
Dynamic Involvement of Telocytes in Modulating Multiple Signaling Pathways in Cardiac Cytoarchitecture

 ,
,  ,
,  and
and 
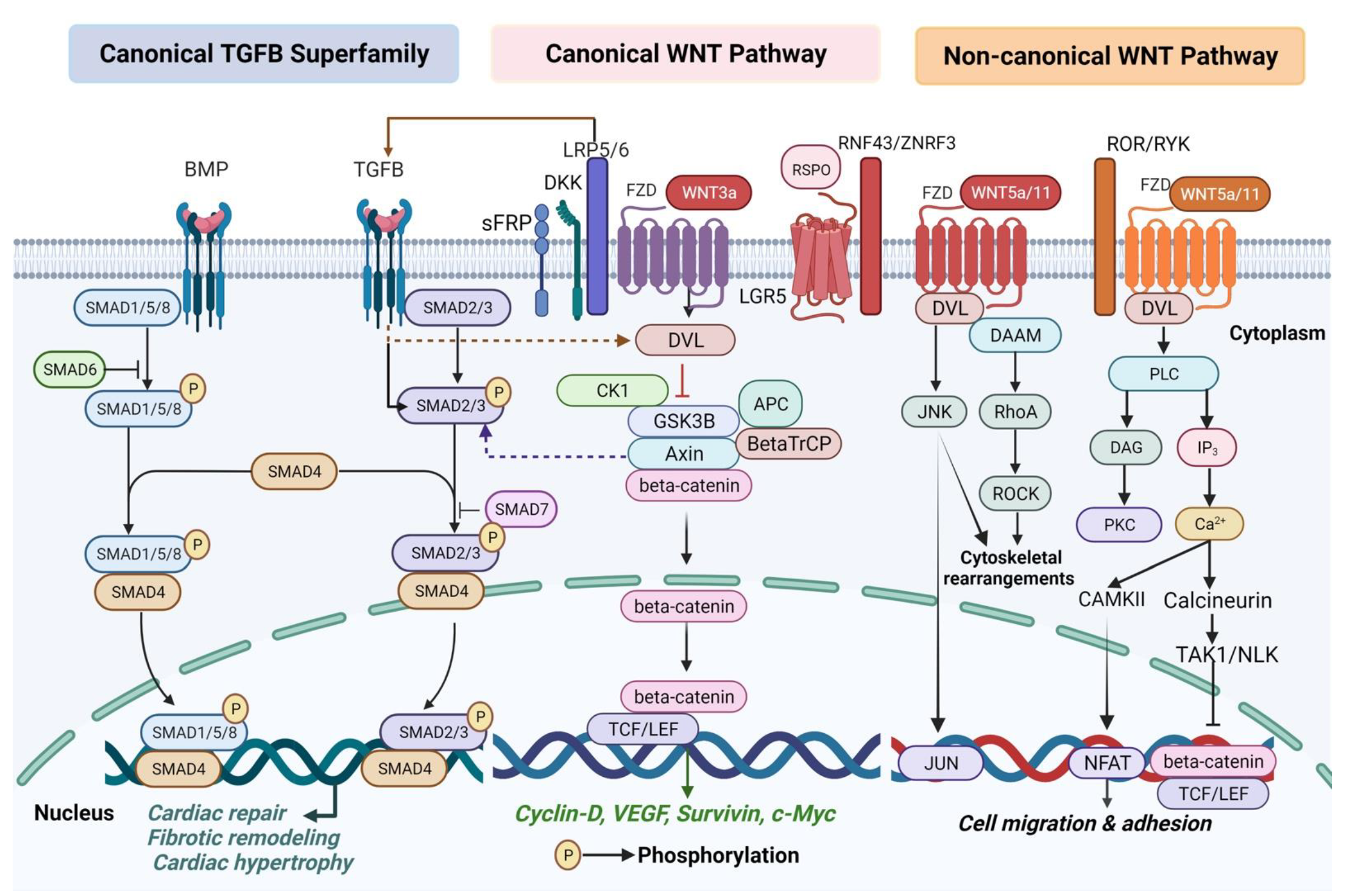{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cardiac Telocytes
3. Network of Interactions between Signaling Pathways
3.1. WNT Signaling Pathway
3.1.1. WNT Signaling in Cardiogenesis and Congenital Heart Diseases
3.1.2. WNT Signaling in Cardiac Remodeling and Heart Failure
3.2. TGFB Signaling Pathway
3.3. PI3K/AKT Signaling Pathway
4. Telocytes Orchestration of Cell-to-Cell Communication to Integrate Molecular Signals
5. Putative Future Cellular Therapies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Popescu, L.M.; Gherghiceanu, M.; Hinescu, M.E.; Cretoiu, D.; Ceafalan, L.; Regalia, T.; Popescu, A.C.; Ardeleanu, C.; Mandache, E. Insights into the interstitium of ventricular myocardium: Interstitial Cajal-like cells (ICLC). J. Cell. Mol. Med. 2006, 10, 429–458. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Joffs, C.; Deschamps, A.M.; Davis, J.; Dowdy, K.; Hendrick, J.; Baicu, S.; Mukherjee, R.; Manhaini, M.; Spinale, F.G. Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H987–H994. [Google Scholar] [CrossRef] [PubMed]
- Iles, L.; Pfluger, H.; Phrommintikul, A.; Cherayath, J.; Aksit, P.; Gupta, S.N.; Kaye, D.M.; Taylor, A.J. Evaluation of diffuse myocardial fibrosis in heart failure with cardiac magnetic resonance contrast-enhanced T1 mapping. J. Am. Coll. Cardiol. 2008, 52, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Popescu, L.M.; Faussone-Pellegrini, M.-S. TELOCYTES—a case of serendipity: The winding way from Interstitial Cells of Cajal (ICC), via Interstitial Cajal-Like Cells (ICLC) to TELOCYTES. J. Cell. Mol. Med. 2010, 14, 729–740. [Google Scholar] [CrossRef]
- Cretoiu, S.M.; Popescu, L.M. Telocytes revisited. Biomol. Concepts 2014, 5, 353–369. [Google Scholar] [CrossRef]
- Popescu, L.M.; Curici, A.; Wang, E.; Zhang, H.; Hu, S.; Gherghiceanu, M. Telocytes and putative stem cells in ageing human heart. J. Cell. Mol. Med. 2015, 19, 31–45. [Google Scholar] [CrossRef]
- Popescu, L.M.; Manole, C.G.; Gherghiceanu, M.; Ardelean, A.; Nicolescu, M.I.; Hinescu, M.E.; Kostin, S. Telocytes in human epicardium. J. Cell. Mol. Med. 2010, 14, 2085–2093. [Google Scholar] [CrossRef]
- Kostin, S. Myocardial telocytes: A specific new cellular entity. J. Cell. Mol. Med. 2010, 14, 1917–1921. [Google Scholar] [CrossRef]
- Gherghiceanu, M.; Manole, C.G.; Popescu, L.M. Telocytes in endocardium: Electron microscope evidence. J. Cell. Mol. Med. 2010, 14, 2330–2334. [Google Scholar] [CrossRef]
- Gherghiceanu, M.; Popescu, L.M. Cardiomyocyte precursors and telocytes in epicardial stem cell niche: Electron microscope images. J. Cell. Mol. Med. 2010, 14, 871–877. [Google Scholar] [CrossRef]
- Suciu, L.; Nicolescu, M.I.; Popescu, L.M. Cardiac telocytes: Serial dynamic images in cell culture. J. Cell. Mol. Med. 2010, 14, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Cretoiu, D.; Hummel, E.; Zimmermann, H.; Gherghiceanu, M.; Popescu, L.M. Human cardiac telocytes: 3D imaging by FIB-SEM tomography. J. Cell. Mol. Med. 2014, 18, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Sun, X.; Zhang, M.; Qian, M.; Zheng, Y.; Li, M.; Cretoiu, S.M.; Chen, C.; Chen, L.; Cretoiu, D.; et al. Variations of chromosomes 2 and 3 gene expression profiles among pulmonary telocytes, pneumocytes, airway cells, mesenchymal stem cells and lymphocytes. J. Cell. Mol. Med. 2014, 18, 2044–2060. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Cretoiu, D.; Yan, G.; Cretoiu, S.M.; Popescu, L.M.; Fang, H.; Wang, X. Protein profiling of human lung telocytes and microvascular endothelial cells using iTRAQ quantitative proteomics. J. Cell. Mol. Med. 2014, 18, 1035–1059. [Google Scholar] [CrossRef]
