
Bone Involvement in Systemic Lupus Erythematosus

 ,
,  , and
, and 
Abstract
:1. Introduction
2. SLE and Osteoporosis
2.1. Epidemiology of Osteoporosis
2.2. Fracture Risk
2.3. Physiopathology of Bone Loss in SLE
3. SLE and Osteonecrosis
3.1. Osteonecrosis in SLE Patients: Epidemiology, Risk Factors and Pathophysiology
3.2. Osteonecrosis Imaging
3.3. Osteonecrosis Management in SLE
4. SLE and Osteomyelitis
4.1. Osteomyelitis in SLE Patients: Epidemiology, Risk factors and Pathophysiology
4.2. Clinical Manifestations of Osteomyelitis in SLE Patients
4.3. Osteomyelitis Imaging
4.4. Osteomyelitis Management in SLE
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1,25(OH)2D | 1,25-dihydroxy vitamin D |
| aCL | Anti-cardiolipin |
| ACPA | Antibody against citrullinated protein |
| ACR | American College of Rheumatology |
| AntiCarP | Anti-carbamylated protein antibody |
| aPL | Anti-phospholipid antibodies |
| APOL1 | Apolipoprotein L1 |
| APS | Anti-Phospholipid Syndrome |
| AVN | Avascular necrosis |
| BAX | Bcl-2 associated X |
| BM | Bone marrow |
| BMD | Bone mineral density |
| BMP | Bone morphogenetic protein |
| CT | Computerized tomography |
| CTLA4 | Cytotoxic T-Lymphocyte antigen 4 |
| CTSG | Cathepsin G |
| DHEA | Dehydroepiandrosterone |
| DKK1 | Dickkopf-1 |
| DNA | Deoxyribo-nucleic acid |
| DXA | Dual-energy X-ray absorptiometry |
| GCs | Glucocorticoids |
| HCQ | Hydroxychloroquine |
| HR | Hazard ratio |
| IGF-1 | Insulin-like growth factor-1 |
| IL1-RN | IL-1 receptor antagonist |
| IL-1 | Interleukin-1 |
| IL-1α | Interleukin-1α |
| IL-1β | Interleukin-1β |
| IL-4 | Interleukin-4 |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| IL-10 | Interleukin-10 |
| IL-12 | Interleukin-12 |
| IL-12β | Interleukin-12β |
| IL-17 | Interleukin-17 |
| IL-21 | Interleukin-21 |
| IFNα | Interferon α |
| INFβ | Interferon β |
| IFNγ | Interferon γ |
| LDL | Low density lipoproteins |
| M-CSF | Macrophage-colony stimulating factor |
| MMP1 | Matrix metalloproteinase 1 |
| MRI | Magnetic resonance imaging |
| MSCs | Mesenchymal stem cells |
| OM | Osteomyelitis |
| OP | Osteoporosis |
| OPG | Osteoprotegerin |
| pDC | Plasmacytoid dendritic cell |
| PLAT | Plasminogen activator, tissue type |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 |
| RANK | Receptor activator of nuclear factor-kB |
| RANKL | Receptor activator of nuclear factor-kB ligand |
| Rx | Radiography |
| Runx2 | Runt-related transcription factor 2 |
| SLE | Systemic lupus erythematosus |
| Smad | Small mother against decapentaplegic |
| SNPs | Single-nucleotide polymorphisms |
| TBS | Trabecular bone score |
| Th1 | T helper 1 cell |
| Th2 | T helper 2 cell |
| Th17 | T helper 17 cell |
| TLR2 | Toll-like receptor 2 |
| TNFα | Tumor necrosis factor α |
| Treg | Regulatory T cell |
| VDR | Vitamin D receptor |
| VF | Vertebral fracture |
