
Dynamic Changes in Ascorbic Acid Content during Fruit Development and Ripening of Actinidia latifolia (an Ascorbate-Rich Fruit Crop) and the Associated Molecular Mechanisms


Abstract
:1. Introduction
2. Results
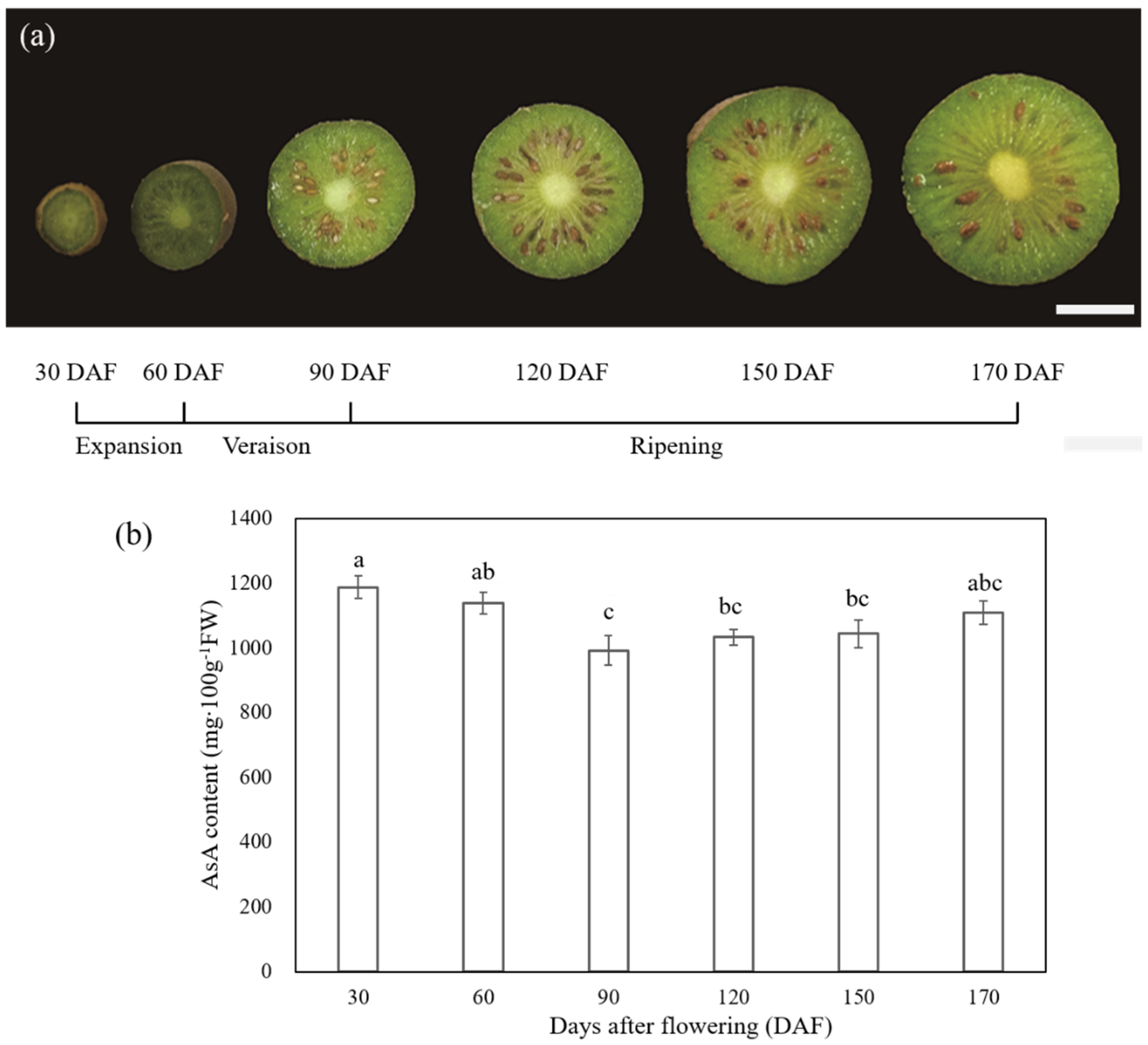
2.1. Morphological Changes and AsA Accumulation at Six Different Stages of the Fruit Development and Ripening of A. latifolia
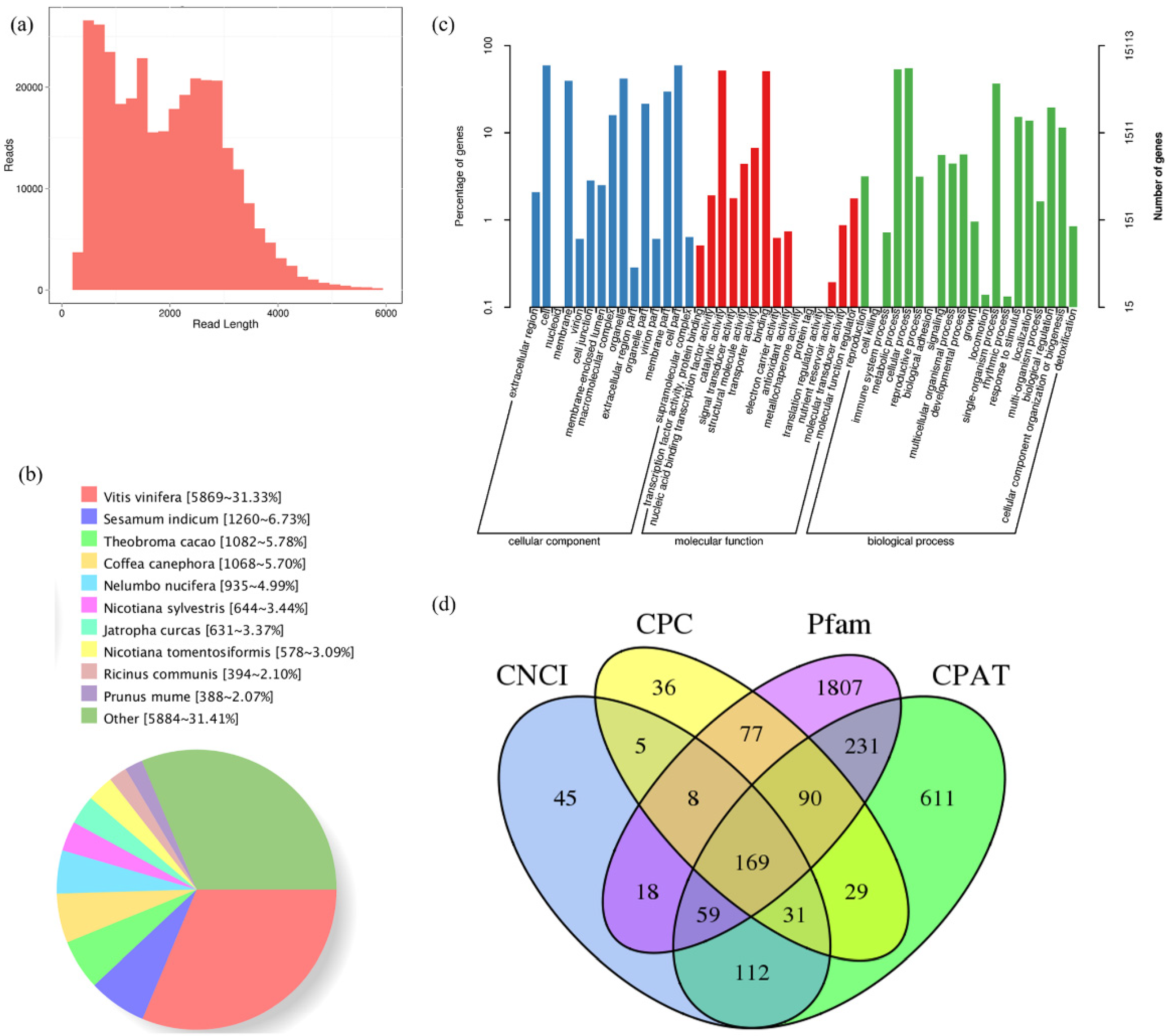
2.2. PacBio SMRT Sequencing-Based FL Transcriptome Atlas of A. latifolia Fruit
2.3. Functional Annotation of A. latifolia Transcripts with Multiple Databases
2.4. Structural Analysis of the FL Transcriptome of A. latifolia
2.5. Differentially Expressed Genes (DEGs) Identified in Comparative Transcriptomic Analysis
2.6. Functional Annotation and Categorization of DEGs
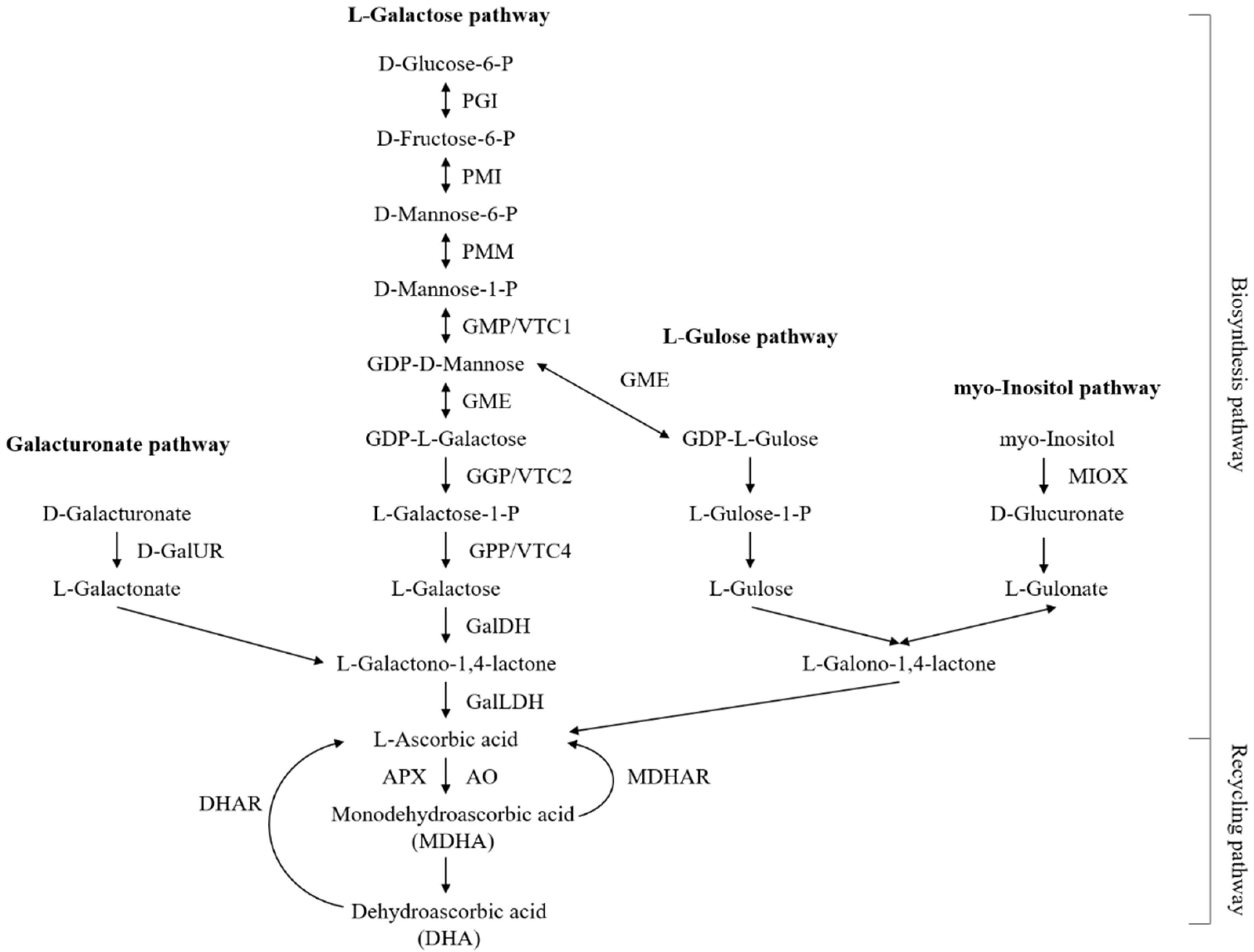
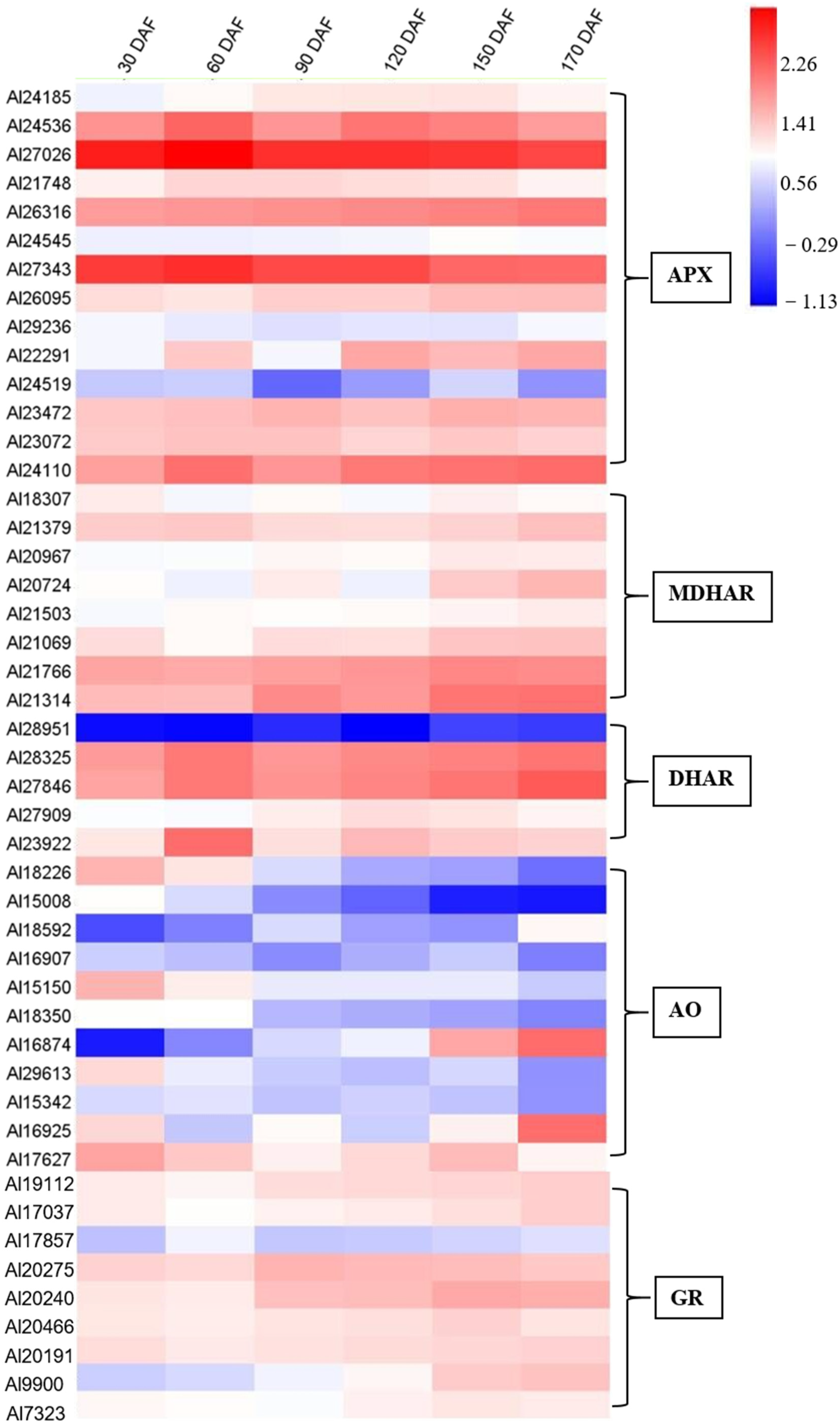
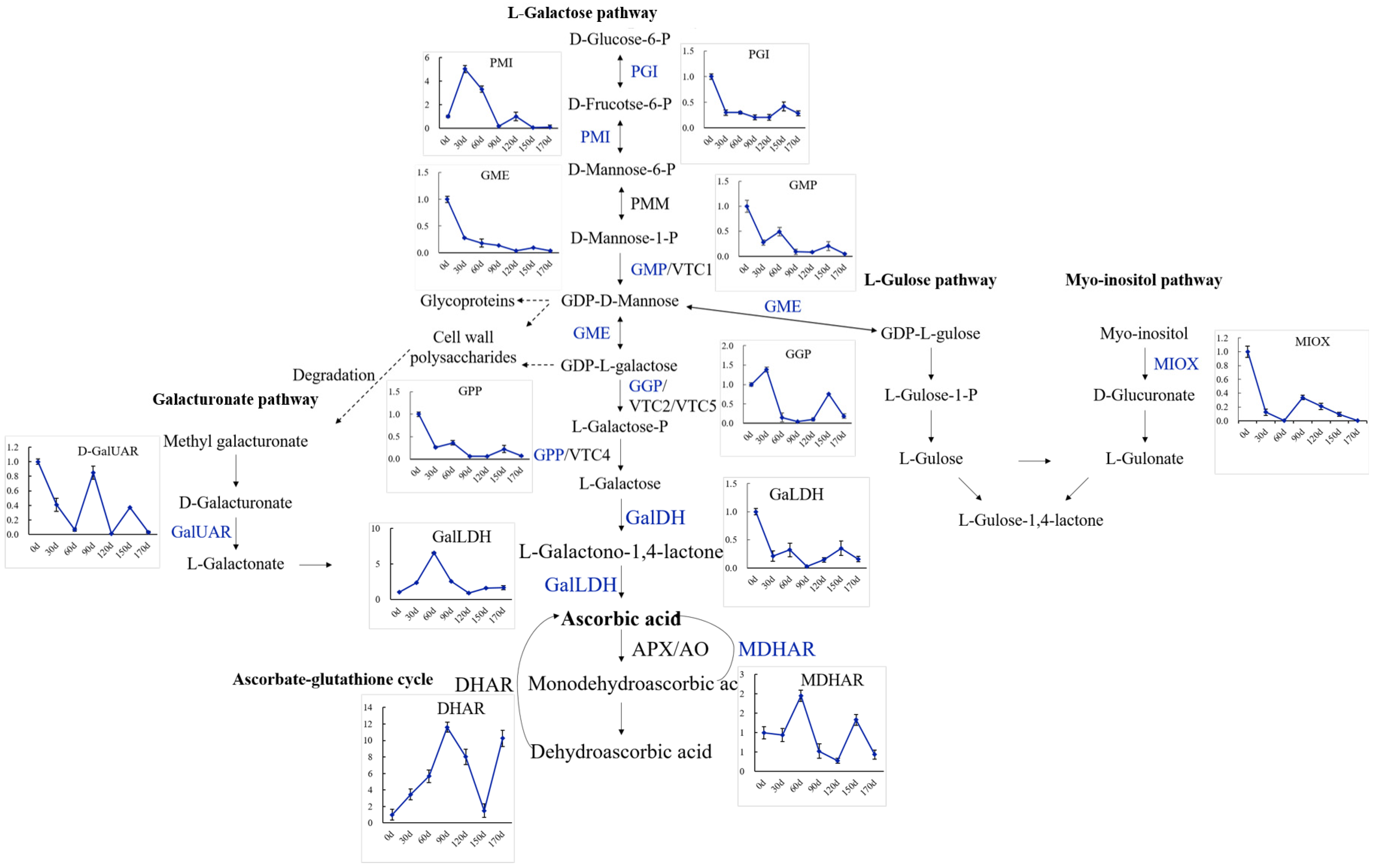
2.7. Identification of Genes in AsA Biosynthesis and Recycling Pathway during the Fruit Development and Ripening of A. latifolia
2.8. LncRNAs Participated in AsA-Related Gene Expression Regulation
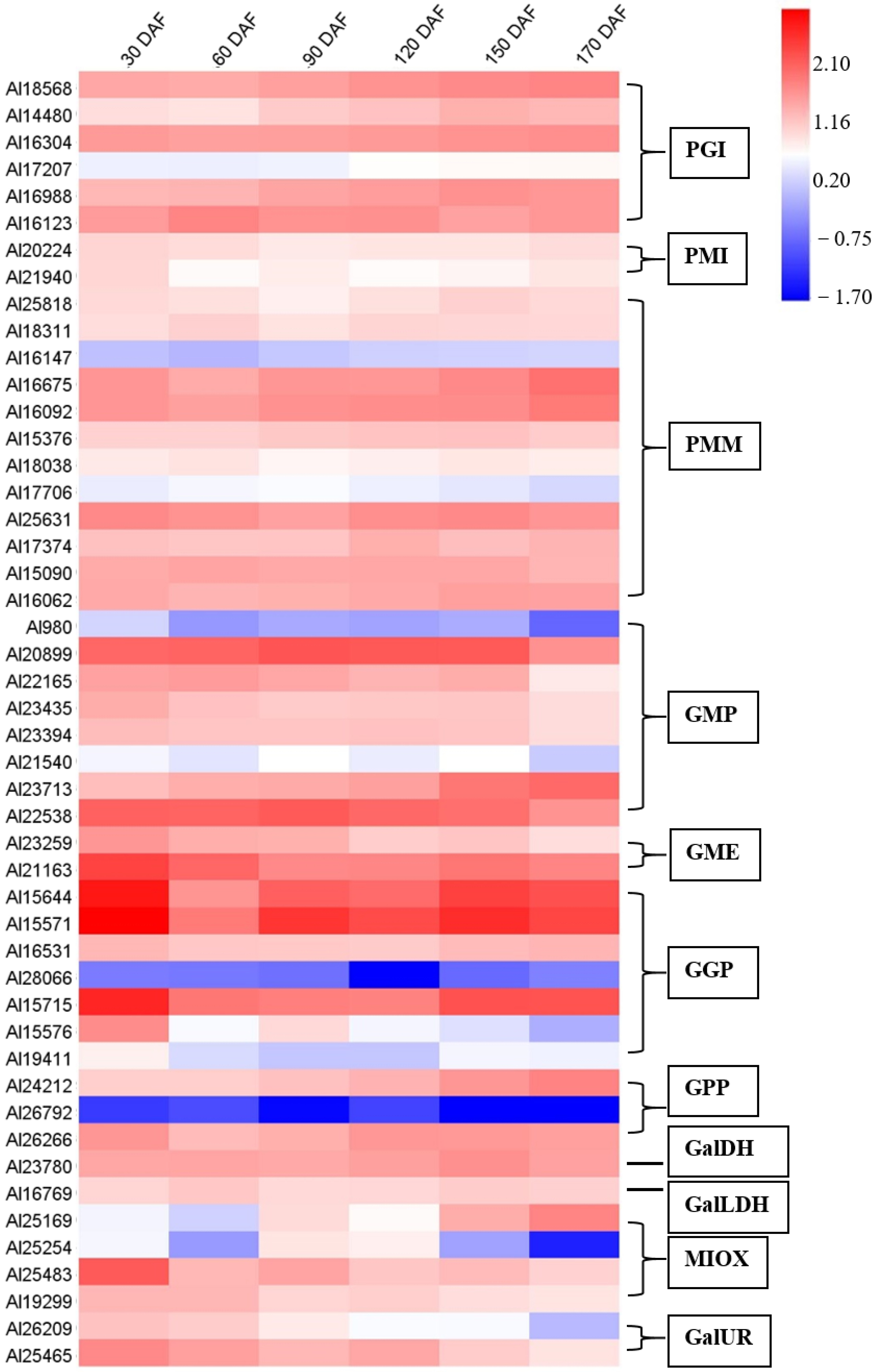
2.9. Expression Patterns of Genes Involved in AsA Biosynthesis and Recycling during Fruit Development and Ripening of A. latifolia Verified by qRT-PCR Analysis
3. Discussion
3.1. The First High-Quality, Functionally Annotated Reference Transcriptome for A. latifolia
3.2. Dynamics of AsA Content Accompanying Fruit Development and Ripening of A. latifolia
3.3. An Elucidation of the Molecular Mechanisms Regulating AsA Accumulation of A. latifolia