- Zheng, Y.; Cretoiu, D.; Yan, G.; Cretoiu, S.M.; Popescu, L.M.; Wang, X. Comparative proteomic analysis of human lung telocytes with fibroblasts. J. Cell. Mol. Med. 2014, 18, 568–589. [Google Scholar] [CrossRef]
- Sheng, J.; Shim, W.; Lu, J.; Lim, S.Y.; Ong, B.H.; Lim, T.S.; Liew, R.; Chua, Y.L.; Wong, P. Electrophysiology of human cardiac atrial and ventricular telocytes. J. Cell. Mol. Med. 2014, 18, 355–362. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, M.; Qian, M.; Wang, L.; Cismasiu, V.B.; Bai, C.; Popescu, L.M.; Wang, X. Genetic comparison of mouse lung telocytes with mesenchymal stem cells and fibroblasts. J. Cell. Mol. Med. 2013, 17, 567–577. [Google Scholar] [CrossRef]
- Manole, C.G.; Simionescu, O. The Cutaneous Telocytes. Adv. Exp. Med. Biol. 2016, 913, 303–323. [Google Scholar] [CrossRef]
- Rusu, M.C.; Poalelungi, C.V.; Vrapciu, A.D.; Nicolescu, M.I.; Hostiuc, S.; Mogoanta, L.; Taranu, T. Endocardial tip cells in the human embryo—facts and hypotheses. PLoS ONE 2015, 10, e0115853. [Google Scholar] [CrossRef]
- Manole, C.G.; Cismaşiu, V.; Gherghiceanu, M.; Popescu, L.M. Experimental acute myocardial infarction: Telocytes involvement in neo-angiogenesis. J. Cell. Mol. Med. 2011, 15, 2284–2296. [Google Scholar] [CrossRef]
- Manole, C.G.; Marinescu, B.G.; Marta, D.; Nicolescu, M.I. Areas of Cartilaginous and Osseous Metaplasia After Experimental Myocardial Infarction in Rats. Anat. Rec. 2019, 302, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Rosa, I.; Messerini, L.; Guiducci, S.; Matucci-Cerinic, M.; Ibba-Manneschi, L. A loss of telocytes accompanies fibrosis of multiple organs in systemic sclerosis. J. Cell. Mol. Med. 2014, 18, 253–262. [Google Scholar] [CrossRef]
- Zhao, M.; Ye, S.; Su, J.; Garg, V. Cardiomyocyte Proliferation and Maturation: Two Sides of the Same Coin for Heart Regeneration. Front. Cell Dev. Biol. 2020, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Jiang, Z.; Zagidullin, N.; Liu, T.; Cai, B. Regulation of cardiomyocyte fate plasticity: A key strategy for cardiac regeneration. Signal Transduct. Target. Ther. 2021, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stylianidis, V.; Hermans, K.; Blankesteijn, W.M. Wnt Signaling in Cardiac Remodeling and Heart Failure. Handb. Exp. Pharmacol. 2015, 243, 251–263. [Google Scholar] [CrossRef]
- Ozhan, G.; Weidinger, G. Wnt/β-catenin signaling in heart regeneration. Cell Regen. 2015, 4, 3. [Google Scholar] [CrossRef]
- Cucu, I.; Nicolescu, M.I. A Synopsis of Signaling Crosstalk of Pericytes and Endothelial Cells in Salivary Gland. Dent. J. 2021, 9, 144. [Google Scholar] [CrossRef]
- Ackers, I.; Malgor, R. Interrelationship of canonical and non-canonical Wnt signalling pathways in chronic metabolic diseases. Diabetes Vasc. Dis. Res. 2017, 15, 3–13. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef]
- Gao, C.; Xiao, G.; Hu, J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci. 2014, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef] [PubMed]
- Taelman, V.F.; Dobrowolski, R.; Plouhinec, J.-L.; Fuentealba, L.C.; Vorwald, P.P.; Gumper, I.; Sabatini, D.D.; De Robertis, E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010, 143, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef]
- Kafka, A.; Bašić-Kinda, S.; Pećina-Šlaus, N. The cellular story of dishevelleds. Croat. Med. J. 2014, 55, 459–467. [Google Scholar] [CrossRef]
- Guan, R.; Zhang, X.; Guo, M. Glioblastoma stem cells and Wnt signaling pathway: Molecular mechanisms and therapeutic targets. Chin. Neurosurg. J. 2020, 6, 25. [Google Scholar] [CrossRef]
- Morgan, R.G.; Mortensson, E.; Williams, A.C. Targeting LGR5 in Colorectal Cancer: Therapeutic gold or too plastic? Br. J. Cancer 2018, 118, 1410–1418. [Google Scholar] [CrossRef]