| Wnt | Wingless/integrated |
References
- Ugarte-Gil, M.F.; González, L.A.; Alarcón, G.S. Lupus: The new epidemic. Lupus 2019, 28, 1031–1050. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidou, M.; Ching, C.L. Systemic Lupus Erythematosus. Ann. Intern. Med. 2020, 172, ITC81–ITC96. [Google Scholar] [CrossRef] [PubMed]
- Fava, A.; Petri, M. Systemic lupus erythematosus: Diagnosis and clinical management. J. Autoimmun. 2019, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, G.; Brennan, M.T. Systemic lupus erythematosus: Epidemiology, pathophysiology, manifestations, and management. Dent. Clin. N. Am. 2013, 57, 631–655. [Google Scholar] [CrossRef] [PubMed]
- Gergianaki, I.; Bortoluzzi, A.; Bertsias, G. Update on the epidemiology, risk factors, and disease outcomes of systemic lupus erythematosus. Best Pract. Res. Clin. Rheumatol. 2018, 32, 188–205. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Zucchi, D.; Elefante, E.; Calabresi, E.; Signorini, V.; Bortoluzzi, A.; Tani, C. One year in review 2019: Systemic lupus erythematosus. Clin. Exp. Rheumatol. 2019, 37, 715–722. [Google Scholar]
- Justiz Vaillant, A.A.; Goyal, A.; Bansal, P.; Varacallo, M. Systemic Lupus Erythematosus; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rúa-Figueroa Fernández de Larrinoa, I. What is new in systemic lupus erythematosus. Reumatol. Clin. 2015, 11, 27–32. [Google Scholar] [CrossRef]
- Corrado, A.; Sanpaolo, E.R.; Di Bello, S.; Cantatore, F.P. Osteoblast as a target of anti-osteoporotic treatment. Postgrad. Med. 2017, 129, 858–865. [Google Scholar] [CrossRef]
- Clynes, M.A.; Harvey, N.C.; Curtis, E.M.; Fuggle, N.R.; Dennison, E.M.; Cooper, C. The epidemiology of osteoporosis. Br. Med. Bull. 2020, 133, 105–117. [Google Scholar] [CrossRef]
- Watts, N.B. Postmenopausal Osteoporosis: A Clinical Review. J. Womens Health 2018, 27, 1093–1096. [Google Scholar] [CrossRef]
- Alejandro, P.; Constantinescu, F. A Review of Osteoporosis in the Older Adult: An Update. Rheum. Dis. Clin. N. Am. 2018, 44, 437–451. [Google Scholar] [CrossRef]
- Bultink, I.E.M.; Vis, M.; van der Horst-Bruinsma, I.E.; Lems, W.F. Inflammatory rheumatic disorders and bone. Curr. Rheumatol. Rep. 2012, 14, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Coury, F.; Peyruchaud, O.; Machuca-Gayet, I. Osteoimmunology of Bone Loss in Inflammatory Rheumatic Diseases. Front. Immunol. 2019, 10, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bultink, I.E.M. Bone Disease in Connective Tissue Disease/Systemic Lupus Erythematosus. Calcif. Tissue Int. 2018, 102, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Maruotti, N.; Corrado, A.; Cantatore, F.P. Osteoporosis and rheumatic diseases. Reumatismo 2014, 66, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Amarasekara, D.S.; Yu, J.; Rho, J. Bone Loss Triggered by the Cytokine Network in Inflammatory Autoimmune Diseases. J. Immunol. Res. 2015, 2015, 832127. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.L.; Huang, W.N.; Chen, H.H.; Chen, J.P.; Chen, D.Y.; Hsieh, T.Y.; Hung, W.T.; Lai, K.L.; Lin, C.T.; Tang, K.T.; et al. Degraded microarchitecture by low trabecular bone score is associated with prevalent vertebral fractures in patients with systemic lupus erythematosus. Arch. Osteoporos. 2020, 15, 54. [Google Scholar] [CrossRef]
- Xia, J.; Luo, R.; Guo, S.; Yang, Y.; Ge, S.; Xu, G.; Zeng, R. Prevalence and Risk Factors of Reduced Bone Mineral Density in Systemic Lupus Erythematosus Patients: A Meta-Analysis. Biomed. Res. Int. 2019, 2019, 3731648. [Google Scholar] [CrossRef] [Green Version]
- Adami, G.; Fassio, A.; Rossini, M.; Caimmi, C.; Giollo, A.; Orsolini, G.; Viapiana, O.; Gatti, D. Osteoporosis in Rheumatic Diseases. Int. J. Mol. Sci. 2019, 20, 5867. [Google Scholar] [CrossRef] [Green Version]
- Adami, G.; Saag, K.G. Glucocorticoid-induced osteoporosis: 2019 concise clinical review. Osteoporos. Int. 2019, 30, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Viapiana, O.; Vitiello, M.; Malavolta, N.; La Montagna, G.; Maddali Bongi, S.; Di Munno, O.; Nuti, R.; Manzini, C.U.; Ferri, C.; et al. Prevalence and incidence of osteoporotic fractures in patients on long-term glucocorticoid treatment for rheumatic diseases: The Glucocorticoid Induced OsTeoporosis TOol (GIOTTO) study. Reumatismo 2017, 69, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevskaya, T.; Gamble, M.P.; Pope, J.E. A meta-analysis of avascular necrosis in systemic lupus erythematosus: Prevalence and risk factors. Clin. Exp. Rheumatol. 2017, 35, 700–710. [Google Scholar] [PubMed]
- Kim, C.S.; Han, K.D.; Jung, J.H.; Choi, H.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. Incidence and risk factors for osteoporotic fractures in patients with systemic lupus erythematosus versus matched controls. Korean J. Intern. Med. 2021, 36, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Pinto, C.; Rojas-Villarraga, A.; Molano-González, N.; Jiménez-Herrera, E.A.; León-Vázquez, M.L.; Montiel-Jarquín, Á.; García-Carrasco, M.; Cervera, R. Bone mineral density and vertebral fractures in patients with systemic lupus erythematosus: A systematic review and meta-regression. PLoS ONE 2018, 13, e0196113. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.H.; Chang, Y.S.; Liu, C.J.; Lai, C.C.; Chen, W.S.; Chen, T.J.; Wang, S.J. Association of systemic lupus erythematosus with a higher risk of cervical but not trochanteric hip fracture: A nationwide population-based study. Arthritis Care Res. 2013, 65, 1674–1681. [Google Scholar] [CrossRef]
- Ruaro, B.; Casabella, A.; Paolino, S.; Alessandri, E.; Patané, M.; Gotelli, E.; Sulli, A.; Cutolo, M. Trabecular Bone Score and Bone Quality in Systemic Lupus Erythematosus Patients. Front. Med. 2020, 7, 574842. [Google Scholar] [CrossRef]