4. Materials and Methods
4.1. Plant Materials and Sampling
4.2. Chemicals and Solvents
4.3. Determination of AsA Using HPLC Coupled with UV Detection
4.4. RNA Extraction and Quality Evaluation
4.5. Construction of Iso-seq cDNA Libraries and PacBio Sequencing
4.6. Reads Processing and Error Collection of PacBio Iso-seq Reads
4.7. Gene Functional Annotation
4.8. Transcript Structure Analysis
4.9. Illumina cDNA Library Construction and Second-Generation Sequencing for Transcriptome of Fruit Development and Ripening Stages
4.10. Validation of DEGs by Quantitative Reverse Transcription PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mellidou, I.; Kanellis, A.K. Genetic control of ascorbic acid biosynthesis and recycling in horticultural crops. Front. Chem. 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallie, D.R. L-Ascorbic Acid: A Multifunctional molecule supporting plant growth and development. Science 2013, 2013, 795964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akram, N.A.; Shafiq, F.; Ashraf, M. Ascorbic acid-a potential oxidant scavenger and its role in plant development and abiotic stress tolerance. Front. Plant Sci. 2017, 8, 613. [Google Scholar] [CrossRef] [PubMed]
- Granger, M.; Eck, P. Dietary vitamin C in human health. Adv. Food Nutr. Res. 2018, 83, 281–310. [Google Scholar] [PubMed]
- Miranda-Massari, J.R.; González, M.J.; Marcial-Vega, V.A.; Soler, J.D. A possible role for ascorbic acid in COVID-19. J. Restor. Med. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Bvenura, C.; Sivakumar, D. The role of wild fruits and vegetables in delivering a balanced and healthy diet. Food Res. Int. 2017, 99, 15–30. [Google Scholar] [CrossRef]
