
Correlation of Immunological and Molecular Profiles with Response to Crizotinib in Alveolar Soft Part Sarcoma: An Exploratory Study Related to the EORTC 90101 “CREATE” Trial

 ,
, 
 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Patient Cohort
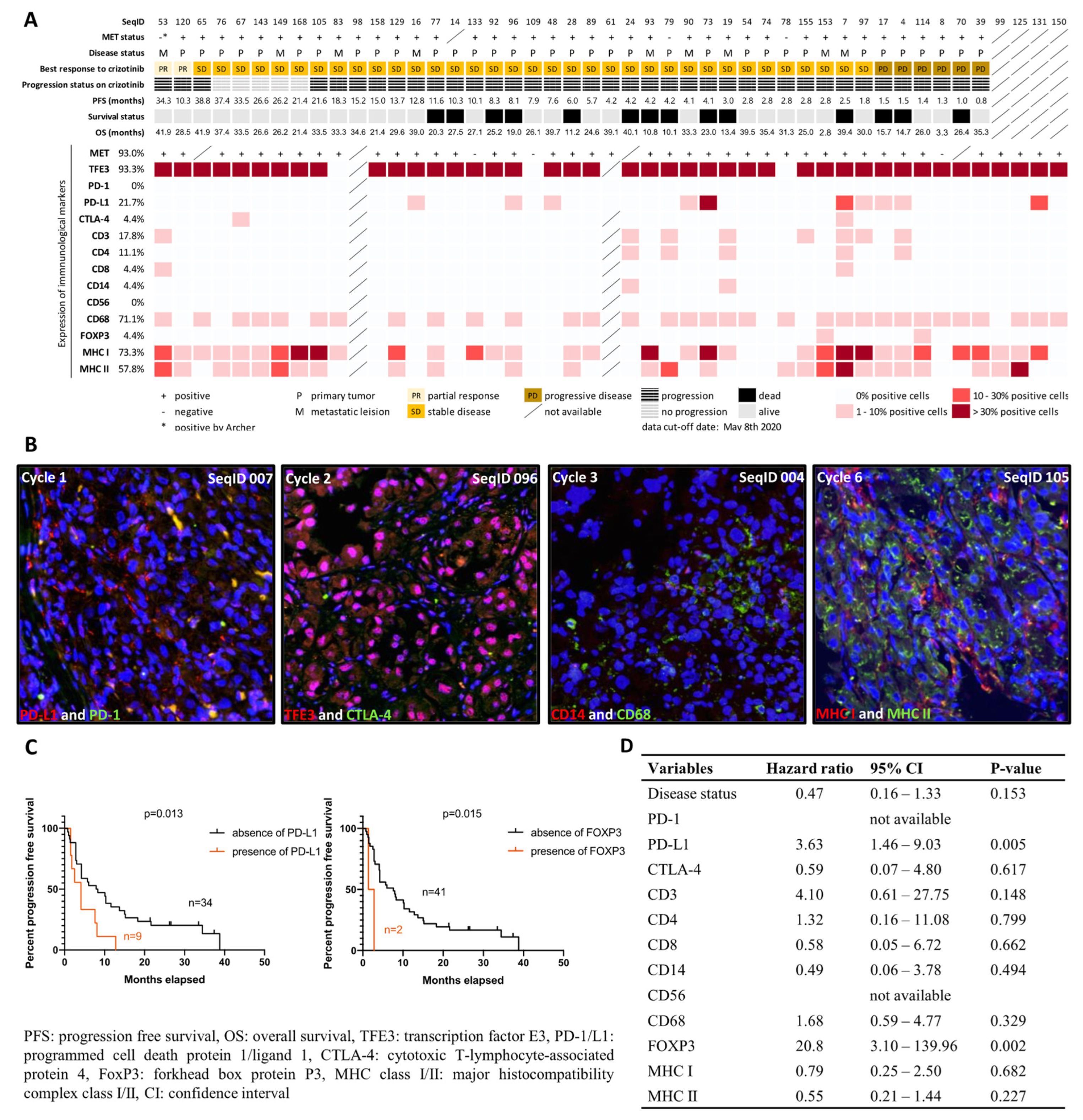
2.2. Characterization of Immunological Components in the Tumor Microenvironment
2.3. MET Status and MET Expression
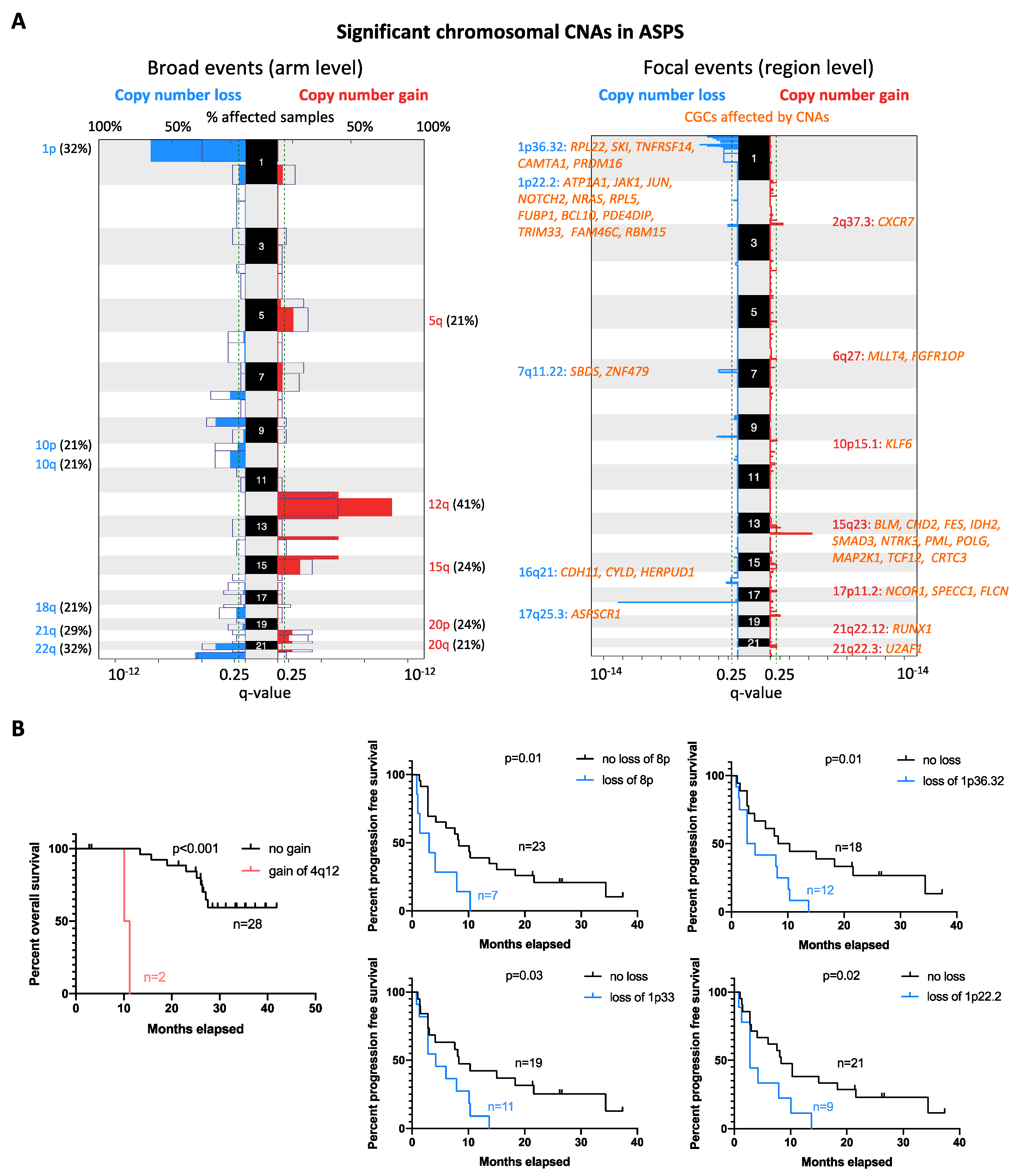
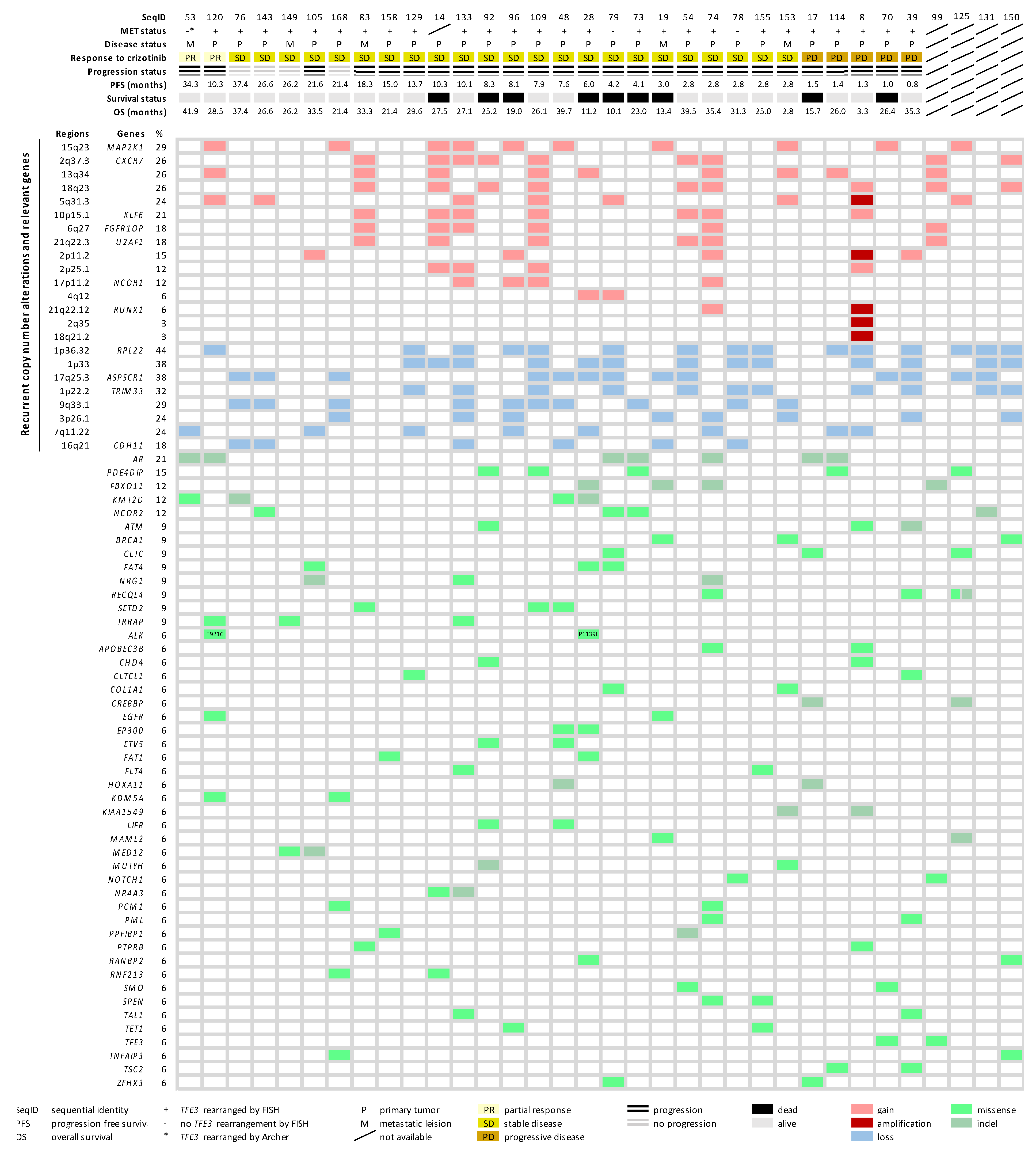
2.4. Copy Number Alteration Profile
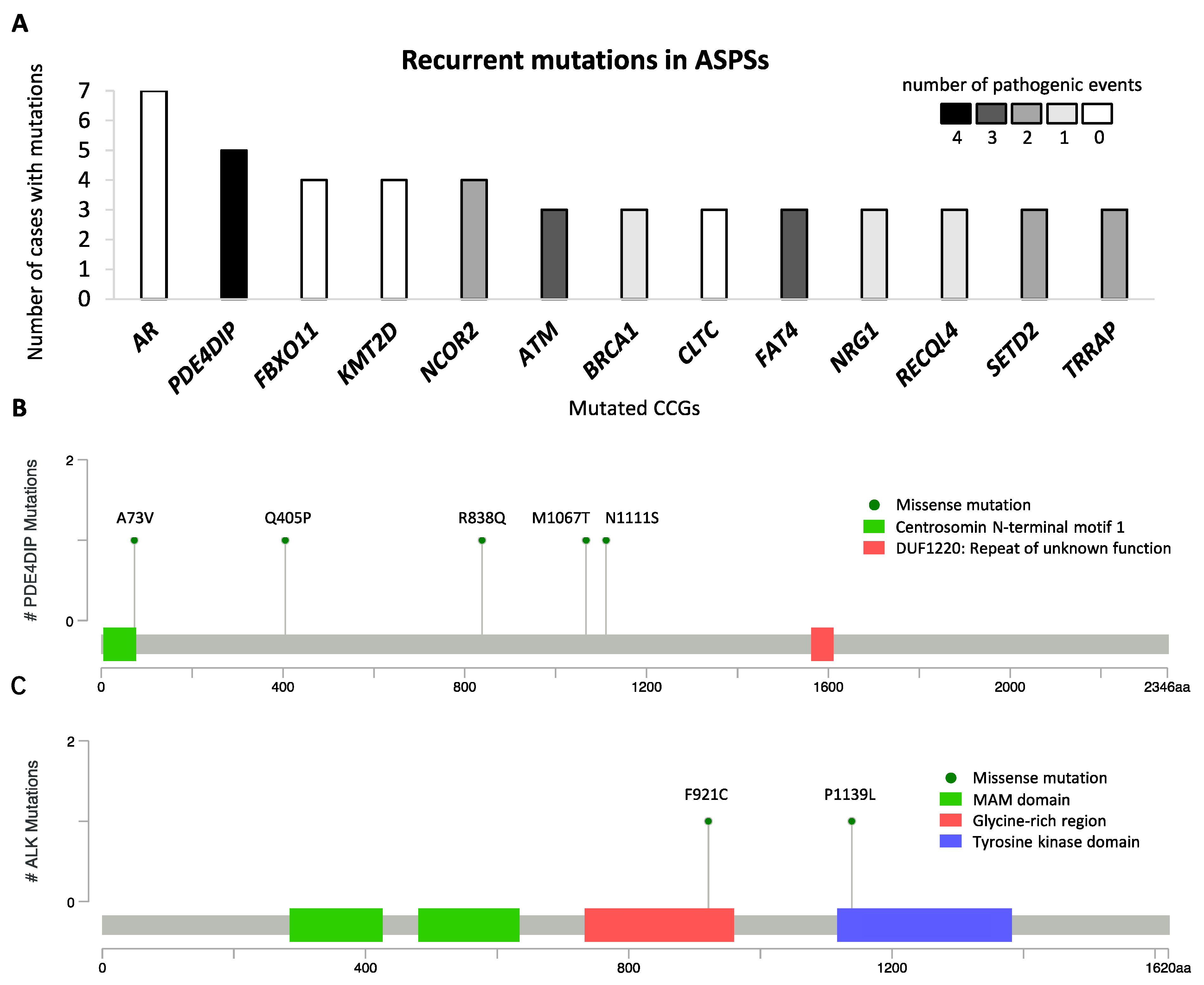
2.5. Mutational Landscape of ASPS
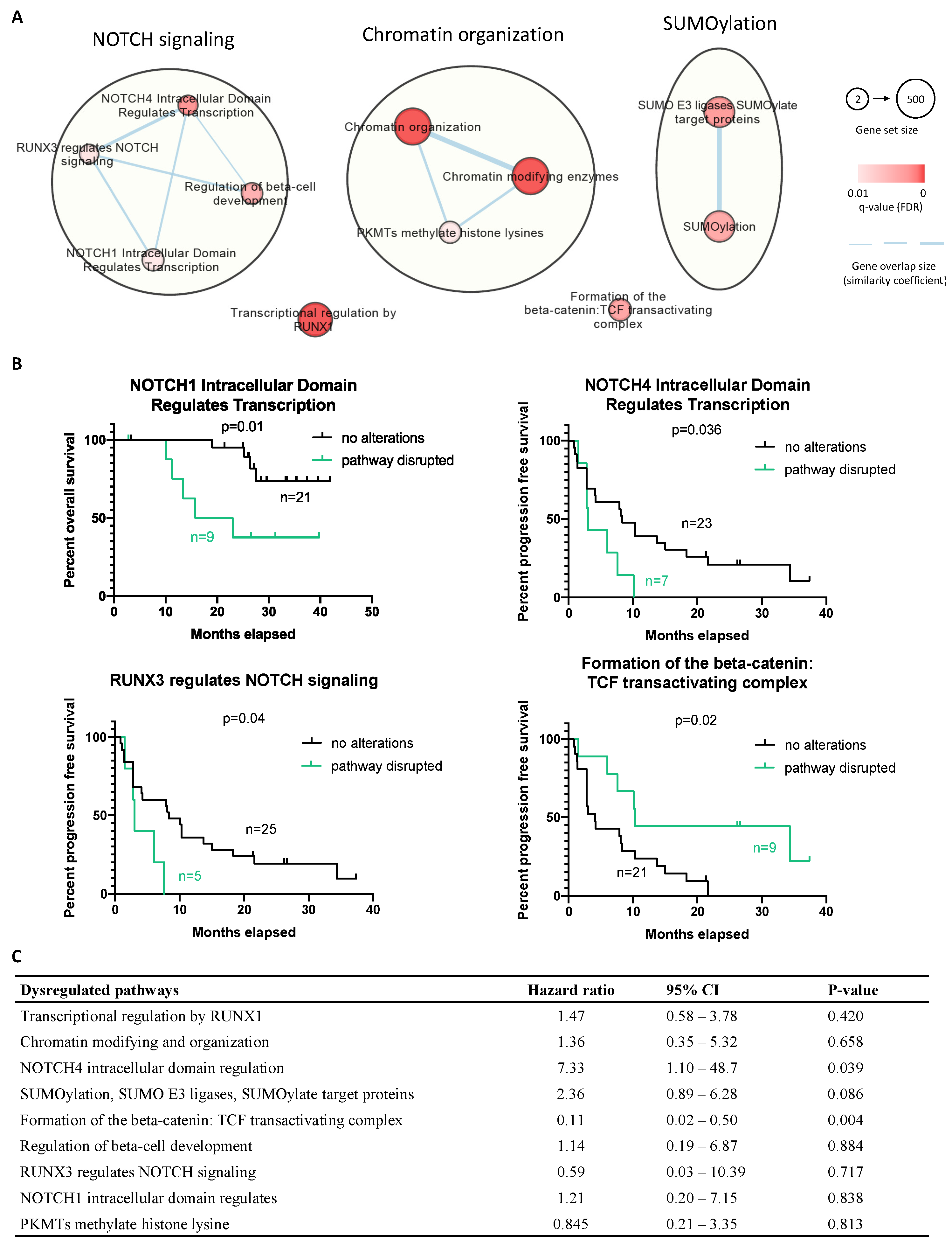
2.6. Molecular Landscape of ASPS
3. Discussion
4. Material and Methods
4.1. Characterization of Immunological Components in the Tumor Microenvironment
4.2. MET Status and MET Expression
4.3. Low-Coverage Whole-Genome Sequencing
4.4. Whole Exome Sequencing
4.5. Pathway Enrichment Analysis
4.6. Clinical Outcome and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASPS | alveolar soft part sarcoma |
| CNA | copy number alteration |
| STS | Soft tissue sarcoma |
| ASPSCR1-TFE3 | alveolar soft part sarcoma critical region 1-transcription factor E3 |
| EORTC | European Organization for Research and Treatment of Cancer |
| FISH | fluorescence in situ hybridization |
| NSCLC | non-small cell lung cancer |
| PI3K | phosphoinositide 3-kinase |
| PD-L1 | programmed cell death protein ligand 1 |
| TIL | tumor-infiltrating lymphocyte |
| EGFR | epidermal growth factor receptor |
| TME | tumor microenvironment |
| FFPE | formalin-fixed, paraffin-embedded |
| TMA | tissue microarray |