- Mendoza-Pinto, C.; García-Carrasco, M.; Sandoval-Cruz, H.; Muñoz-Guarneros, M.; Escárcega, R.O.; Jiménez-Hernández, M.; Munguía-Realpozo, P.; Sandoval-Cruz, M.; Delezé-Hinojosa, M.; López-Colombo, A.; et al. Risk factors of vertebral fractures in women with systemic lupus erythematosus. Clin. Rheumatol. 2009, 28, 579–585. [Google Scholar] [CrossRef]
- Li, E.K.; Tam, L.S.; Griffith, J.F.; Zhu, T.Y.; Li, T.K.; Li, M.; Wong, K.C.; Chan, M.; Lam, C.W.; Chu, F.S.; et al. High prevalence of asymptomatic vertebral fractures in Chinese women with systemic lupus erythematosus. J. Rheumatol. 2009, 36, 1646–1652. [Google Scholar] [CrossRef]
- Ruaro, B.; Casabella, A.; Molfetta, L.; Salton, F.; Confalonieri, P.; Confalonieri, M.; Baratella, E.; De Tanti, A.; Bruni, C. What Role Does Trabecular Bone Score Play in Chronic Inflammatory Rheumatic Diseases? Front. Med. 2020, 7, 600697. [Google Scholar] [CrossRef]
- Katz, P.P.; Andrews, J.; Yazdany, J.; Schmajuk, G.; Trupin, L.; Yelin, E. Is frailty a relevant concept in SLE? Lupus Sci. Med. 2017, 4, e000186. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.; Guyatt, G.; Fink, H.A.; Cannon, M.; Grossman, J.; Hansen, K.E.; Humphrey, M.B.; Lane, N.E.; Magrey, M.; Miller, M.; et al. 2017 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis. Arthritis Care Res. 2017, 69, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B. TNF and Bone Remodeling. Curr. Osteoporos. Rep. 2017, 15, 126–134. [Google Scholar] [CrossRef]
- Corrado, A.; Maruotti, N.; Cantatore, F.P. Osteoblast Role in Rheumatic Diseases. Int. J. Mol. Sci. 2017, 18, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Fan, P.; Luo, J.; Wu, S.; Sun, H.; He, L.; Zhou, B. Assessment of bone mineral density and bone metabolism in young male adults recently diagnosed with systemic lupus erythematosus in China. Lupus 2017, 26, 289–293. [Google Scholar] [CrossRef]
- Teichmann, J.; Lange, U.; Stracke, H.; Federlin, K.; Bretzel, R.G. Bone metabolism and bone mineral density of systemic lupus erythematosus at the time of diagnosis. Rheumatol. Int. 1999, 18, 137–140. [Google Scholar] [CrossRef]
- Sarkissian, A.; Sivaraman, V.; Bout-Tabaku, S.; Ardoin, S.P.; Moore-Clingenpeel, M.; Mruk, V.; Steigelman, H.; Morris, K.; Bowden, S.A. Bone turnover markers in relation to vitamin D status and disease activity in adults with systemic lupus erythematosus. Lupus 2019, 28, 156–162. [Google Scholar] [CrossRef]
- Gao, L.; Liesveld, J.; Anolik, J.; Mcdavid, A.; Looney, R.J. IFNβ signaling inhibits osteogenesis in human SLE bone marrow. Lupus 2020, 29, 1040–1049. [Google Scholar] [CrossRef]
- Corrado, A.; Rotondo, C.; Mele, A.; Cici, D.; Maruotti, N.; Sanpaolo, E.; Colia, R.; Cantatore, F.P. Influence of glucocorticoid treatment on trabecular bone score and bone remodeling regulators in early rheumatoid arthritis. Arthritis Res. Ther. 2021, 23, 180. [Google Scholar] [CrossRef]
