate (TFA) or TiCl4, respectively.
3-Hydroxy-6-phenylquinazoline-2,4(1H,3H)-dione (21a). According to the above general procedure, 14a was first subjected to the Suzuki coupling reaction with phenylboronic acid to afford crude 20a. MS (ESI) m/z: 343.3 [M−H]−. Subsequent debenzylation of 20a by catalytic hydrogenation gave 21a as a white solid (28 mg, 30% over two steps). MS (ESI) m/z: 253.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 10.63 (s, 1H), 8.15 (s, 1H), 8.00 (d, J = 6.4 Hz, 1H), 7.69 (d, J = 5.4 Hz, 2H), 7.48 (d, J = 6.0 Hz, 2H), 7.38 (t, J = 6.8 Hz, 1H), 7.30 (d, J = 6.9 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 159.24, 148.51, 138.47, 137.53, 134.31, 133.04, 128.95 (2C), 127.44, 126.25 (2C), 124.23, 115.97, 114.43.
6-(4-Fluorophenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21b). According to the above general procedure, 14a was first subjected to the Suzuki coupling reaction with (4-fluorophenyl)boronic acid to afford crude 20b. MS (ESI) m/z: 361.1 [M−H]−. Subsequent debenzylation of 20b by catalytic hydrogenation gave 21b as a white solid (38.9 mg, 29% over two steps). MS (ESI) m/z: 271.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 10.63 (s, 1H), 8.13 (s, 1H), 8.01–7.90 (m, 1H), 7.74 (dd, J = 7.7, 4.3 Hz, 2H), 7.30 (t, J = 6.8 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 161.75 (d, J = 244.8 Hz), 159.19, 148.49, 137.49, 134.98, 133.32, 132.98, 128.35, 128.30, 124.22, 115.97, 115.79, 115.65, 114.42.
3-Hydroxy-6-(4-methoxyphenyl)quinazoline-2,4(1H,3H)-dione (21c). According to the above general procedure, 14a was first subjected to the Suzuki coupling reaction with (4-methoxyphenyl)boronic acid to afford crude 20c. MS (ESI) m/z: 373.1 [M−H]−. Subsequent debenzylation of 20c by catalytic hydrogenation gave 21c as a white solid (25 mg, 18% over two steps). MS (ESI) m/z: 283.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 10.61 (s, 1H), 8.10 (s, 1H), 7.95 (d, J = 8.6 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.6 Hz, 1H), 7.04 (d, J = 8.3 Hz, 2H), 3.81 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 159.27, 158.83, 148.49, 136.96, 134.10, 132.62, 130.83, 127.39 (2C), 123.47, 115.88, 114.39, 114.35 (2C), 55.03.
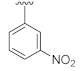
3-Hydroxy-6-(3-nitrophenyl)quinazoline-2,4(1H,3H)-dione (21d). According to the above general procedure, 14a was first subjected to the Suzuki coupling reaction with (3-nitrophenyl)boronic acid to afford crude 20d. MS (ESI) m/z: 388.1 [M−H]−. Subsequent debenzylation of 20d by catalytic hydrogenation gave 21d as a white solid (25 mg, 29% over two steps). MS (ESI) m/z: 298.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.72 (s, 1H), 10.68 (s, 1H), 8.49–8.42 (m, 1H), 8.30–8.16 (m, 3H), 8.12 (dd, J = 8.7, 2.3 Hz, 1H), 7.77 (t, J = 8.0 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 159.15, 148.54, 148.38, 140.12, 138.39, 133.26, 132.91, 131.92, 130.53, 125.05, 122.13, 120.76, 116.26, 114.64.
3-Hydroxy-7-phenylquinazoline-2,4(1H,3H)-dione (21e). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with phenylboronic acid to afford crude 20e. MS (ESI) m/z: 343.1 [M−H]−. Subsequent debenzylation of 20e by catalytic hydrogenation gave 21e as an off-white solid (42 mg, 42% over two steps). MS (ESI) m/z: 253.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 10.63 (s, 1H), 8.02 (d, J = 8.2 Hz, 1H), 7.68 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 3H), 7.48 (d, J = 6.7 Hz, 1H), 7.41 (d, J = 6.2 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 159.14, 148.77, 146.17, 138.75, 138.55, 129.14, 128.69, 127.76, 126.90, 121.23, 113.07, 112.83.
7-(4-Fluorophenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21f). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (4-fluorophenyl)boronic acid to afford crude 20f. MS (ESI) m/z: 361.1 [M−H]−. Subsequent debenzylation of 20f by catalytic hydrogenation gave 21f as a white solid (20 mg, 15% over two steps). MS (ESI) m/z: 271.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 10.61 (s, 1H), 8.01 (d, J = 7.6 Hz, 1H), 7.73 (dd, J = 8.6, 5.0 Hz, 2H), 7.50 (d, J = 8.3 Hz, 1H), 7.36 (d, J = 10.6 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 162.36 (d, J = 246.2 Hz), 158.99, 148.63, 144.96, 138.62, 134.91, 128.97, 128.92, 127.67, 121.06, 115.98, 115.84, 112.92, 112.65.
7-(2-Aminophenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21g). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (2-aminophenyl)boronic acid to afford crude 20g. MS (ESI) m/z: 360.1 [M + H]+. Subsequent debenzylation of 20g by catalytic hydrogenation gave 21g as a white solid (34.5 mg, 37% over two steps). MS (ESI) m/z: 268.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H), 10.65 (s, 1H), 7.98 (d, J = 8.6 Hz, 1H), 7.26 (d, J = 5.8 Hz, 2H), 7.09 (t, J = 7.0 Hz, 1H), 7.01 (d, J = 7.7 Hz, 1H), 6.77 (d, J = 8.2 Hz, 1H), 6.65 (t, J = 7.3 Hz, 1H), 4.97 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 159.18, 148.77, 146.12, 145.13, 138.57, 129.66, 129.03, 127.41, 123.82, 123.22, 116.56, 115.41, 114.99, 112.44.
3-Hydroxy-7-(4-(trifluoromethyl)phenyl)quinazoline-2,4(1H,3H)-dione (21h). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (4-(trifluoromethyl)phenyl)boronic acid to afford crude 20h. MS (ESI) m/z: 413.1 [M + H]+. Subsequent debenzylation of 20h by catalytic hydrogenation gave 21h as a white solid (21 mg, 12% over two steps). MS (ESI) m/z: 321.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.62 (s, 1H), 10.67 (s, 1H), 8.05 (dt, J = 10.8, 5.2 Hz, 1H), 7.88 (d, J = 8.3 Hz, 4H), 7.57 (d, J = 8.7 Hz, 1H), 7.44 (d, J = 10.0 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 158.97, 148.65, 144.41, 142.52, 138.72, 128.78 (d, J = 31.9 Hz), 127.90, 127.76, 125.94 (d, J = 3.9 Hz), 124.03 (d, J = 271.8 Hz), 121.34, 113.75, 113.33.