| MILAN | Multiple Iteractive Labeling by Antibody Neodeposition |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| PD-1 | programmed cell death protein 1 |
| MHC | major histocompatibility complex class |
| FOXP3 | forkhead box protein P3 |
| GISTIC | Genomic Identification of Significant Targets in Cancer |
| CGC | Cancer Gene Consensus |
| COSMIC | Catalogue of Somatic Mutations in Cancer databases |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| PFS | progression-free |
| OS | overall survival |
| PR | partial response |
| SD | stable disease |
| PD | progressive disease |
| DAPI | 4′,6-diamidino-2-phenylindole |
| TCF | T-cell factor |
| VSC | Flemish Supercomputer Center |
| FWO | Research Foundation—Flanders |
References
- de Pinieux, G.; Karanian, M.; Loarer, F.L.; Guellec, S.L.; Chabaud, S.; Terrier, P.; Bouvier, C.; Batistella, M.; Neuville, A.; Robin, Y.-M.; et al. Nationwide Incidence of Sarcomas and Connective Tissue Tumors of Intermediate Malignancy over Four Years Using an Expert Pathology Review Network. PLoS ONE 2021, 16, e0246958. [Google Scholar] [CrossRef] [PubMed]
- Folpe, A.L.; Deyrup, A.T. Alveolar Soft-part Sarcoma: A Review and Update. J. Clin. Pathol. 2006, 59, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Paoluzzi, L.; Maki, R.G. Diagnosis, Prognosis, and Treatment of Alveolar Soft-Part Sarcoma: A Review. JAMA Oncol. 2019, 5, 254–260. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. World Health Organization Classification of Tumours, 5th ed.; IARC Press: Lyon, France, 2020; Volume 3. [Google Scholar]