- Wallace, T.C.; Bailey, R.L.; Blumberg, J.B.; Burton-Freeman, B.; Chen, C.O.; Crowe-White, K.M.; Drewnowski, A.; Hooshmand, S.; Johnson, E.; Lewis, R.; et al. Fruits, vegetables, and health: A comprehensive narrative, umbrella review of the science and recommendations for enhanced public policy to improve intake. Crit. Rev. Food Sci. Nutr. 2020, 60, 2174–2211. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Ding, J.; Deng, D.; Tang, W.; Sun, H.; Liu, D.; Zhang, L.; Niu, X.; Zhang, X.; Meng, M.; et al. Draft genome of the kiwifruit Actinidia chinensis. Nat. Commun. 2013, 4, 2640. [Google Scholar] [CrossRef]
- Liang, D.; Deng, H.; Deng, Q.; Lin, L.; Lv, X.; Wang, J.; Wang, Z.; Xiong, B.; Zhao, X.; Xia, H. Dynamic changes of phenolic compounds and their associated gene expression profiles occurring during fruit development and ripening of the donghong kiwifruit. J. Agric. Food Chem. 2020, 68, 11421–11433. [Google Scholar] [CrossRef]
- Xia, H.; Wang, X.; Su, W.; Jiang, L.; Lin, L.; Deng, Q.; Wang, J.; Deng, H.; Hu, R.; Liao, M.; et al. Changes in the carotenoids profile of two yellow-fleshed kiwifruit cultivars during storage. Postharvest Biol. Technol. 2020, 164, 111162. [Google Scholar] [CrossRef]
- Vissers, M.C.M.; Carr, A.C.; Pullar, J.M.; Bozonet, S.M. The bioavailability of vitamin C from kiwifruit. Adv. Food Nutr. Res. 2013, 68, 125–147. [Google Scholar] [PubMed]