- Barker, N.; Tan, S.; Clevers, H. Lgr proteins in epithelial stem cell biology. Development 2013, 140, 2484–2494. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Sahara, M.; Santoro, F.; Sohlmér, J.; Zhou, C.; Witman, N.; Leung, C.Y.; Mononen, M.; Bylund, K.; Gruber, P.; Chien, K.R. Population and Single-Cell Analysis of Human Cardiogenesis Reveals Unique LGR5 Ventricular Progenitors in Embryonic Outflow Tract. Dev. Cell 2019, 48, 475–490.e7. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.-L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.-M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.-K.; Li, V.S.W.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Brahmbhatt, A.; Deng, S.X.; Zheng, J.J. Wnt signaling activation: Targets and therapeutic opportunities for stem cell therapy and regenerative medicine. RSC Chem. Biol. 2021, 2, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.-M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef]
- Bahar Halpern, K.; Massalha, H.; Zwick, R.K.; Moor, A.E.; Castillo-Azofeifa, D.; Rozenberg, M.; Farack, L.; Egozi, A.; Miller, D.R.; Averbukh, I.; et al. Lgr5+ telocytes are a signaling source at the intestinal villus tip. Nat. Commun. 2020, 11, 3–14. [Google Scholar] [CrossRef]
- Shoshkes-Carmel, M.; Wang, Y.J.; Wangensteen, K.J.; Tóth, B.; Kondo, A.; Massasa, E.E.; Itzkovitz, S.; Kaestner, K.H. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 2018, 557, 242–246. [Google Scholar] [CrossRef]
- Jha, R.; Singh, M.; Wu, Q.; Gentillon, C.; Preininger, M.K.; Xu, C. Downregulation of LGR5 Expression Inhibits Cardiomyocyte Differentiation and Potentiates Endothelial Differentiation from Human Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 513–527. [Google Scholar] [CrossRef]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Jia, Y.; Liu, H.; He, M.; Yang, Y.; Xiao, W.; Li, Y. RhoA/ROCK pathway: Implication in osteoarthritis and therapeutic targets. Am. J. Transl. Res. 2019, 11, 5324–5331. [Google Scholar] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Weerackoon, N.; Gunawardhana, K.L.; Mani, A. Wnt Signaling Cascades and Their Role in Coronary Artery Health and Disease. J. Cell. Signal. 2021, 2, 52–62. [Google Scholar] [CrossRef]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell. Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.V.; Berry, C.T.; Kim, K.; Sen, P.; Kim, T.; Carrer, A.; Trefely, S.; Zhao, S.; Fernandez, S.; Barney, L.E.; et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca(2+)-NFAT signaling. Genes Dev. 2018, 32, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.A.; El-Badri, N.; Zaher, A. Wnt Signaling: The double-edged sword diminishing the potential of stem cell therapy in congenital heart disease: New Hopes for Congenital Heart Disease. Life Sci. 2019, 239, 116937. [Google Scholar] [CrossRef]
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart fields and cardiac morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a015750. [Google Scholar] [CrossRef]
- Kostin, S. Cardiac telocytes in normal and diseased hearts. Semin. Cell Dev. Biol. 2016, 55, 22–30. [Google Scholar] [CrossRef]
- Bei, Y.; Zhou, Q.; Fu, S.; Lv, D.; Chen, P.; Chen, Y.; Wang, F.; Xiao, J. Cardiac telocytes and fibroblasts in primary culture: Different morphologies and immunophenotypes. PLoS ONE 2015, 10, 1–11. [Google Scholar] [CrossRef]
- Bei, Y.; Zhou, Q.; Sun, Q.; Xiao, J. Telocytes in cardiac regeneration and repair. Semin. Cell Dev. Biol. 2016, 55, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Cyganek, L.; Chen, S.; Borchert, T.; Guan, K. Cardiac Progenitor Cells and their Therapeutic Application for Cardiac Repair. J. Clin. Exp. Cardiolog. 2013, S11, 008. [Google Scholar] [CrossRef]
- Touma, M.; Kang, X.; Gao, F.; Zhao, Y.; Cass, A.A.; Biniwale, R.; Xiao, X.; Eghbali, M.; Coppola, G.; Reemtsen, B.; et al. Wnt11 regulates cardiac chamber development and disease during perinatal maturation. JCI Insight 2017, 2, e94904. [Google Scholar] [CrossRef] [PubMed]