- Tsuda, M.; Davis, I.J.; Argani, P.; Shukla, N.; McGill, G.G.; Nagai, M.; Saito, T.; Laé, M.; Fisher, D.E.; Ladanyi, M. TFE3 Fusions Activate MET Signaling by Transcriptional Up-Regulation, Defining Another Class of Tumors as Candidates for Therapeutic MET Inhibition. Cancer Res. 2007, 67, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Wozniak, A.; Kasper, B.; Aamdal, S.; Leahy, M.G.; Rutkowski, P.; Bauer, S.; Gelderblom, H.; Italiano, A.; Lindner, L.H.; et al. Activity and Safety of Crizotinib in Patients with Alveolar Soft Part Sarcoma with Rearrangement of TFE3: European Organization for Research and Treatment of Cancer (EORTC) Phase II Trial 90101 ‘CREATE’. Ann. Oncol 2018, 29, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.-H.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor Activity of Crizotinib in Lung Cancers Harboring a MET Exon 14 Alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef]
- Jamme, P.; Fernandes, M.; Copin, M.-C.; Descarpentries, C.; Escande, F.; Morabito, A.; Grégoire, V.; Jamme, M.; Baldacci, S.; Tulasne, D.; et al. Alterations in the PI3K Pathway Drive Resistance to MET Inhibitors in NSCLC Harboring MET Exon 14 Skipping Mutations. J. Thorac. Oncol. 2020, 15, 741–751. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Sawa, K.; Fukui, M.; Oyanagi, J.; Izumi, M.; Ogawa, K.; Suzumura, T.; Watanabe, T.; Kaneda, H.; Mitsuoka, S.; et al. Impact of Tumor Microenvironment on the Efficacy of Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Non-Small Cell Lung Cancer. Cancer Sci. 2019, 110, 3244–3254. [Google Scholar] [CrossRef]
- Naqash, A.R.; O’Sullivan Coyne, G.H.; Moore, N.; Sharon, E.; Takebe, N.; Fino, K.K.; Ferry-Galow, K.V.; Hu, J.S.; Van Tine, B.A.; Burgess, M.A.; et al. Phase II Study of Atezolizumab in Advanced Alveolar Soft Part Sarcoma (ASPS). J. Clin. Oncol. 2021, 39, 11519. [Google Scholar] [CrossRef]