- Ferguson, A.R. Botanical description. In The Kiwifruit Genome, Compendium of Plant Genomes; Testolin, R., Huang, H., Ferguson, A.R., Eds.; Springer: Cham, Switzerland, 2016; pp. 1–14. [Google Scholar]
- Du, G.; Li, M.; Ma, F.; Liang, D. Antioxidant capacity and the relationship with polyphenol and vitamin C in Actinidia fruits. Food Chem. 2009, 113, 557–562. [Google Scholar] [CrossRef]
- Rassam, M.; Laing, W. Variation in ascorbic acid and oxalate levels in the fruit of Actinidia chinensis tissues and genotypes. J. Agric. Food Chem. 2005, 53, 2322–2326. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, I.; Yamashita, Y.; Yamanaka, M.; Shimohashi, A.; Fukuda, T.; Oota, T. Varietal difference in vitamin C content in the fruit of kiwifruit and other Actinidia species. J. Agric. Food Chem. 2004, 52, 5472–5475. [Google Scholar] [CrossRef] [PubMed]
- Latocha, P.; Krupa, T.; Wołosiak, R.; Worobiej, E.; Wilczak, J. Antioxidant activity and chemical difference in fruit of different Actinidia sp. Int. J. Food Sci. Nutr. 2010, 61, 381–394. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Liu, S.; Qi, H.; Cai, H.; Xu, M. Research progress on plant long non-coding RNA. Plants 2020, 9, 408. [Google Scholar] [CrossRef] [Green Version]
- Fraser, L.G.; Harvey, C.F.; Crowhurst, R.N.; De Silva, H.N. EST-derived microsatellites from Actinidia species and their potential for mapping. Theor. Appl. Genet. 2004, 108, 1010–1016. [Google Scholar] [CrossRef]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Deng, K.; Deng, R.; Fan, J.; Chen, E. Transcriptome analysis and development of simple sequence repeat (SSR) markers in Zingiber striolatum Diels. Physiol. Mol. Biol. Plants 2018, 24, 125–134. [Google Scholar] [CrossRef]