3-Hydroxy-7-(4-methoxyphenyl)quinazoline-2,4(1H,3H)-dione (21i). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (4-methoxyphenyl)boronic acid to afford crude 20i. MS (ESI) m/z: 373.1 [M−H]−. Subsequent debenzylation of 20i by catalytic hydrogenation gave 21i as a white solid (14.2 mg, 17% over two steps). MS (ESI) m/z: 283.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.56 (s, 1H), 10.57 (s, 1H), 8.07–7.89 (m, 1H), 7.69–7.60 (m, 2H), 7.54–7.45 (m, 1H), 7.36 (d, J = 2.1 Hz, 1H), 7.14–7.05 (m, 2H), 3.82 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 159.82, 159.16, 148.78, 145.80, 138.77, 130.66, 128.11 (2C), 127.68, 120.74, 114.57 (2C), 112.43, 111.96, 55.19.
7-(3-Aminophenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21j). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (3-aminophenyl)boronic acid to afford crude 20j. MS (ESI) m/z: 358.1 [M−H]−. Subsequent debenzylation of 20j in the mixture of hydrobromic acid/glacial acetic acid gave 21j as a white solid (32.4 mg, 42% over two steps). MS (ESI) m/z: 268.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.58 (s, 1H), 7.98 (d, J = 7.5 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.33 (s, 1H), 7.16 (t, J = 7.0 Hz, 1H), 6.86 (s, 1H), 6.79 (d, J = 7.4 Hz, 1H), 6.66 (d, J = 7.8 Hz, 1H), 5.42 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 159.11, 148.85, 148.73, 147.08, 139.23, 138.59, 129.56, 127.53, 121.00, 114.50, 114.34, 112.76, 112.43, 112.08.
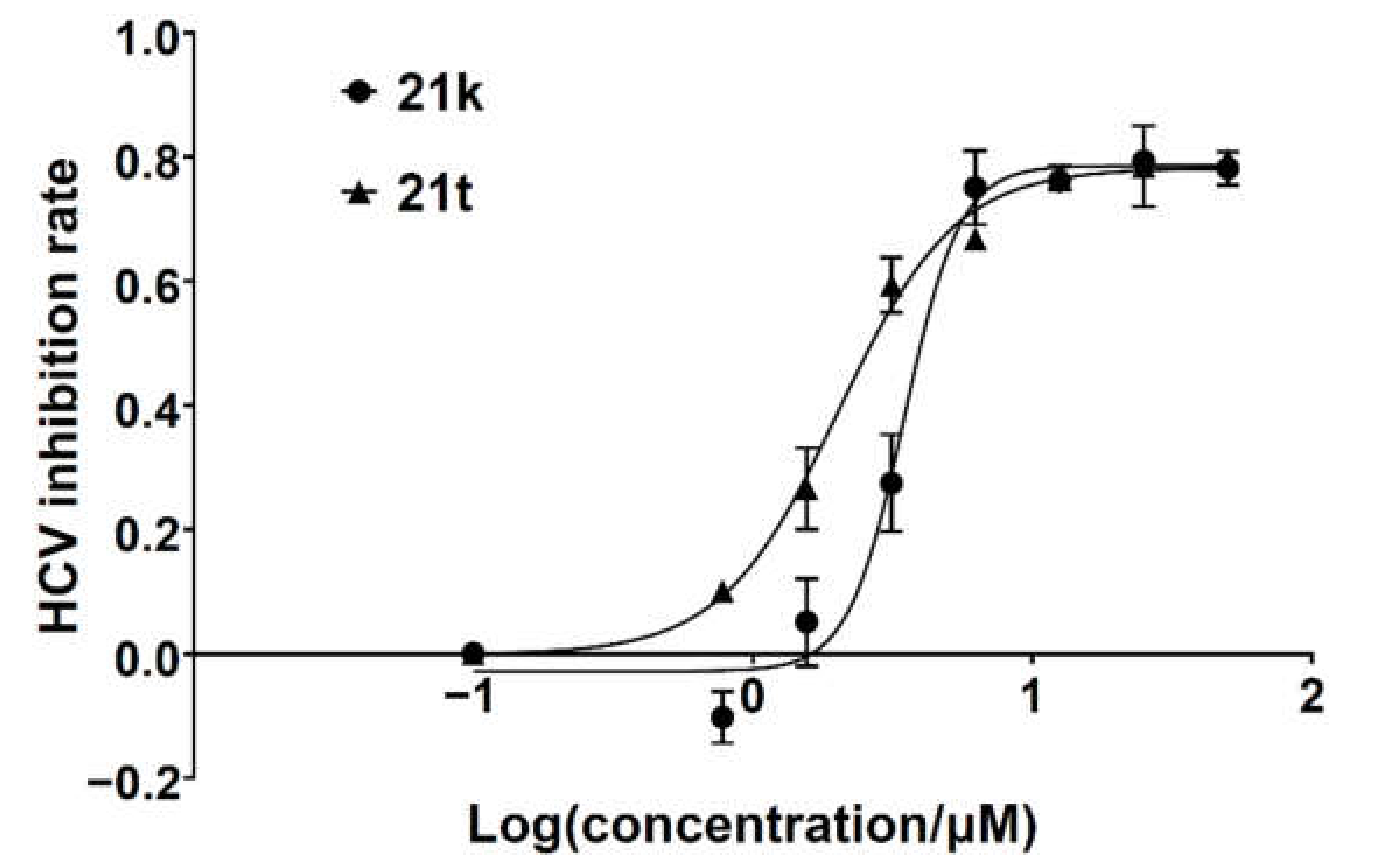
3-Hydroxy-7-(3-nitrophenyl)quinazoline-2,4(1H,3H)-dione (21k). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (3-nitrophenyl)boronic acid to afford crude 20k. MS (ESI) m/z: 388.1 [M−H]−. Subsequent debenzylation of 20k in the mixture of hydrobromic acid/glacial acetic acid gave 21k as a white solid (16 mg, 13% over two steps). MS (ESI) m/z: 298.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.65 (s, 1H), 8.45 (s, 1H), 8.31 (s, 1H), 8.16 (s, 1H), 8.07 (d, J = 8.1 Hz, 1H), 7.84 (s, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.50 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 159.00, 148.67, 148.33, 143.60, 140.06, 138.81, 133.44, 130.78, 128.07, 123.31, 121.41, 121.35, 113.90, 113.37.