- Lewin, J.; Davidson, S.; Anderson, N.D.; Lau, B.Y.; Kelly, J.; Tabori, U.; Salah, S.; Butler, M.O.; Aung, K.L.; Shlien, A.; et al. Response to Immune Checkpoint Inhibition in Two Patients with Alveolar Soft-Part Sarcoma. Cancer Immunol. Res. 2018, 6, 1001–1007. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive Biomarkers for Checkpoint Inhibitor-Based Immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune Infiltration and PD-L1 Expression in the Tumor Microenvironment Are Prognostic in Osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Huang, H. Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces. Int. J. Mol. Sci. 2018, 19, 3448. [Google Scholar] [CrossRef]
- Wei, Z.-W.; Wu, J.; Huang, W.-B.; Li, J.; Lu, X.-F.; Yuan, Y.-J.; Xiong, W.-J.; Zhang, X.-H.; Wang, W.; He, Y.-L.; et al. Immune-Infiltration Based Signature as a Novel Prognostic Biomarker in Gastrointestinal Stromal Tumour. EBioMedicine 2020, 57, 102850. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, L.; Xia, Y.; Chen, X.; Xiong, Y.; Qu, Y.; Wang, J.; Bai, Q.; Guo, J.; Xu, J. Tumor Infiltrating CD19+ B Lymphocytes Predict Prognostic and Therapeutic Benefits in Metastatic Renal Cell Carcinoma Patients Treated with Tyrosine Kinase Inhibitors. Oncoimmunology 2018, 7, e1477461. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.-Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-Associated Macrophages and Macrophage-Related Immune Checkpoint Expression in Sarcomas. OncoImmunology 2020, 9, 1747340. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Perrot, D.; Leroux, A.; Collin, F.; Le Cesne, A.; Coindre, J.-M.; Blay, J.-Y.; Birnbaum, D.; Mamessier, E. PDL1 Expression Is a Poor-Prognosis Factor in Soft-Tissue Sarcomas. Oncoimmunology 2017, 6, e1278100. [Google Scholar] [CrossRef]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef]