- Sukhacheva, T.V.; Nizyaeva, N.V.; Samsonova, M.V.; Chernyaev, A.L.; Shchegolev, A.I.; Serov, R.A. Telocytes in the Myocardium of Children with Congenital Heart Disease Tetralogy of Fallot. Bull. Exp. Biol. Med. 2020, 169, 137–146. [Google Scholar] [CrossRef]
- Dimopoulos, A.; Sicko, R.J.; Kay, D.M.; Rigler, S.L.; Druschel, C.M.; Caggana, M.; Browne, M.L.; Fan, R.; Romitti, P.A.; Brody, L.C.; et al. Rare copy number variants in a population-based investigation of hypoplastic right heart syndrome. Birth Defects Res. 2017, 109, 8–15. [Google Scholar] [CrossRef]
- Kobayashi, J.; Yoshida, M.; Tarui, S.; Hirata, M.; Nagai, Y.; Kasahara, S.; Naruse, K.; Ito, H.; Sano, S.; Oh, H. Directed Differentiation of Patient-Specific Induced Pluripotent Stem Cells Identifies the Transcriptional Repression and Epigenetic Modification of NKX2-5, HAND1, and NOTCH1 in Hypoplastic Left Heart Syndrome. PLoS ONE 2014, 9, e102796. [Google Scholar] [CrossRef]
- Qyang, Y.; Martin-Puig, S.; Chiravuri, M.; Chen, S.; Xu, H.; Bu, L.; Jiang, X.; Lin, L.; Granger, A.; Moretti, A.; et al. The Renewal and Differentiation of Isl1+ Cardiovascular Progenitors Are Controlled by a Wnt/β-Catenin Pathway. Cell Stem Cell 2007, 1, 165–179. [Google Scholar] [CrossRef]
- Ai, D.; Fu, X.; Wang, J.; Lu, M.-F.; Chen, L.; Baldini, A.; Klein, W.H.; Martin, J.F. Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc. Natl. Acad. Sci. USA 2007, 104, 9319–9324. [Google Scholar] [CrossRef]
- van Vliet, P.P.; Lin, L.; Boogerd, C.J.; Martin, J.F.; Andelfinger, G.; Grossfeld, P.D.; Evans, S.M. Tissue specific requirements for WNT11 in developing outflow tract and dorsal mesenchymal protrusion. Dev. Biol. 2017, 429, 249–259. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef]
- Ahuja, P.; Sdek, P.; MacLellan, W.R. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 2007, 87, 521–544. [Google Scholar] [CrossRef]
- Ma, L.-L.; Ding, Z.-W.; Yin, P.-P.; Wu, J.; Hu, K.; Sun, A.-J.; Zou, Y.-Z.; Ge, J.-B. Hypertrophic preconditioning cardioprotection after myocardial ischaemia/reperfusion injury involves ALDH2-dependent metabolism modulation. Redox Biol. 2021, 43, 101960. [Google Scholar] [CrossRef] [PubMed]
- Oikonomopoulos, A.; Sereti, K.-I.; Conyers, F.; Bauer, M.; Liao, A.; Guan, J.; Crapps, D.; Han, J.-K.; Dong, H.; Bayomy, A.F.; et al. Wnt signaling exerts an antiproliferative effect on adult cardiac progenitor cells through IGFBP3. Circ. Res. 2011, 109, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Luo, M.; Zhang, Y.; Wilkes, D.C.; Ge, G.; Grieskamp, T.; Yamada, C.; Liu, T.-C.; Huang, G.; Basson, C.T.; et al. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat. Cell Biol. 2009, 11, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.W.; Kühl, M. WNT Signaling in Adult Cardiac Hypertrophy and Remodeling. Circ. Res. 2010, 107, 1198–1208. [Google Scholar] [CrossRef]
- Alfaro, M.P.; Pagni, M.; Vincent, A.; Atkinson, J.; Hill, M.F.; Cates, J.; Davidson, J.M.; Rottman, J.; Lee, E.; Young, P.P. The Wnt modulator sFRP2 enhances mesenchymal stem cell engraftment, granulation tissue formation and myocardial repair. Proc. Natl. Acad. Sci. USA 2008, 105, 18366–18371. [Google Scholar] [CrossRef]
- Schumann, H.; Holtz, J.; Zerkowski, H.R.; Hatzfeld, M. Expression of secreted frizzled related proteins 3 and 4 in human ventricular myocardium correlates with apoptosis related gene expression. Cardiovasc. Res. 2000, 45, 720–728. [Google Scholar] [CrossRef]
- Askevold, E.T.; Aukrust, P.; Nymo, S.H.; Lunde, I.G.; Kaasbøll, O.J.; Aakhus, S.; Florholmen, G.; Ohm, I.K.; Strand, M.E.; Attramadal, H.; et al. The cardiokine secreted Frizzled-related protein 3, a modulator of Wnt signalling, in clinical and experimental heart failure. J. Intern. Med. 2014, 275, 621–630. [Google Scholar] [CrossRef]