- Taheri, S.; Abdullah, T.L.; Yusop, M.R.; Hanafi, M.M.; Sahebi, M.; Azizi, P.; Shamshiri, R.R. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules 2018, 23, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulley, S.M.; Rassam, M.; Hoser, D.; Otto, W.; Schünemann, N.; Wright, M.; MacRae, E.; Gleave, A.; Laing, W. Gene expression studies in kiwifruit and gene over-expression in Arabidopsis indicates that GDP-L-galactose guanyltransferase is a major control point of vitamin C biosynthesis. J. Exp. Bot. 2009, 60, 765–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Ma, F.; Liang, D.; Li, J.; Wang, Y. Ascorbate biosynthesis during early fruit development is the main reason for its accumulation in kiwi. PLoS ONE 2010, 5, e14281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.Y.; Pan, D.L.; Jia, Z.H.; Wang, T.; Wang, G.; Guo, Z.R. Chlorophyll, carotenoid and vitamin C metabolism regulation in Actinidia chinensis “Hongyang” outer pericarp during fruit development. PLoS ONE 2018, 13, e0194835. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Amaya, I.; Valpuesta, V.; Botella, M.A. Vitamin C content in fruits: Biosynthesis and regulation. Front. Plant Sci. 2019, 9, 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Chen, X.; Wang, P.; Ma, F. Ascorbic acid accumulation and expression of genes involved in its biosynthesis and recycling in developing apple fruit. J. Am. Soc. Hortic. Sci. 