7-(3-Chloro-4-fluorophenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21l). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (3-chloro-4-fluorophenyl)boronic acid to afford crude 20l. MS (ESI) m/z: 395.0 [M−H]−. Subsequent debenzylation of 20l in the mixture of hydrobromic acid/glacial acetic acid gave 21l as a white solid (34 mg, 42% over two steps). MS (ESI) m/z: 306.8 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (s, 1H), 7.90 (s, 1H), 7.69 (s, 1H), 7.56 (d, J = 18.8 Hz, 2H), 7.37 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 158.97, 157.37 (d, J = 248.7 Hz), 148.68, 143.61, 138.68, 136.40, 128.97, 128.07–126.56 (m), 121.21, 120.19 (d, J = 17.7 Hz), 117.59, 117.45, 113.40, 113.10.
7-(3,5-Difluorophenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21m). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (3,5-difluorophenyl)boronic acid to afford crude 20m. MS (ESI) m/z: 379.1 [M−H]−. Subsequent debenzylation of 20m by catalytic hydrogenation gave 21m as a white solid (30 mg, 48% over two steps). MS (ESI) m/z: 290.8 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 10.67 (s, 1H), 8.02 (d, J = 8.3 Hz, 1H), 7.61–7.52 (m, 1H), 7.48–7.31 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 162.67 (dd, J = 246.4, 13.4 Hz, 2C), 158.91, 148.62, 143.43, 142.17, 138.62, 127.78, 121.28, 113.88, 113.34, 110.30 (d, J = 5.8 Hz), 110.17 (d, J = 5.7 Hz), 103.91 (t, J = 25.8 Hz).
7-(2,4-Difluorophenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21n). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (2,4-difluorophenyl)boronic acid to afford crude 20n. MS (ESI) m/z: 379.1 [M−H]−. Subsequent debenzylation of 20n by catalytic hydrogenation gave 21n as a white solid (39.5 mg, 62% over two steps). MS (ESI) m/z: 290.8 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.62 (s, 1H), 10.63 (s, 1H), 8.03 (d, J = 7.8 Hz, 1H), 7.64 (q, J = 8.2 Hz, 1H), 7.44 (t, J = 10.3 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.33 (s, 1H), 7.25 (t, J = 8.8 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 163.18–161.23 (m), 158.99 (dd, J = 249.7, 12.7 Hz), 158.96, 148.64, 140.04, 138.33, 134.90–130.51 (m), 127.40, 123.26 (d, J = 12.9 Hz), 122.90, 115.08, 113.37, 112.31 (d, J = 21.2 Hz), 104.69 (t, J = 26.4 Hz).
7-(3-Fluoro-4-methoxyphenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21o). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (3-fluoro-4-methoxyphenyl)boronic acid to afford crude 20o. MS (ESI) m/z: 391.1 [M−H]−. Subsequent debenzylation of 20o by catalytic hydrogenation gave 21o as a white solid (20.6 mg, 32% over two steps). MS (ESI) m/z: 302.8 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.56 (s, 1H), 10.60 (s, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.63–7.54 (m, 1H), 7.54–7.46 (m, 2H), 7.40–7.35 (m, 1H), 7.33 (t, J = 8.4 Hz, 1H), 3.91 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 159.02, 151.56 (d, J = 244.3 Hz), 148.68, 147.54 (d, J = 10.5 Hz), 144.51, 138.67, 131.16 (d, J = 6.2 Hz), 127.64, 123.19, 120.79, 114.26 (d, J = 6.6 Hz), 114.15, 112.79, 112.25, 55.97.
7-(5-Fluoro-2-methylphenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21p). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (5-fluoro-2-methylphenyl)boronic acid to afford crude 20p. MS (ESI) m/z: 375.1 [M−H]−. Subsequent debenzylation of 20p by catalytic hydrogenation gave 21p as a white solid (22.6 mg, 39% over two steps). MS (ESI) m/z: 286.8 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H), 10.62 (s, 1H), 8.00 (dd, J = 8.1, 2.4 Hz, 1H), 7.38 (t, J = 7.0 Hz, 1H), 7.26–7.14 (m, 2H), 7.10 (d, J = 12.7 Hz, 2H), 2.19 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 160.20 (d, J = 242.5 Hz), 159.03, 148.65, 146.07, 141.31 (d, J = 7.7 Hz), 138.15, 132.21 (d, J = 7.9 Hz), 130.66, 127.04, 123.28, 115.51 (d, J = 21.7 Hz), 115.24, 114.68 (d, J = 20.3 Hz), 113.03, 19.05.
3-Hydroxy-7-(3,4,5-trifluorophenyl)quinazoline-2,4(1H,3H)-dione (21q). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with (3,4,5-trifluorophenyl)boronic acid to afford crude 20q. MS (ESI) m/z: 397.1 [M−H]−. Subsequent debenzylation of 20q by catalytic hydrogenation gave 21q as a white solid (30.5 mg, 37% over two steps). MS (ESI) m/z: 307.0 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 10.64 (s, 1H), 8.06–7.97 (m, 1H), 7.68 (td, J = 6.9, 2.8 Hz, 2H), 7.54 (dd, J = 8.4, 1.9 Hz, 1H), 7.36 (d, J = 1.9 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 158.89, 150.47 (d, J = 246.6 Hz), 148.61, 142.81, 138.90 (d, J = 251.3 Hz), 138.58, 135.37, 127.76, 121.25, 113.77, 113.31, 111.86 (d, J = 4.6 Hz), 111.75 (d, J = 4.4 Hz).
3-Hydroxy-7-(pyridin-3-yl)quinazoline-2,4(1H,3H)-dione (21r). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with pyridin-3-ylboronic acid to afford crude 20r. MS (ESI) m/z: 344.1 [M−H]−. To 20r (90 mg, 0.261 mmol, 1.0 equiv.) was added TFA (3 mL) and the suspension was refluxed at 80 °C for 12 h. The reaction mixture was evaporated to dryness and diethyl ether (5 mL) was added. The suspension was well stirred for 15 min and then filtrated. The filtrate was discarded and the residue was washed with diethyl ether (20 mL), dried, and recrystallized with methanol to give the trifluoroacetate 21r as a white solid (20 mg, 10% over two steps). MS (ESI) m/z: 254.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.70 (s, 1H), 8.98 (d, J = 7.9 Hz, 1H), 8.74 (d, J = 6.0 Hz, 1H), 8.28 (d, J = 9.2 Hz, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.70 (d, J = 6.7 Hz, 1H), 7.60 (d, J = 8.6 Hz, 1H), 7.45 (d, J = 7.9 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 163.70, 153.37, 152.43, 150.72 (d, J = 33.6 Hz), 146.95, 143.48, 141.35, 139.63, 132.70, 129.49, 126.06, 118.55, 118.06 (d, J = 4.7 Hz).