- Hou, N.; Ye, B.; Li, X.; Margulies, K.B.; Xu, H.; Wang, X.; Li, F. Transcription Factor 7-like 2 Mediates Canonical Wnt/β-Catenin Signaling and c-Myc Upregulation in Heart Failure. Circ. Heart Fail. 2016, 9, e003010. [Google Scholar] [CrossRef]
- Zhou, J.; Ahmad, F.; Parikh, S.; Hoffman, N.E.; Rajan, S.; Verma, V.K.; Song, J.; Yuan, A.; Shanmughapriya, S.; Guo, Y.; et al. Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy. Circ. Res. 2016, 118, 1208–1222. [Google Scholar] [CrossRef]
- Wolke, C.; Antileo, E.; Lendeckel, U. Minireview WNT signaling in atrial fibrillation. Exp. Biol. Med. 2021, 246, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Działo, E.; Rudnik, M.; Koning, R.I.; Czepiel, M.; Tkacz, K.; Baj-Krzyworzeka, M.; Distler, O.; Siedlar, M.; Kania, G.; Błyszczuk, P. WNT3a and WNT5a Transported by Exosomes Activate WNT Signaling Pathways in Human Cardiac Fibroblasts. Int. J. Mol. Sci. 2019, 20, 1436. [Google Scholar] [CrossRef] [PubMed]
- Sukhacheva, T.V.; Nizyaeva, N.V.; Samsonova, M.V.; Cherniaev, A.L.; Burov, A.A.; Iurova, M.V.; Shchegolev, A.I.; Serov, R.A.; Sukhikh, G.T. Morpho-functional changes of cardiac telocytes in isolated atrial amyloidosis in patients with atrial fibrillation. Sci. Rep. 2021, 11, 3563. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Tang, Y.; Chen, X.; Yang, Y. Telocytes-derived extracellular vesicles alleviate aortic valve calcification by carrying miR-30b. ESC Hear. Fail. 2021, 8, 3935–3946. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef]
- Vilahur, G.; Juan-Babot, O.; Peña, E.; Oñate, B.; Casaní, L.; Badimon, L. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J. Mol. Cell. Cardiol. 2011, 50, 522–533. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-β Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-β superfamily signaling pathways in human disease. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 197–228. [Google Scholar] [CrossRef]
- Lai, P.F.H.; Courtman, D.W.; Stewart, D.J. NO to Small Mothers Against Decapentaplegic (Smad). Circ. Res. 2005, 97, 1087–1089. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yousefi, F.; Shabaninejad, Z.; Vakili, S.; Derakhshan, M.; Movahedpour, A.; Dabiri, H.; Ghasemi, Y.; Mahjoubin-Tehran, M.; Nikoozadeh, A.; Savardashtaki, A.; et al. TGF-β and WNT signaling pathways in cardiac fibrosis: Non-coding RNAs come into focus. Cell Commun. Signal. 2020, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Kulessa, H.; Tompkins, K.; Zhou, Y.; Batts, L.; Baldwin, H.S.; Hogan, B.L.M. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003, 17, 2362–2367. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Selever, J.; Wang, D.; Lu, M.-F.; Moses, K.A.; Schwartz, R.J.; Martin, J.F. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc. Natl. Acad. Sci. USA 2004, 101, 4489–4494. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef]
- Alexandra, K.; Yumiko, S.; Taketo, M.M.; Eldad, T.; Walter, B. Distinct roles of Wnt/β-catenin and Bmp signaling during early cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18531–18536. [Google Scholar] [CrossRef]
- Schultheiss, T.M.; Burch, J.B.; Lassar, A.B. A role for bone morphogenetic proteins in the induction of cardiac myogenesis. Genes Dev. 1997, 11, 451–462. [Google Scholar] [CrossRef]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef]
- Song, D.; Tang, L.; Huang, J.; Wang, L.; Zeng, T.; Wang, X. Roles of transforming growth factor-β and phosphatidylinositol 3-kinase isoforms in integrin β1-mediated bio-behaviors of mouse lung telocytes. J. Transl. Med. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Tang, L.; Song, D.; Qi, R.; Zhu, B.; Wang, X. Roles of pulmonary telocytes in airway epithelia to benefit experimental acute lung injury through production of telocyte-driven mediators and exosomes. Cell Biol. Toxicol. 2022, 3, 1–15. [Google Scholar] [CrossRef]