- Kwai, A.; Nishikawa, T.; Kawasaki, M.; Tomatsuri, S.; Okamura, N.; Ogawa, G.; Hirakawa, A.; Shibata, T.; Nakamura, T.; Kakunaga, S.; et al. Efficacy and Safety of Nivolumab Monothereapy in Patients with Unresectable Clear Cell Sarcoma and Alveolar Soft Part Sarcoma (OSARC TRIAL, NCCH1510): A Muticenter, Phase 2 Clinical Trial. In Proceedings of the CTOS Annual Meeting, Vancouver, BC, Canada, 18–21 November 2020. [Google Scholar]
- Mariuk-Jarema, A.; Koseła-Paterczyk, H.; Rogala, P.; Klimczak, A.; Wągrodzki, M.; Maksymiuk, B.; Rutkowski, P. A Durable Complete Response to Immunotherapy in a Patient with Metastatic Alveolar Soft Part Sarcoma. Tumori 2020, 106, NP9–NP13. [Google Scholar] [CrossRef]
- Hodge, J.C.; Pearce, K.E.; Wang, X.; Wiktor, A.E.; Oliveira, A.M.; Greipp, P.T. Molecular Cytogenetic Analysis for TFE3 Rearrangement in Xp11.2 Renal Cell Carcinoma and Alveolar Soft Part Sarcoma: Validation and Clinical Experience with 75 Cases. Mod. Pathol. 2014, 27, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Aulmann, S.; Longerich, T.; Schirmacher, P.; Mechtersheimer, G.; Penzel, R. Detection of the ASPSCR1–TFE3 Gene Fusion in Paraffin-Embedded Alveolar Soft Part Sarcomas. Histopathology 2007, 50, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Brady, O.A.; Jeong, E.; Martina, J.A.; Pirooznia, M.; Tunc, I.; Puertollano, R. The Transcription Factors TFE3 and TFEB Amplify P53 Dependent Transcriptional Programs in Response to DNA Damage. eLife 2018, 7, e40856. [Google Scholar] [PubMed]