2011, 136, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Dowdle, J.; Ishikawa, T.; Gatzek, S.; Rolinski, S.; Smirnoff, N. Two genes in Arabidopsis thaliana encoding GDP-L-galactose phosphorylase are required for ascorbate biosynthesis and seedling viability. Plant J. 2007, 52, 673–689. [Google Scholar] [CrossRef]
- Mellidou, I.; Keulemans, J.; Kanellis, A.K.; Davey, M.W. Regulation of fruit ascorbic acid concentrations during ripening in high and low vitamin C tomato cultivars. BMC Plant Biol. 2012, 12, 239. [Google Scholar] [CrossRef] [Green Version]
- Mellidou, I.; Chagné, D.; Laing, W.A.; Keulemans, J.; Davey, M.W. Allelic variation in paralogs of GDP-L-galactose phosphorylase is a major determinant of vitamin C concentrations in apple fruit. Plant Physiol. 2012, 160, 1613–1629. [Google Scholar] [CrossRef] [Green Version]
- Laing, W.A.; Martínez-Sánchez, M.; Wright, M.A.; Bulley, S.M.; Brewster, D.; Dare, A.P.; Rassam, M.; Wang, D.; Storey, R.; Macknight, R.C.; et al. An upstream open reading frame is essential for feedback regulation of ascorbate biosynthesis in arabidopsis. Plant Cell 2015, 27, 772–786. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, Z.W.; Wu, Z.J.; Wang, Y.X.; Teng, R.M.; Zhuang, J. Differentially expressed protein and gene analysis revealed the effects of temperature on changes in ascorbic acid metabolism in harvested tea leaves. Hortic. Res. 2018, 5, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 2015, 10, 30132628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yangyang, D.; Jiangqi, L.; Songfeng, W.; Yunping, Z.; Chen, Y.; Fuchu, H. Integrated Nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Control | DAF60 | DAF90 | DAF120 | DAF150 | DAF170 |