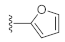
7-(Furan-2-yl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21s). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with furan-2-ylboronic acid to afford crude 20s. MS (ESI) m/z: 333.1 [M−H]−. To a suspension of 20s (100 mg, 0.299 mmol, 1.0 equiv.) in anhydrous DCM (5 mL) was added a solution of TiCl4 (75 μL) in anhydrous DCM (750 μL) dropwise at 0 °C, and the mixture was stirred at 0 °C for 1 h. 1 mol/L potassium sodium tartrate aqueous solution (3 mL) was subsequently added, and the mixture was stirred for 30 min at room temperature. After completion of the reaction, DCM was evaporated, and water (10 mL) was added. The resulting suspension was extracted with ethyl acetate (4 × 10 mL). The combined organic layer was dried by Na2SO4, filtrated, concentrated, and recrystallized in petroleum ether/ethyl acetate to obtain 21s as a pale yellow solid (9.7 mg, 10% over two steps). MS (ESI) m/z: 243.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H), 10.57 (s, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.88 (s, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.46 (s, 1H), 7.16 (d, J = 3.8 Hz, 1H), 6.68 (d, J = 3.8 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 158.98, 151.32, 148.73, 144.57, 138.92, 135.39, 127.79, 117.95, 112.62, 112.51, 109.12, 108.63.
7-(Benzofuran-2-yl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21t). According to the above general procedure, 14b was first subjected to the Suzuki coupling reaction with benzofuran-2-ylboronic acid to afford crude 20t. MS (ESI) m/z: 383.1 [M−H]−. Subsequent debenzylation of 20t by catalytic hydrogenation gave 21t as a white solid (21 mg, 21% over two steps). MS (ESI) m/z: 293.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 11.67 (s, 1H), 10.64 (s, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.73 (d, J = 7.3 Hz, 1H), 7.69 (t, J = 3.6 Hz, 2H), 7.66–7.62 (m, 1H), 7.40 (t, J = 7.3 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 158.93, 154.48, 153.28, 148.71, 138.91, 134.94, 128.34, 127.87, 125.62, 123.51, 121.72, 118.96, 113.72, 111.25, 110.17, 105.10.
3-Hydroxy-8-phenylquinazoline-2,4(1H,3H)-dione (21u). According to the above general procedure, 14c was first subjected to the Suzuki coupling reaction with phenylboronic acid to afford crude 20u. MS (ESI) m/z: 342.8 [M−H]−. Subsequent debenzylation of 20u by catalytic hydrogenation gave 21u as a white solid (17.6 mg, 23% over two steps). MS (ESI) m/z: 253.0 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 10.16 (s, 1H), 8.01 (dd, J = 7.4, 2.2 Hz, 1H), 7.57–7.47 (m, 3H), 7.47–7.38 (m, 3H), 7.31 (t, J = 7.8 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 159.08, 148.36, 136.10, 135.72, 135.28, 129.07 (2C), 128.89, 128.81 (2C), 127.93, 126.41, 122.59, 114.93.
8-(4-Fluorophenyl)-3-hydroxyquinazoline-2,4(1H,3H)-dione (21v). According to the above general procedure, 14c was first subjected to the Suzuki coupling reaction with (4-fluorophenyl)boronic acid to afford crude 20v. MS (ESI) m/z: 361.1 [M−H]−. Subsequent debenzylation of 20v by catalytic hydrogenation gave 21v as a white solid (18 mg, 32% over two steps). MS (ESI) m/z: 270.9 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.62 (d, J = 8.1 Hz, 1H), 10.34 (d, J = 6.9 Hz, 1H), 8.00 (d, J = 8.1 Hz, 1H), 7.55–7.48 (m, 1H), 7.45 (ddd, J = 8.4, 5.7, 2.3 Hz, 2H), 7.36–7.21 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 162.06 (d, J = 244.0 Hz), 159.04, 148.45, 135.73, 135.57, 132.52, 131.36, 131.31, 128.01, 126.52, 122.48, 115.69, 115.55, 114.92.
3-Hydroxy-8-(4-methoxyphenyl)quinazoline-2,4(1H,3H)-dione (21w). According to the above general procedure, 14c was first subjected to the Suzuki coupling reaction with (4-methoxyphenyl)boronic acid to afford crude 20w. MS (ESI) m/z: 373.1 [M−H]−. Subsequent debenzylation of 20w by catalytic hydrogenation gave 21w as a white solid (45.1 mg, 61% over two steps). MS (ESI) m/z: 282.9 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 10.07 (s, 1H), 8.07–7.92 (m, 1H), 7.57–7.47 (m, 1H), 7.35 (d, J = 8.8 Hz, 2H), 7.29 (t, J = 7.8 Hz, 1H), 7.13–7.03 (m, 2H), 3.82 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 159.12, 159.06, 148.33, 135.60, 135.41, 130.30 (2C), 128.70, 128.23, 126.00, 122.55, 114.84, 114.30 (2C), 55.03.
3-Hydroxy-8-(4-(trifluoromethyl)phenyl)quinazoline-2,4(1H,3H)-dione (21x). According to the above general procedure, 14c was first subjected to the Suzuki coupling reaction with (4-(trifluoromethyl)phenyl)boronic acid to afford crude 20x. MS (ESI) m/z: 411.1 [M−H]−. Subsequent debenzylation of 20x by catalytic hydrogenation gave 21x as a white solid (28.5 mg, 20% over two steps). MS (ESI) m/z: 320.8 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 10.58 (s, 1H), 8.13–7.98 (m, 1H), 7.84 (d, J = 7.9 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 7.60–7.51 (m, 1H), 7.34 (dd, J = 9.4, 6.4 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 158.97, 148.53, 140.51, 135.67, 135.50, 130.16, 128.33 (d, J = 31.5 Hz), 127.57, 127.10, 125.59 (d, J = 4.1 Hz), 124.26 (d, J = 272.0 Hz), 122.54, 115.07.