- Zhang, X.G.; Wei, Y.; Jiang, J.; Wang, L.; Liang, H.Y.; Lei, C.B. Effect of TGF-β1 on myocardial cell apoptosis in rats with acute myocardial infarction via MAPK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1350–1356. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.T.; Akazawa, H.; Takano, H.; Minamino, T.; Nagai, T.; Aburatani, H.; Komuro, I. Phosphatidylinositol 3-kinase-Akt pathway plays a critical role in early cardiomyogenesis by regulating canonical Wnt signaling. Circ. Res. 2005, 97, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Działo, E.; Czepiel, M.; Tkacz, K.; Siedlar, M.; Kania, G.; Błyszczuk, P. WNT/β-Catenin Signaling Promotes TGF-β-Mediated Activation of Human Cardiac Fibroblasts by Enhancing IL-11 Production. Int. J. Mol. Sci. 2021, 22, 10072. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.P.; Herazo-Maya, J.D.; Sennello, J.A.; Flozak, A.S.; Russell, S.; Mutlu, G.M.; Budinger, G.R.S.; DasGupta, R.; Varga, J.; Kaminski, N.; et al. Wnt coreceptor Lrp5 is a driver of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Yagi, K.; Yamamoto, H.; Furukawa, Y.; Shimada, S.; Nakamura, Y.; Kikuchi, A.; Miyazono, K.; Kato, M. Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol. Cell. Biol. 2001, 21, 5132–5141. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.R.; Greene, R.M.; Pisano, M.M. Interaction between Smad 3 and Dishevelled in murine embryonic craniofacial mesenchymal cells. Orthod. Craniofac. Res. 2005, 8, 123–130. [Google Scholar] [CrossRef]
- Kumawat, K.; Menzen, M.H.; Slegtenhorst, R.M.; Halayko, A.J.; Schmidt, M.; Gosens, R. TGF-β-activated kinase 1 (TAK1) signaling regulates TGF-β-induced WNT-5A expression in airway smooth muscle cells via Sp1 and β-catenin. PLoS ONE 2014, 9, e94801. [Google Scholar] [CrossRef]
- Blyszczuk, P.; Müller-Edenborn, B.; Valenta, T.; Osto, E.; Stellato, M.; Behnke, S.; Glatz, K.; Basler, K.; Lüscher, T.F.; Distler, O.; et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur. Heart J. 2017, 38, 1413–1425. [Google Scholar] [CrossRef]
- Haudek, S.B.; Gupta, D.; Dewald, O.; Schwartz, R.J.; Wei, L.; Trial, J.; Entman, M.L. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc. Res. 2009, 83, 511–518. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Bo, J.; Taffet, G.E.; Chang, J.; Shi, J.; Reddy, A.K.; Michael, L.H.; Schneider, M.D.; Entman, M.L.; Schwartz, R.J.; et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Lauriol, J.; Keith, K.; Jaffré, F.; Couvillon, A.; Saci, A.; Goonasekera, S.A.; McCarthy, J.R.; Kessinger, C.W.; Wang, J.; Ke, Q.; et al. RhoA signaling in cardiomyocytes protects against stress-induced heart failure but facilitates cardiac fibrosis. Sci. Signal. 2014, 7, ra100. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Matsumori, A.; Shioi, T.; Furukawa, Y.; Sasayama, S. Cytokine gene expression after myocardial infarction in rat hearts: Possible implication in left ventricular remodeling. Circulation 1998, 98, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Tang, L.; Wang, L.; Huang, J.; Zeng, T.; Fang, H.; Wang, X. Roles of TGFβ1 in the expression of phosphoinositide 3-kinase isoform genes and sensitivity and response of lung telocytes to PI3K inhibitors. Cell Biol. Toxicol. 2020, 36, 51–64. [Google Scholar] [CrossRef]
- Zheng, L.; Li, L.; Qi, G.; Hu, M.; Hu, C.; Wang, S.; Li, J.; Zhang, M.; Zhang, W.; Zeng, Y.; et al. Transplantation of Telocytes Attenuates Unilateral Ureter Obstruction-Induced Renal Fibrosis in Rats. Cell. Physiol. Biochem. 2018, 46, 2056–2071. [Google Scholar] [CrossRef]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Cao, L.; Massey, I.Y. Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol. Cell. Biochem. 2021, 476, 4045–4059. [Google Scholar] [CrossRef]