- Buttgereit, F. Views on glucocorticoid therapy in rheumatology: The age of convergence. Nat. Rev. Rheumatol. 2020, 16, 239–246. [Google Scholar] [CrossRef]
- Floris, A.; Piga, M.; Chessa, E.; Congia, M.; Erre, G.L.; Angioni, M.M.; Mathieu, A.; Cauli, A. Long-term glucocorticoid treatment and high relapse rate remain unresolved issues in the real-life management of polymyalgia rheumatica: A systematic literature review and meta-analysis. Clin. Rheumatol. 2022, 41, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Bultink, I.E.M.; Lems, W.F. Systemic lupus erythematosus and fractures. RMD Open 2015, 1 (Suppl. S1), e000069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallas, R.; Li, J.; Petri, M. Predictors of osteonecrosis in systemic lupus erythematosus: A prospective cohort study. Arthritis Care Res. 2020. published online ahead of print. [Google Scholar] [CrossRef]
- Hussein, S.; Suitner, M.; Béland-Bonenfant, S.; Baril-Dionne, A.; Vandermeer, B.; Santesso, N.; Keeling, S.; Pope, J.E.; Fifi-Mah, A.; Bourré-Tessier, J. Monitoring of Osteonecrosis in Systemic Lupus Erythematosus: A Systematic Review and Metaanalysis. J. Rheumatol. 2018, 45, 1462–1476. [Google Scholar] [CrossRef]
- Faezi, S.T.; Hoseinian, A.S.; Paragomi, P.; Akbarian, M.; Esfahanian, F.; Gharibdoost, F.; Akhlaghi, M.; Nadji, A.; Jamshidi, A.R.; Shahram, F.; et al. Non-corticosteroid risk factors of symptomatic avascular necrosis of bone in systemic lupus erythematosus: A retrospective case-control study. Mod. Rheumatol. 2015, 25, 590–594. [Google Scholar] [CrossRef]
- Shaharir, S.S.; Chua, S.H.; Mohd, R.; Mustafar, R.; Noh, M.M.; Shahril, N.S.; Said, M.S.M.; Rajalingham, S. Risk factors for symptomatic Avascular Necrosis (AVN) in a multi-ethnic Systemic Lupus Erythematosus (SLE) cohort. PLoS ONE 2021, 16, e0248845. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, Q.; Gong, Y.; Qi, X.; Liu, J. Multifocal osteonecrosis affecting all four limbs in systemic lupus erythematosus: A case report. Exp. Ther. Med. 2019, 18, 2475–2478. [Google Scholar] [CrossRef]
- Lespasio, M.J.; Sodhi, N.; Mont, M.A. Osteonecrosis of the Hip: A Primer. Perm. J. 2019, 23, 18–100. [Google Scholar] [CrossRef] [Green Version]
- Lalani, T.A.; Kanne, J.P.; Hatfield, G.A.; Chen, P. Imaging findings in systemic lupus erythematosus. Radiographics 2004, 24, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Torrente-Segarra, V.; Bonet, M. Multifocal osteonecrosis in systemic lupus erythematosus: Two case reports and literature review. Eur. J. Rheumatol. 2021, 8, 46–47. [Google Scholar] [CrossRef]