3.1.6. Synthesis of Compounds 23a–j
To compound 20a, 20d–f, 20i or 20s (0.436 mmol, 1.0 equiv.) was added K2CO3 (0.872 mmol, 2.0 equiv.), the corresponding halide (0.872 mmol, 2.0 equiv.), and DMF (4 mL). The suspension was heated at 80 °C for 2 h or stirred at room temperature for 2 h when the halide was iodomethane. After cooling, the reaction was quenched with water (10 mL), and the precipitate was collected by filtration, washed with water, and dried to give the crude product 22a–j. Debenzylation reactions of 22a–j were subsequently conducted using three different conditions to obtain 23a–j, respectively. For 22a, the same method of debenzylation using TFA as described in the preparation of 21r was performed to give 23a. For 22b or 22c, the same procedure of debenzylation in the mixture of hydrobromic acid/glacial acetic acid as described in the preparation of 10b–e was conducted to afford 23b or 23c, respectively. For 22d–j, the method of catalytic hydrogenation described in the preparation of 10f–q was used to give 23d–j, respectively.
3-Hydroxy-1-methyl-6-(3-nitrophenyl)quinazoline-2,4(1H,3H)-dione (23a). According to the above general procedure, 20d was subjected to the alkylation reaction with iodomethane to afford crude 22a. MS (ESI) m/z: 404.1 [M + H]+. Subsequent debenzylation of 22a in TFA gave 23a as a grey solid (53.4 mg, 56% over two steps). MS (ESI) m/z: 312.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.61 (s, 1H), 8.31 (d, J = 8.2 Hz, 2H), 8.16 (d, J = 8.1 Hz, 1H), 7.90–7.80 (m, 1H), 7.76 (s, 1H), 7.71 (d, J = 8.0 Hz, 1H), 3.66 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 158.06, 149.24, 148.30, 143.98, 140.26, 139.81, 133.95, 130.45, 128.24, 123.21, 121.87, 121.60, 114.47, 113.30, 30.80.
3-Hydroxy-1-methyl-7-phenylquinazoline-2,4(1H,3H)-dione (23b). According to the above general procedure, 20e was subjected to the alkylation reaction with iodomethane to afford crude 22b. MS (ESI) m/z: 359.1 [M + H]+. Subsequent debenzylation of 22b in the mixture of hydrobromic acid/glacial acetic acid gave 23b as a grey solid (209 mg, 73% over two steps). MS (ESI) m/z: 267.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.12 (d, J = 8.1 Hz, 1H), 7.83 (d, J = 7.4 Hz, 2H), 7.61 (d, J = 7.6 Hz, 2H), 7.54 (t, J = 7.4 Hz, 2H), 7.47 (t, J = 7.2 Hz, 1H), 3.64 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 158.19, 149.30, 146.46, 139.73, 138.65, 128.93 (2C), 128.63, 128.01, 127.27 (2C), 121.36, 113.65, 112.61, 30.65.
7-(4-Fluorophenyl)-3-hydroxy-1-methylquinazoline-2,4(1H,3H)-dione (23c). According to the above general procedure, 20f was subjected to the alkylation reaction with iodomethane to afford crude 22c. MS (ESI) m/z: 377.1 [M + H]+. Subsequent debenzylation of 22c in the mixture of hydrobromic acid/glacial acetic acid gave 23c as a white solid (30.8 mg, 45% over two steps). MS (ESI) m/z: 285.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.98–7.85 (m, 2H), 7.60 (d, J = 9.4 Hz, 2H), 7.37 (t, J = 8.4 Hz, 2H), 3.64 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 163.36, 161.73, 158.21, 149.35, 145.39, 139.79, 135.15, 129.56, 129.50, 128.09, 121.34, 115.91, 115.76, 113.68, 112.64, 30.73.
3-Hydroxy-7-(4-methoxyphenyl)-1-methylquinazoline-2,4(1H,3H)-dione (23d). According to the above general procedure, 20i was subjected to the alkylation reaction with iodomethane to afford crude 22d. MS (ESI) m/z: 389.2 [M + H]+. Subsequent debenzylation of 22d by catalytic hydrogenation gave 23d as a white solid (20 mg, 19% over two steps). MS (ESI) m/z: 297.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 8.08 (s, 1H), 7.81 (s, 2H), 7.58 (s, 2H), 7.09 (s, 2H), 3.83 (s, 3H), 3.64 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 159.86, 158.27, 149.39, 146.15, 139.80, 130.86, 128.58 (2C), 128.00, 120.89, 114.40 (2C), 113.06, 111.80, 55.21, 39.94, 39.81, 39.68, 39.54, 39.40, 39.26, 39.12, 38.98, 30.68.
1-Ethyl-3-hydroxy-7-phenylquinazoline-2,4(1H,3H)-dione (23e). According to the above general procedure, 20e was subjected to the alkylation reaction with bromoethane to afford crude 22e. MS (ESI) m/z: 373.2 [M + H]+. Subsequent debenzylation of 22e by catalytic hydrogenation gave 23e as a white solid (6.8 mg, 8% over two steps). MS (ESI) m/z: 281.2 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.14 (d, J = 8.7 Hz, 1H), 7.82 (d, J = 7.2 Hz, 2H), 7.56 (td, J = 30.5, 26.6, 9.5 Hz, 5H), 4.40–4.18 (m, 2H), 1.28 (dt, J = 11.9, 5.8 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 158.11, 148.86, 146.71, 138.68, 138.62, 128.93 (2C), 128.62, 128.40, 127.36 (2C), 121.43, 113.92, 112.13, 38.24, 12.40.
3-Hydroxy-6-phenyl-1-propylquinazoline-2,4(1H,3H)-dione (23f). According to the above general procedure, 20a was subjected to the alkylation reaction with 1-bromopropane to afford crude 22f. MS (ESI) m/z: 387.0 [M + H]+. Subsequent debenzylation of 22f by catalytic hydrogenation gave 23f as a white solid (22.3 mg, 41% over two steps). MS (ESI) m/z: 295.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.28 (s, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 7.2 Hz, 1H), 7.51 (t, J = 7.3 Hz, 2H), 7.41 (d, J = 7.1 Hz, 1H), 4.12 (t, J = 7.5 Hz, 2H), 1.69 (q, J = 7.6 Hz, 2H), 0.97 (t, J = 6.8 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 158.26, 148.91, 138.08, 137.74, 134.42, 133.08, 128.99 (2C), 127.59, 126.30 (2C), 124.87, 115.50, 115.36, 44.60, 20.14, 10.70.