- Carnero, A.; Paramio, J.M. The PTEN/PI3K/AKT Pathway in vivo, Cancer Mouse Models. Front. Oncol. 2014, 4, 252. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Sun, H.; Kerfant, B.-G.; Crackower, M.A.; Penninger, J.M.; Backx, P.H. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol. 2004, 37, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Shioi, T.; McMullen, J.R.; Tarnavski, O.; Converso, K.; Sherwood, M.C.; Manning, W.J.; Izumo, S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 2003, 107, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006, 20, 3347–3365. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Sato, K.; Izumiya, Y.; Schiekofer, S.; Ito, M.; Liao, R.; Colucci, W.S.; Walsh, K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Investig. 2005, 115, 2108–2118. [Google Scholar] [CrossRef]
- Guo, Y.; Gupte, M.; Umbarkar, P.; Singh, A.P.; Sui, J.Y.; Force, T.; Lal, H. Entanglement of GSK-3β, β-catenin and TGF-β1 signaling network to regulate myocardial fibrosis. J. Mol. Cell. Cardiol. 2017, 110, 109–120. [Google Scholar] [CrossRef]
- Seoane, J.; Le, H.-V.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and Forkhead Pathways in the Control of Neuroepithelial and Glioblastoma Cell Proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Ma, Z.-G.; Yuan, Y.-P.; Zhang, X.; Xu, S.-C.; Wang, S.-S.; Tang, Q.-Z. Piperine Attenuates Pathological Cardiac Fibrosis Via PPAR-γ/AKT Pathways. EBioMedicine 2017, 18, 179–187. [Google Scholar] [CrossRef]
- Chow, J.Y.C.; Quach, K.T.; Cabrera, B.L.; Cabral, J.A.; Beck, S.E.; Carethers, J.M. RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis 2007, 28, 2321–2327. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Crackower, M.A.; Eriksson, U.; Sarao, R.; Kozieradzki, I.; Sasaki, T.; Irie-Sasaki, J.; Gidrewicz, D.; Rybin, V.O.; Wada, T.; et al. Phosphoinositide 3-kinase gamma-deficient mice are protected from isoproterenol-induced heart failure. Circulation 2003, 108, 2147–2152. [Google Scholar] [CrossRef]
- Onose, G.; Anghelescu, A.; Blendea, D.; Ciobanu, V.; Daia, C.; Firan, F.; Oprea, M.; Spinu, A.; Popescu, C.; Ionescu, A.; et al. Cellular and Molecular Targets for Non-Invasive, Non-Pharmacological Therapeutic/Rehabilitative Interventions in Acute Ischemic Stroke. Int. J. Mol. Sci. 2022, 23, 907. [Google Scholar] [CrossRef]
- Gherghiceanu, M.; Popescu, L.M. Cardiac telocytes—Their junctions and functional implications. Cell Tissue Res. 2012, 348, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Fertig, E.T.; Gherghiceanu, M.; Popescu, L.M. Extracellular vesicles release by cardiac telocytes: Electron microscopy and electron tomography. J. Cell. Mol. Med. 2014, 18, 1938–1943. [Google Scholar] [CrossRef] [PubMed]
- Popescu, L.M.; Fertig, E.T.; Gherghiceanu, M. Reaching out: Junctions between cardiac telocytes and cardiac stem cells in culture. J. Cell. Mol. Med. 2016, 20, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Kaestner, K.H. Emerging diverse roles of telocytes. Development 2019, 146, dev175018. [Google Scholar] [CrossRef]
- Hao, J.; Ju, H.; Zhao, S.; Junaid, A.; Scammell-La Fleur, T.; Dixon, I.M. Elevation of expression of Smads 2, 3, and 4, decorin and TGF-beta in the chronic phase of myocardial infarct scar healing. J. Mol. Cell. Cardiol. 1999, 31, 667–678. [Google Scholar] [CrossRef]
- Carthy, J.M.; Garmaroudi, F.S.; Luo, Z.; McManus, B.M. Wnt3a Induces Myofibroblast Differentiation by Upregulating TGF-β Signaling Through SMAD2 in a β-Catenin-Dependent Manner. PLoS ONE 2011, 6, e19809. [Google Scholar] [CrossRef]
- Cohen, E.D.; Miller, M.F.; Wang, Z.; Moon, R.T.; Morrisey, E.E. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development 2012, 139, 1931–1940. [Google Scholar] [CrossRef]
- Song, H.-P.; Chu, Z.-G.; Zhang, D.-X.; Dang, Y.-M.; Zhang, Q. PI3K-AKT Pathway Protects Cardiomyocytes Against Hypoxia-Induced Apoptosis by MitoKATP-Mediated Mitochondrial Translocation of pAKT. Cell. Physiol. Biochem. 2018, 49, 717–727. [Google Scholar] [CrossRef]