|---|---|---|---|---|---|
| DAF30 | 1871/1917 | 1628/1857 | 1931/2384 | 2373/2892 | 2632/3447 |
| DAF60 | 1768/2147 | 1687/2288 | 2425/2911 | 2676/3441 | |
| DAF90 | 410/813 | 866/1513 | 1491/2412 | ||
| DAF120 | 434/689 | 1167/1866 | |||
| DAF150 | 643/1152 |
| DEG_set a | Annotated b | COG c | GO d | KEGG e | KOG f | Pfam g | Swiss-Prot h | eggNOG i | Nr j |
|---|---|---|---|---|---|---|---|---|---|
| DAF 30_vs_60 | 3718 | 1736 | 2940 | 1455 | 2053 | 3306 | 2983 | 3664 | 3709 |
| DAF 30_vs_90 | 3405 | 1542 | 2704 | 1234 | 1760 | 3045 | 2756 | 3326 | 3381 |
| DAF 30_vs_120 | 4232 | 1993 | 3435 | 1687 | 2207 | 3823 | 3474 | 4142 | 4209 |
| DAF 30_vs_150 | 5176 | 2477 | 4187 | 2093 | 2771 | 4663 | 4232 | 5093 | 5156 |
| DAF 30_vs_170 | 5991 | 2832 | 4841 | 2386 | 3305 | 5383 | 4807 | 5893 | 5964 |
| Gene Name | Number of Genes | ||||
|---|---|---|---|---|---|
| Hong Yang (A. chinensis) v2 | Hong Yang (A. chinensis) v3 | Red 5 (A. chinensis) | White (A. eriantha) | A. latifolia | |
| PGI | 5 | 4 | 4 | 6 | 6 |
| PMI | 2 | 5 | 4 | 5 | 2 |
| PMM | 10 | 8 | 7 | 8 | 14 |
| GMP | 3 | 1 | 3 | 3 | 8 |
| GME | 2 | 3 | 2 | 4 | 3 |
| GGP | 3 | 4 | 3 | 5 | 9 |
| GPP | 2 | 2 | 2 | 2 | 3 |
| GalDH | 1 | 1 | 1 | 1 | 1 |
| GalLDH | 1 | 1 | 1 | 1 | 1 |
| MIOX | 7 | 8 | 6 | 10 | 4 |
| GalUR | 5 | 5 | 3 | 6 | 2 |
| AO | 3 | 2 | 7 | 4 | 11 |
| APX | 12 | 4 | 11 | 17 | 17 |
| DHAR | 5 | 4 | 4 | 5 | 5 |