- Noureldine, M.H.A.; Uthman, I. Antiphospholipid (Hughes) syndrome: Insights for orthopedics. Lupus 2018, 27, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zheng, Y.; Jia, J.; Ding, J.; Wu, Z. Systemic lupus erythematosus patients with high disease activity are associated with accelerated incidence of osteonecrosis: A systematic review and meta-analysis. Clin. Rheumatol. 2018, 37, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Mont, M.A.; Pivec, R.; Banerjee, S.; Issa, K.; Elmallah, R.K.; Jones, L.C. High-Dose Corticosteroid Use and Risk of Hip Osteonecrosis: Meta-Analysis and Systematic Literature Review. J. Arthroplast. 2015, 30, 1506–1512.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cai, Q.; Xu, Y.; Fu, Q.; Feng, Y.; Chen, X.; Dai, S.; Zhao, D.; Zhan, C.; Xu, W.; et al. Associations between glucocorticoids, antiphospholipid antibodies and femur head necrosis in patients with SLE: A directed acyclic graph-based multicentre study. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211002677. [Google Scholar] [CrossRef]
- Blazer, A.; Wang, B.; Simpson, D.; Kirchhoff, T.; Heffron, S.; Clancy, R.M.; Heguy, A.; Ray, K.; Snuderl, M.; Buyon, J.P. Apolipoprotein L1 risk variants associate with prevalent atherosclerotic disease in African American systemic lupus erythematosus patients. PLoS ONE 2017, 12, e0182483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Matteo, A.; Smerilli, G.; Cipolletta, E.; Salaffi, F.; De Angelis, R.; Di Carlo, M.; Filippucci, E.; Grassi, W. Imaging of Joint and Soft Tissue Involvement in Systemic Lupus Erythematosus. Curr. Rheumatol. Rep. 2021, 23, 73. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, K.; Tripathy, S.K.; Sen, R.K.; Santhosh, S.; Bhattacharya, A. Nuclear medicine imaging in osteonecrosis of hip: Old and current concepts. World J. Orthop. 2017, 8, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Chang, Y.S.; Chen, W.S.; Tsao, Y.P.; Wang, W.H.; Liao, H.T.; Tsai, C.Y.; Lai, C.C. Incidence and risk factors of osteomyelitis in adult and pediatric systemic lupus erythematosus: A nationwide, population-based cohort study. Lupus 2019, 28, 19–26. [Google Scholar] [CrossRef]
- Herrinton, L.J.; Liu, L.; Goldfien, R.; Michaels, M.A.; Tran, T.N. Risk of Serious Infection for Patients with Systemic Lupus Erythematosus Starting Glucocorticoids with or without Antimalarials. J. Rheumatol. 2016, 43, 1503–1509. [Google Scholar] [CrossRef]
- Rajeev, A.; Mavrotas, J.; Taribagil, S.; Loughead, J. Acute Pyogenic Osteomyelitis of the Pubic Bone in a Patient with Systemic Lupus Erythematosus Mimicking Fracture of the Pubic Bone and Periprosthetic Joint Infection of the Hip. Case Rep. Orthop. 2021, 2021, 6665938. [Google Scholar] [CrossRef]
- Kedves, M.; Kósa, F.; Kunovszki, P.; Takács, P.; Szabó, M.Z.; Karyekar, C.; Lofland, J.H.; Nagy, G. Large-scale mortality gap between SLE and control population is associated with increased infection-related mortality in lupus. Rheumatology 2020, 59, 3443–3451. [Google Scholar] [CrossRef]