- Carll, A.P.; Willis, M.S.; Lust, R.M.; Costa, D.L.; Farraj, A.K. Merits of non-invasive rat models of left ventricular heart failure. Cardiovasc. Toxicol. 2011, 11, 91–112. [Google Scholar] [CrossRef]
- Wang, J.; Bo, H.; Meng, X.; Wu, Y.; Bao, Y.; Li, Y. A simple and fast experimental model of myocardial infarction in the mouse. Texas Hear. Inst. J. 2006, 33, 290–293. [Google Scholar]
- Dani, S.S.; Lone, A.N.; Javed, Z.; Khan, M.S.; Zia Khan, M.; Kaluski, E.; Virani, S.S.; Shapiro, M.D.; Cainzos-Achirica, M.; Nasir, K.; et al. Trends in Premature Mortality From Acute Myocardial Infarction in the United States, 1999 to 2019. J. Am. Heart Assoc. 2022, 11, e021682. [Google Scholar] [CrossRef] [PubMed]
- Wöhrle, J.; Merkle, N.; Mailänder, V.; Nusser, T.; Schauwecker, P.; von Scheidt, F.; Schwarz, K.; Bommer, M.; Wiesneth, M.; Schrezenmeier, H.; et al. Results of intracoronary stem cell therapy after acute myocardial infarction. Am. J. Cardiol. 2010, 105, 804–812. [Google Scholar] [CrossRef]
- Müller, P.; Lemcke, H.; David, R. Stem Cell Therapy in Heart Diseases—Cell Types, Mechanisms and Improvement Strategies. Cell. Physiol. Biochem. 2018, 48, 2607–2655. [Google Scholar] [CrossRef] [PubMed]
- Popescu, L.M.; Gherghiceanu, M.; Manole, C.G.; Faussone-Pellegrini, M.S. Cardiac renewing: Interstitial Cajal-like cells nurse cardiomyocyte progenitors in epicardial stem cell niches. J. Cell. Mol. Med. 2009, 13, 866–886. [Google Scholar] [CrossRef]
- Venugopal, H.; Hanna, A.; Humeres, C.; Frangogiannis, N.G. Properties and Functions of Fibroblasts and Myofibroblasts in Myocardial Infarction. Cells 2022, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, W.; Fang, Y.; Hu, H.; Chang, N.; Pang, M.; Hu, Y.-F.; Li, X.; Long, H.; Xiong, J.-W.; et al. Inhibition of TGF-β/Smad3 Signaling Disrupts Cardiomyocyte Cell Cycle Progression and Epithelial-Mesenchymal Transition-Like Response During Ventricle Regeneration. Front. cell Dev. Biol. 2021, 9, 632372. [Google Scholar] [CrossRef] [PubMed]
- Chablais, F.; Jazwinska, A. The regenerative capacity of the zebrafish heart is dependent on TGFβ signaling. Development 2012, 139, 1921–1930. [Google Scholar] [CrossRef]
- Lin, Z.; Zhou, P.; von Gise, A.; Gu, F.; Ma, Q.; Chen, J.; Guo, H.; van Gorp, P.R.R.; Wang, D.-Z.; Pu, W.T. Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ. Res. 2015, 116, 35–45. [Google Scholar] [CrossRef]
- Walkowski, B.; Kleibert, M.; Majka, M.; Wojciechowska, M. Insight into the Role of the PI3K/Akt Pathway in Ischemic Injury and Post-Infarct Left Ventricular Remodeling in Normal and Diabetic Heart. Cells 2022, 11, 1553. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucu, I.; Nicolescu, M.I.; Busnatu, Ș.-S.; Manole, C.G. Dynamic Involvement of Telocytes in Modulating Multiple Signaling Pathways in Cardiac Cytoarchitecture. Int. J. Mol. Sci. 2022, 23, 5769. https://doi.org/10.3390/ijms23105769
Cucu I, Nicolescu MI, Busnatu Ș-S, Manole CG. Dynamic Involvement of Telocytes in Modulating Multiple Signaling Pathways in Cardiac Cytoarchitecture. International Journal of Molecular Sciences. 2022; 23(10):5769. https://doi.org/10.3390/ijms23105769
Chicago/Turabian StyleCucu, Ioana, Mihnea Ioan Nicolescu, Ștefan-Sebastian Busnatu, and Cătălin Gabriel Manole. 2022. "Dynamic Involvement of Telocytes in Modulating Multiple Signaling Pathways in Cardiac Cytoarchitecture" International Journal of Molecular Sciences 23, no. 10: 5769. https://doi.org/10.3390/ijms23105769
APA StyleCucu, I., Nicolescu, M. I., Busnatu, Ș.-S., & Manole, C. G. (2022). Dynamic Involvement of Telocytes in Modulating Multiple Signaling Pathways in Cardiac Cytoarchitecture. International Journal of Molecular Sciences, 23(10), 5769. https://doi.org/10.3390/ijms23105769