| MDHAR | 7 | 6 | 7 | 8 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, H.; Xia, H.; Guo, Y.; Liu, X.; Lin, L.; Wang, J.; Xu, K.; Lv, X.; Hu, R.; Liang, D. Dynamic Changes in Ascorbic Acid Content during Fruit Development and Ripening of Actinidia latifolia (an Ascorbate-Rich Fruit Crop) and the Associated Molecular Mechanisms. Int. J. Mol. Sci. 2022, 23, 5808. https://doi.org/10.3390/ijms23105808
Deng H, Xia H, Guo Y, Liu X, Lin L, Wang J, Xu K, Lv X, Hu R, Liang D. Dynamic Changes in Ascorbic Acid Content during Fruit Development and Ripening of Actinidia latifolia (an Ascorbate-Rich Fruit Crop) and the Associated Molecular Mechanisms. International Journal of Molecular Sciences. 2022; 23(10):5808. https://doi.org/10.3390/ijms23105808
Chicago/Turabian StyleDeng, Honghong, Hui Xia, Yuqi Guo, Xinling Liu, Lijin Lin, Jin Wang, Kunfu Xu, Xiulan Lv, Rongping Hu, and Dong Liang. 2022. "Dynamic Changes in Ascorbic Acid Content during Fruit Development and Ripening of Actinidia latifolia (an Ascorbate-Rich Fruit Crop) and the Associated Molecular Mechanisms" International Journal of Molecular Sciences 23, no. 10: 5808. https://doi.org/10.3390/ijms23105808
APA StyleDeng, H., Xia, H., Guo, Y., Liu, X., Lin, L., Wang, J., Xu, K., Lv, X., Hu, R., & Liang, D. (2022). Dynamic Changes in Ascorbic Acid Content during Fruit Development and Ripening of Actinidia latifolia (an Ascorbate-Rich Fruit Crop) and the Associated Molecular Mechanisms. International Journal of Molecular Sciences, 23(10), 5808. https://doi.org/10.3390/ijms23105808