- Morozov, V.M.; Massoll, N.A.; Vladimirova, O.V.; Maul, G.G.; Ishov, A.M. Regulation of C-Met Expression by Transcription Repressor Daxx. Oncogene 2008, 27, 2177–2186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, J.; Babic, A. Regulation of the MET Oncogene: Molecular Mechanisms. Carcinogenesis 2016, 37, 345–355. [Google Scholar] [CrossRef]
- van Echten, J.; van den Berg, E.; van Baarlen, J.; van Noort, G.; Vermey, A.; Dam, A.; Molenaar, W.M. An Important Role for Chromosome 17, Band Q25, in the Histogenesis of Alveolar Soft Part Sarcoma. Cancer Genet. Cytogenet 1995, 82, 57–61. [Google Scholar] [CrossRef]
- Sreekantaiah, C.; Li, F.P.; Weidner, N.; Sandberg, A.A. Multiple and Complex Abnormalities in a Case of Alveolar Soft-Part Sarcoma. Cancer Genet. Cytogenet 1991, 55, 167–171. [Google Scholar] [CrossRef]
- Heidenblad, M.; Hallor, K.H.; Staaf, J.; Jönsson, G.; Borg, Å.; Höglund, M.; Mertens, F.; Mandahl, N. Genomic Profiling of Bone and Soft Tissue Tumors with Supernumerary Ring Chromosomes Using Tiling Resolution Bacterial Artificial Chromosome Microarrays. Oncogene 2006, 25, 7106–7116. [Google Scholar] [CrossRef][Green Version]
- Italiano, A.; Cardot, N.; Dupré, F.; Monticelli, I.; Keslair, F.; Piche, M.; Mainguené, C.; Coindre, J.-M.; Pedeutour, F. Gains and Complex Rearrangements of the 12q13-15 Chromosomal Region in Ordinary Lipomas: The “Missing Link” between Lipomas and Liposarcomas? Int. J. Cancer Res. 2007, 121, 308–315. [Google Scholar] [CrossRef]