- Jung, J.Y.; Suh, C.H. Infection in systemic lupus erythematosus, similarities, and differences with lupus flare. Korean J. Intern. Med. 2017, 32, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maffulli, N.; Papalia, R.; Zampogna, B.; Torre, G.; Albo, E.; Denaro, V. The management of osteomyelitis in the adult. Surgeon 2016, 14, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.K. Osteomyelitis. Infect. Dis Clin. N. Am. 2017, 31, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Li, J.; Gu, F.; Zhang, K.; Su, Z.; Wen, Q.; Sui, Z.; Zhou, P.; Yu, T. Genetic Determinants for Bacterial Osteomyelitis: A Focused Systematic Review of Published Literature. Front. Genet. 2021, 12, 654792. [Google Scholar] [CrossRef]
- Canella, C.; Costa, F.; Danowisk, A.; de Melo, A.S.A.; Marchiori, E. Stress fracture and osteomyelitis in a patient with systemic lupus erythematosus. Radiol. Bras. 2018, 51, 277–278. [Google Scholar] [CrossRef]
- Kim, S.S.; Perino, G.; Boettner, F.; Miller, A.; Goodman, S. Salmonella septic arthritis of the knees in a patient with systemic lupus erythematosus. Lupus 2013, 22, 740–743. [Google Scholar] [CrossRef]
- Gray, M.E.; Liu, P.W.; Wispelwey, B. Mycobacterium Avium complex vertebral osteomyelitis in the absence of HIV infection: A case report and review. BMC Infect. Dis. 2018, 18, 235. [Google Scholar] [CrossRef]
- Sato, K.; Yazawa, H.; Ikuma, D.; Maruyama, T.; Kajiyama, H.; Mimura, T. Osteomyelitis due to methicillin-resistant Staphylococcus aureus successfully treated by an oral combination of minocycline and trimethoprim-sulfamethoxazole. SAGE Open Med. Case Rep. 2019, 7, 2050313X19841465. [Google Scholar] [CrossRef] [Green Version]
- Mandell, J.C.; Khurana, B.; Smith, J.T.; Czuczman, G.J.; Ghazikhanian, V.; Smith, S.E. Osteomyelitis of the lower extremity: Pathophysiology, imaging, and classification, with an emphasis on diabetic foot infection. Emerg. Radiol. 2018, 25, 175–188. [Google Scholar] [CrossRef]
- Dym, H.; Zeidan, J. Microbiology of Acute and Chronic Osteomyelitis and Antibiotic Treatment. Dent. Clin. N. Am. 2017, 61, 271–282. [Google Scholar] [CrossRef]
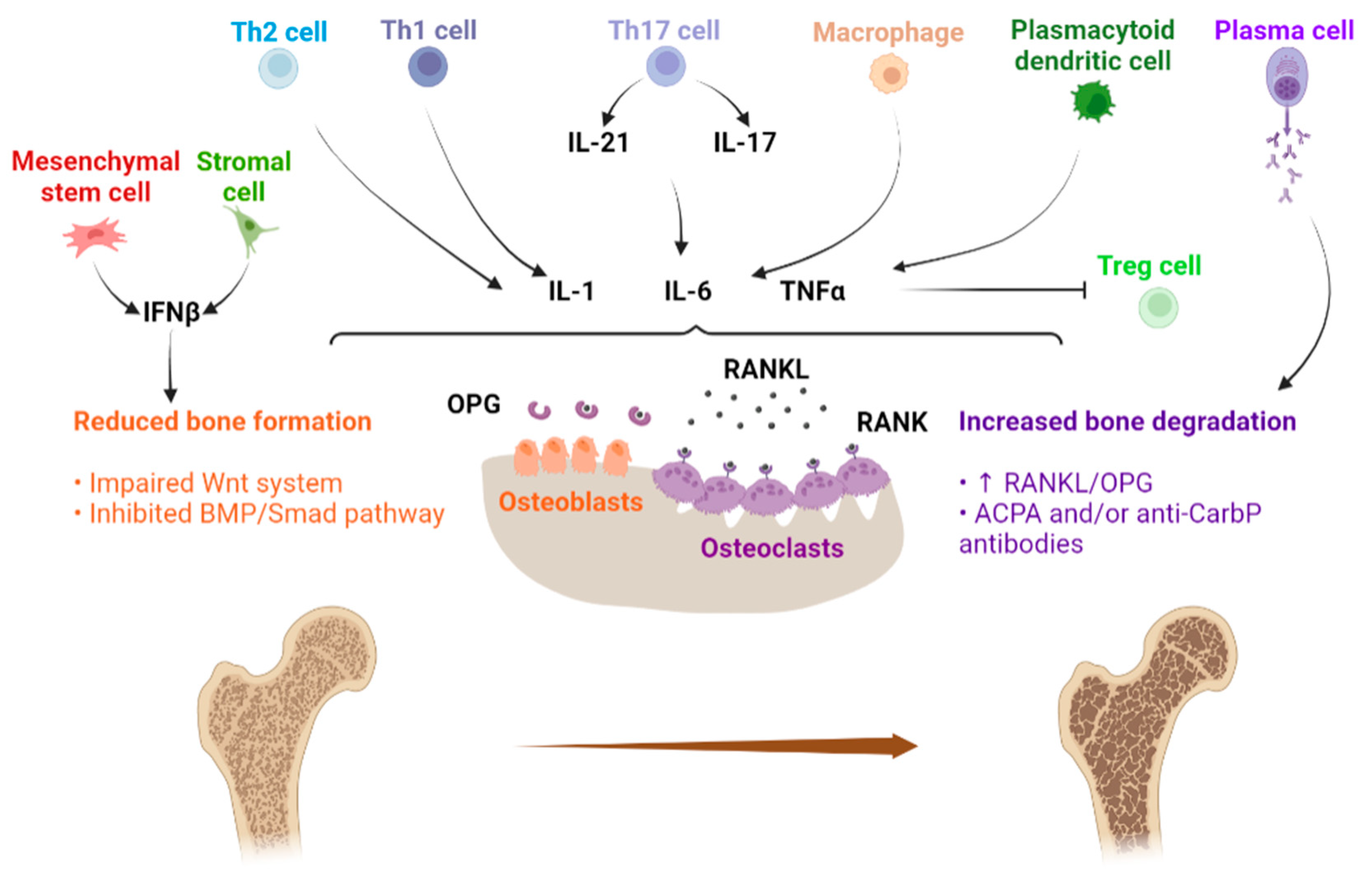

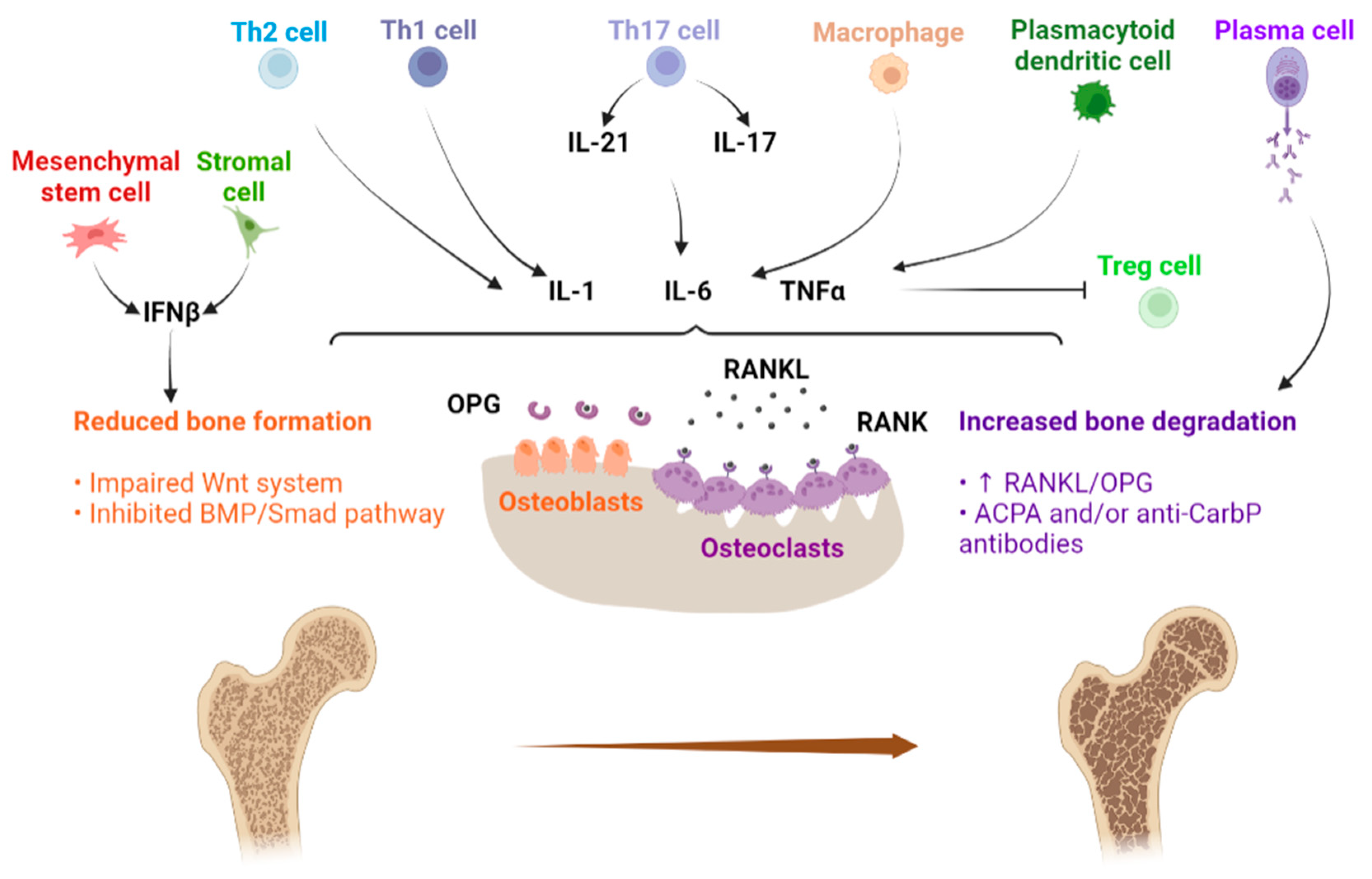{kind=link}
| Traditional Independent Risk Factors | Disease-Related Risk Factors |
|---|---|
| Low body mass index [12,13,14,16] | Systemic inflammation [14,16,17,18,19] |
| Old age [11,12,13,14,16] | Lupus nephritis [14,16,17,21] |
| Female gender [11,12,13,14,16] | High levels of oxidized LDL [14,16,17] |
| Postmenopausal status [11,12,13,14,16] | Hormonal abnormalities [14,16,17,21] |
| Smoking [13,14,16] | Presence of anti-Ro, ACPA and/or antiCarP antibodies [14,15,16,21] |
| Alcohol abuse [13,14,16] | Medication [12,13,14,16,17,19,22,23,24] |
| Low level of vitamin D [12,13,14,16] | Immobility [14,15,16,17] |
| Low intake of calcium [12,13,14,16] | |