3-Hydroxy-7-phenyl-1-propylquinazoline-2,4(1H,3H)-dione (23g). According to the above general procedure, 20e was subjected to the alkylation reaction with 1-bromopropane to afford crude 22g. MS (ESI) m/z: 387.2 [M + H]+. Subsequent debenzylation of 22g by catalytic hydrogenation gave 23g as a white solid (25 mg, 25% over two steps). MS (ESI) m/z: 295.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.19–8.10 (m, 1H), 7.80 (d, J = 4.7 Hz, 2H), 7.66–7.57 (m, 2H), 7.57–7.51 (m, 2H), 7.52–7.43 (m, 1H), 4.23 (t, J = 8.0 Hz, 2H), 1.78–1.61 (m, 2H), 0.96 (t, J = 6.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 158.08, 149.18, 146.65, 138.90, 138.71, 128.96 (2C), 128.60, 128.35, 127.33 (2C), 121.48, 113.85, 112.35, 44.32, 20.16, 10.82.
1-Benzyl-3-hydroxy-6-phenylquinazoline-2,4(1H,3H)-dione (23h). According to the above general procedure, 20a was subjected to the alkylation reaction with benzyl bromide to afford crude 22h. MS (ESI) m/z: 435.0 [M + H]+. Subsequent debenzylation of 22h by catalytic hydrogenation gave 23h as a white solid (20 mg, 33% over two steps). MS (ESI) m/z: 343.1 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.92 (s, 1H), 8.31 (d, J = 8.6 Hz, 1H), 7.99 (d, J = 9.1 Hz, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.48 (t, J = 7.6 Hz, 2H), 7.45–7.22 (m, 7H), 5.44 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 158.41, 149.61, 138.05, 137.87, 136.00, 134.78, 133.09, 129.04 (2C), 128.66 (2C), 127.69, 127.25, 126.38 (2C), 126.32 (2C), 124.93, 115.90, 115.62, 46.40.
1-Benzyl-3-hydroxy-7-phenylquinazoline-2,4(1H,3H)-dione (23i). According to the above general procedure, 20e was subjected to the alkylation reaction with benzyl bromide to afford crude 22i. MS (ESI) m/z: 435.1 [M + H]+. Subsequent debenzylation of 22i by catalytic hydrogenation gave 23i as a white solid (26 mg, 29% over two steps). MS (ESI) m/z: 345.2 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (d, J = 20.9 Hz, 1H), 8.24–8.10 (m, 1H), 7.74–7.20 (m, 12H), 5.57 (d, J = 21.3 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 158.23, 149.87, 146.26, 138.95, 138.49, 136.27, 129.03 (2C), 128.72 (2C), 128.67, 128.39, 127.25, 127.07 (2C), 126.42 (2C), 121.61, 114.09, 112.96, 46.18.
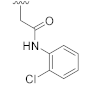