- Agaram, N.P.; Zhang, L.; Yun-Shao, S.; Singer, S.; Stevens, T.; Prieto-Granada, C.N.; Bishop, J.A.; Wood, B.A.; Swanson, D.; Dickson, B.C.; et al. GLI1-Amplifications Expands the Spectrum of Soft Tissue Neoplasms Defined by GLI1 Gene Fusions. Mod. Pathol. 2019, 32, 1617–1626. [Google Scholar] [CrossRef]
- Bagchi, A.; Mills, A.A. The Quest for the 1p36 Tumor Suppressor. Cancer Res. 2008, 68, 2551–2556. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Weiss, M.J.; Guo, C.; Gerbing, R.B.; Stram, D.O.; White, P.S.; Hogarty, M.D.; Sulman, E.P.; Thompson, P.M.; Lukens, J.N.; et al. Loss of Heterozygosity at 1p36 Independently Predicts for Disease Progression but Not Decreased Overall Survival Probability in Neuroblastoma Patients: A Children’s Cancer Group Study. J. Clin. Oncol. 2000, 18, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Fee, B.E.; Rivas, M.V.; Lin, J.; Adamson, D.C. Adherens Junctional Associated Protein-1: A Novel 1p36 Tumor Suppressor Candidate in Gliomas (Review). Int. J. Oncol. 2014, 45, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Roubin, R.; Acquaviva, C.; Chevrier, V.; Sedjaï, F.; Zyss, D.; Birnbaum, D.; Rosnet, O. Myomegalin Is Necessary for the Formation of Centrosomal and Golgi-Derived Microtubules. Biol. Open 2012, 2, 238–250. [Google Scholar] [CrossRef]
- Er, T.-K.; Su, Y.-F.; Wu, C.-C.; Chen, C.-C.; Wang, J.; Hsieh, T.-H.; Herreros-Villanueva, M.; Chen, W.-T.; Chen, Y.-T.; Liu, T.-C.; et al. Targeted Next-Generation Sequencing for Molecular Diagnosis of Endometriosis-Associated Ovarian Cancer. J. Mol. Med. 2016, 94, 835–847. [Google Scholar] [CrossRef]
- da Costa, L.T.; Dos Anjos, L.G.; Kagohara, L.T.; Torrezan, G.T.; De Paula, C.A.A.; Baracat, E.C.; Carraro, D.M.; Carvalho, K.C. The Mutational Repertoire of Uterine Sarcomas and Carcinosarcomas in a Brazilian Cohort: A Preliminary Study. Clinics 2021, 76, e2324. [Google Scholar] [CrossRef]