| Low physical activity [12,13,14,16] | |
| Hypogonadism [12,13] Family history of osteoporosis [11,12,13,14,16] |
| Independent Risk Factors | Disease-Related Risk Factors | Medication-Induced Adverse Effects | Bone Impairment |
|---|---|---|---|
| Old age [11,12,13,14,16] | Disease duration [14,16,17,18] | Glucocorticoids [14,16,17,22,23,33] | Low BMD [11,12,13,14,16,17,19] |
| Female gender [11,12,14,16] | Seizures [16,17,18] | Antiepileptic drugs [16] | Low TBS [19,28,31] |
| Smoking [13,14,16] | History of stroke [16,18] | Anticoagulants [16] | Previous fragility fractures [13,16,18] |
| Alcohol abuse [13,16] | Renal failure [14,16,21] | Cyclophosphamide [16] | |
| Postmenopausal status [11,12,14,16] | Presence of lupus anticoagulant [14,16] | ||
| Frailty [16,32] | |||
| Obesity [14,16] | |||
| Low level of vitamin D [12,13,14,16] |
| Independent Risk Factors | Disease-Related Factors | |
|---|---|---|
| Traumatic-Associated Risk Factors | Atraumatic-Associated Risk Factors | |
| Fractures [49,52] | Hyperlipidemia [44,45,49] | High disease activity [24,44,45,46,47,53] |
| Dislocation or fracture- dislocation [48,49] | Chronic renal failure/ hemodialysis [44,47,48,49] | Medication-induced adverse effects [44,45,46,47,48,51,53,54] |
| Radiation [49] | Smoking [44,47,48,49] | Antiphospholipid syndrome [24,44,45,46,47,52,55] |
| Other comorbidities (i.e., hematologic diseases) [45,48,49] | Alcohol use [44,45,48,49] | Vasculopathy and abnormal endothelial function [44,45,46,47] |
| Organ transplantation [49] | Clinical features [24,44,45,46,53] | |
| Hyperuricemia/gout [49] | Genetic factors (APOL1 variant alleles) [44,47,56] | |
| HIV [49] | Younger age of disease onset [24,44,45,46] | |
| Intravascular coagulation [44,49] | ||
| Thrombophlebitis [49] | ||
| Cushing disease [44,49] | ||
| Drugs (GCs) [45,47,48,49,51] | ||
| Pancreatitis [49] | ||
| Pregnancy [49] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rella, V.; Rotondo, C.; Altomare, A.; Cantatore, F.P.; Corrado, A. Bone Involvement in Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2022, 23, 5804. https://doi.org/10.3390/ijms23105804
Rella V, Rotondo C, Altomare A, Cantatore FP, Corrado A. Bone Involvement in Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2022; 23(10):5804. https://doi.org/10.3390/ijms23105804
Chicago/Turabian StyleRella, Valeria, Cinzia Rotondo, Alberto Altomare, Francesco Paolo Cantatore, and Addolorata Corrado. 2022. "Bone Involvement in Systemic Lupus Erythematosus" International Journal of Molecular Sciences 23, no. 10: 5804. https://doi.org/10.3390/ijms23105804
APA StyleRella, V., Rotondo, C., Altomare, A., Cantatore, F. P., & Corrado, A. (2022). Bone Involvement in Systemic Lupus Erythematosus. International Journal of Molecular Sciences, 23(10), 5804. https://doi.org/10.3390/ijms23105804