N-(2-Chlorophenyl)-2-(7-(furan-2-yl)-3-hydroxy-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (23j). According to the above general procedure, 20s was subjected to the alkylation reaction with 2-chloro-N-(2-chlorophenyl)acetamide to afford crude 22j. MS (ESI) m/z: 502.1 [M + H]+. Subsequent debenzylation of 22j by catalytic hydrogenation gave 23j as a white solid (26 mg, 13% over two steps). MS (ESI) m/z: 410.0 [M−H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 10.10 (s, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.88 (s, 1H), 7.67 (d, J = 8.2 Hz, 1H), 7.65–7.56 (m, 2H), 7.51 (d, J = 7.7 Hz, 1H), 7.32 (t, J = 6.5 Hz, 1H), 7.27 (s, 1H), 7.24 (d, J = 10.8 Hz, 1H), 6.70 (s, 1H), 5.17 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 165.98, 158.16, 151.37, 149.59, 139.66, 135.80, 134.22, 129.52, 128.45, 127.47, 127.22, 126.89, 126.46, 118.21, 113.40, 112.63, 112.54, 109.56, 108.37, 46.22.


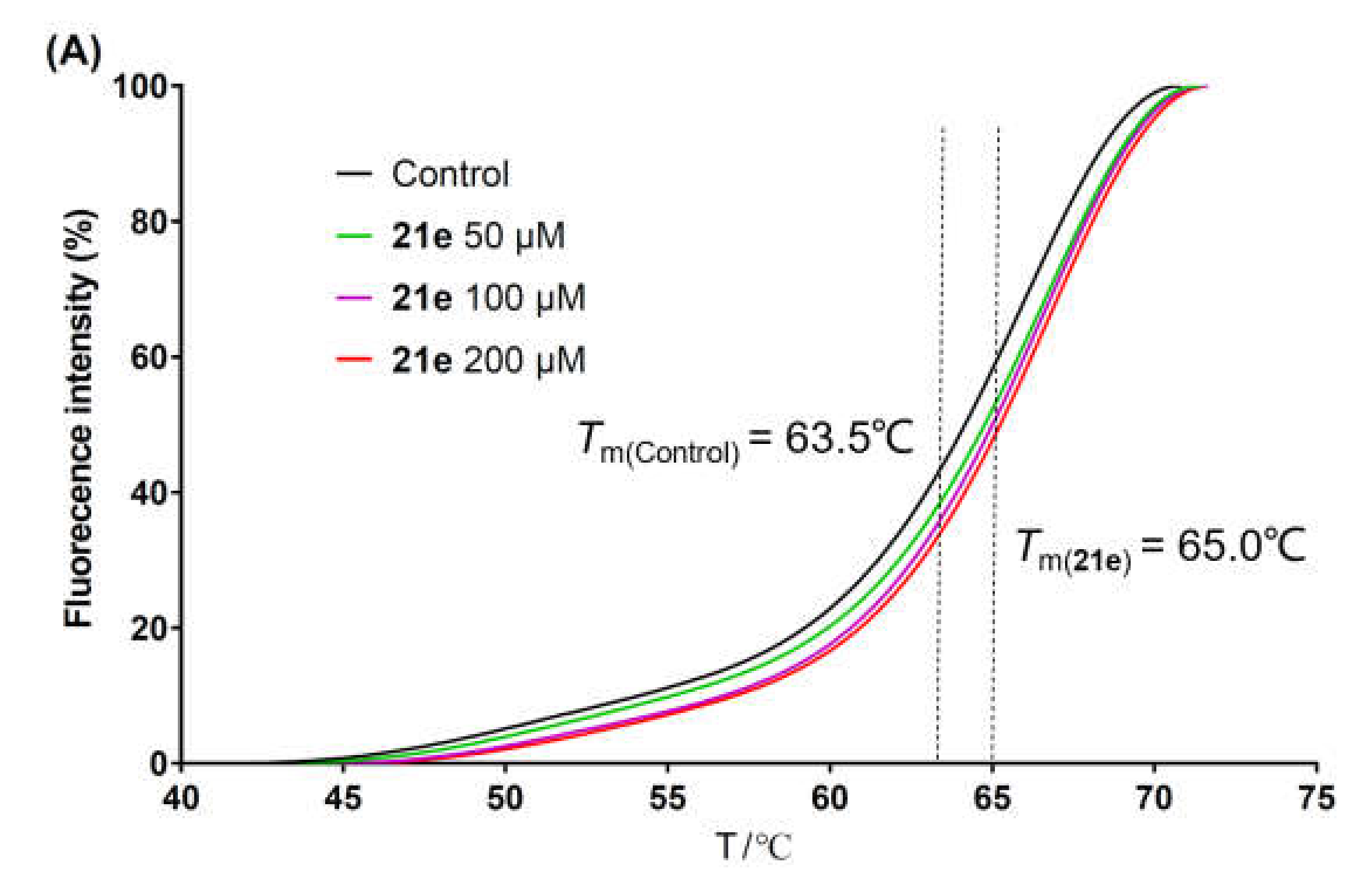
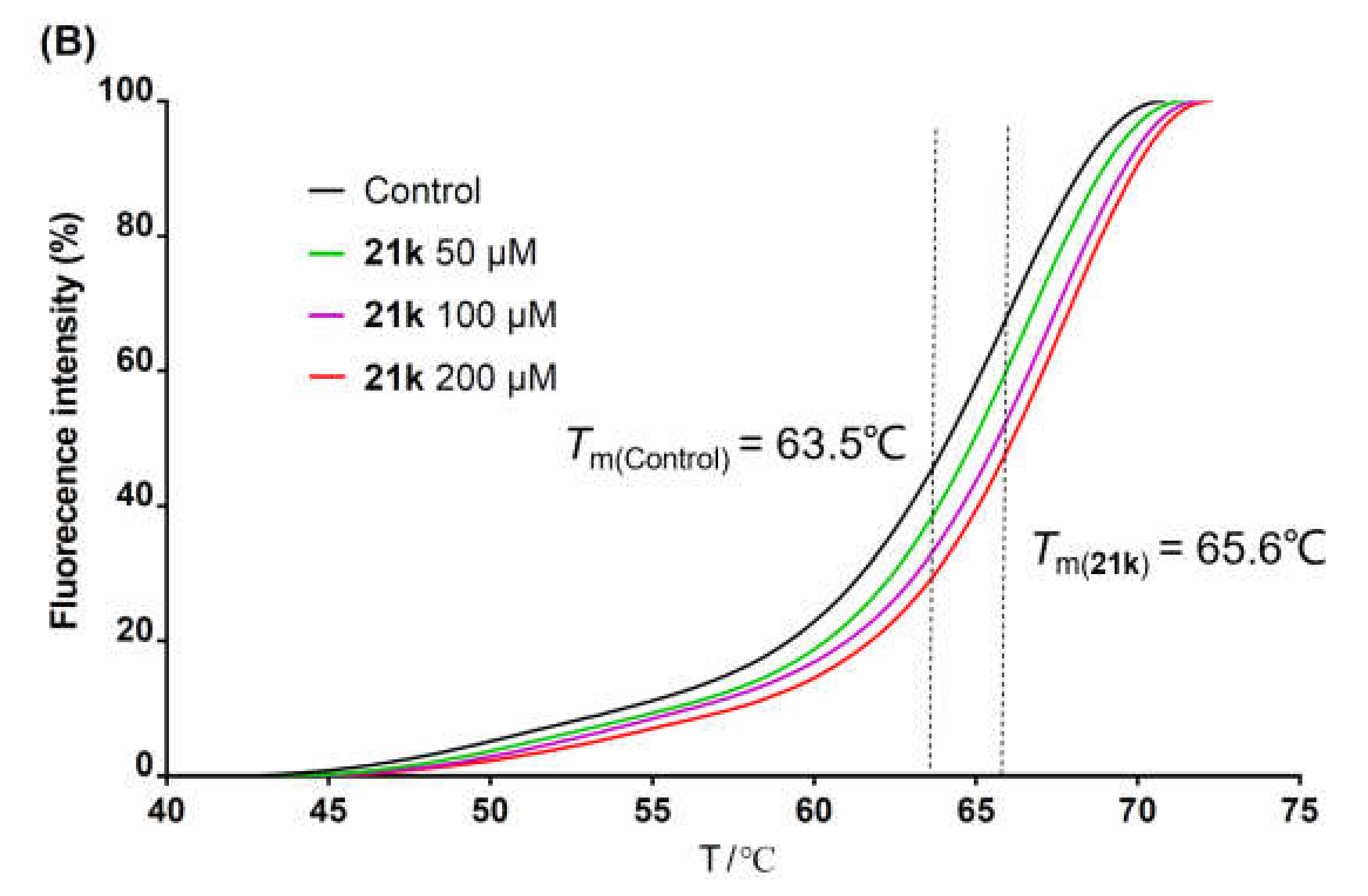
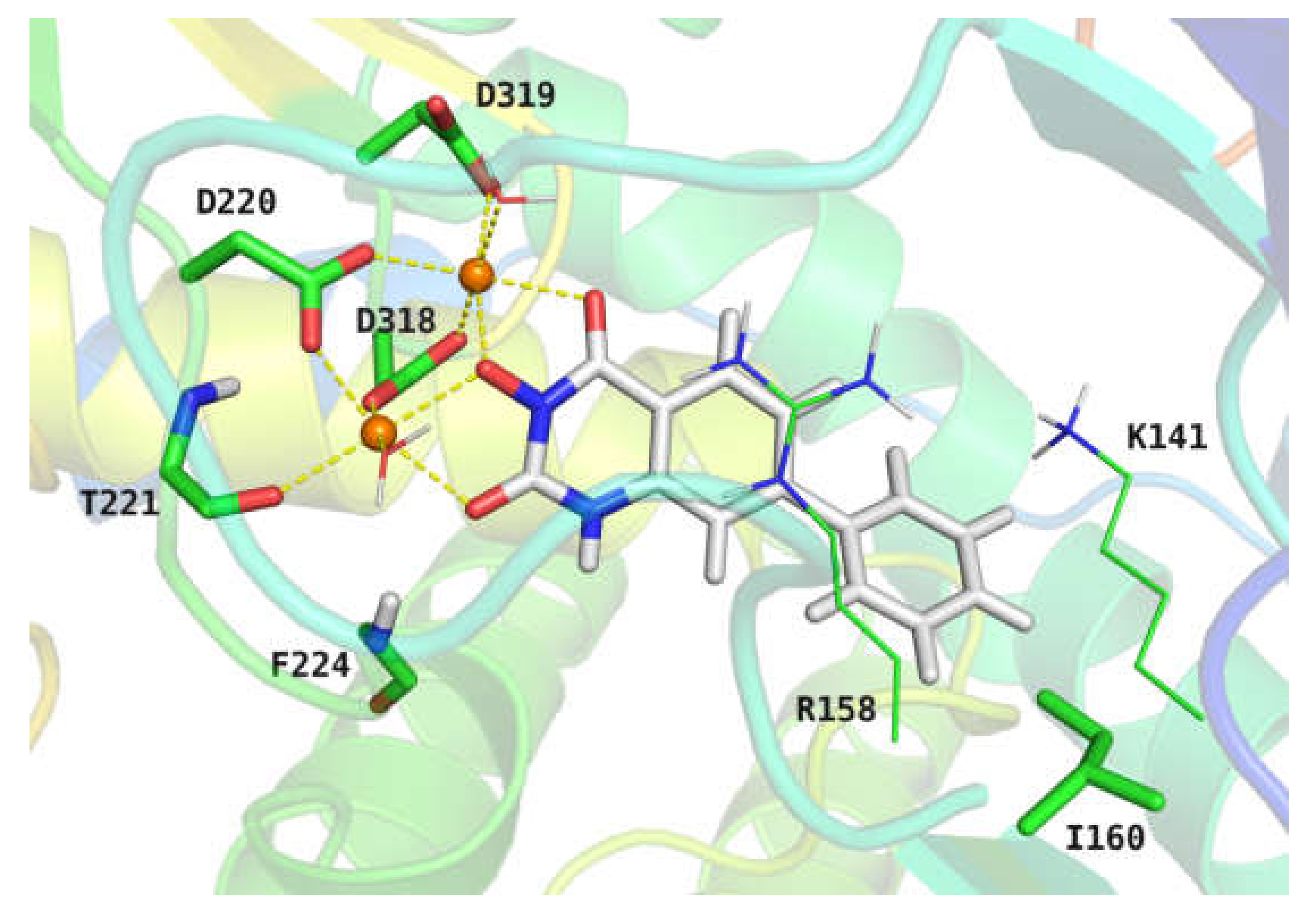

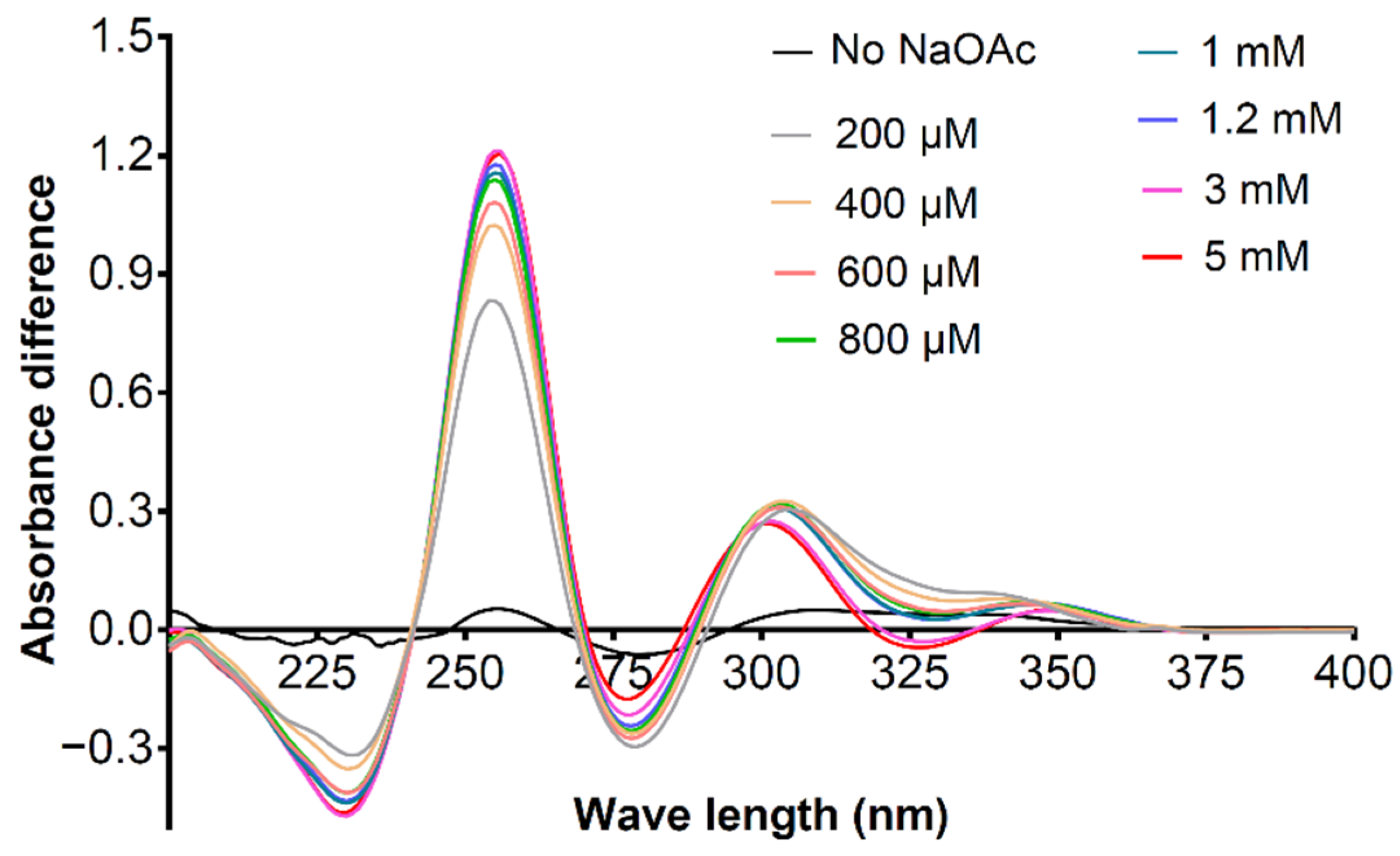

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}