- Groisberg, R.; Roszik, J.; Conley, A.P.; Lazar, A.J.; Portal, D.E.; Hong, D.S.; Naing, A.; Herzog, C.E.; Somaiah, N.; Zarzour, M.A.; et al. Genomics, Morphoproteomics, and Treatment Patterns of Patients with Alveolar Soft Part Sarcoma and Response to Multiple Experimental Therapies. Mol. Cancer Ther. 2020, 19, 1165–1172. [Google Scholar] [CrossRef]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 Lysine 4 Methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb Perspect Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Sun, J.; Wang, Y.; Xia, Y.; Xu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Mutations in RECQL Gene Are Associated with Predisposition to Breast Cancer. PLoS Genet. 2015, 11, e1005228. [Google Scholar] [CrossRef]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Holla, V.R.; Elamin, Y.Y.; Bailey, A.M.; Johnson, A.M.; Litzenburger, B.C.; Khotskaya, Y.B.; Sanchez, N.S.; Zeng, J.; Shufean, M.A.; Shaw, K.R.; et al. ALK: A Tyrosine Kinase Target for Cancer Therapy. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001115. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Shim, H.S.; Gingipally, S.; Mino-Kenudson, M.; Le, L.; Gainor, J.F.; Zheng, Z.; Aryee, M.; Xia, J.; Jia, P.; et al. MET Exon 14 Skipping in Non-Small Cell Lung Cancer. Oncologist 2016, 21, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhang, J.; Lu, X.; Wang, W.; Chen, H.; Robinson, M.K.; Cheng, J.; Tang, G.; Medeiros, L.J. Coexistent Genetic Alterations Involving ALK, RET, ROS1 or MET in 15 Cases of Lung Adenocarcinoma. Mod. Pathol. 2018, 31, 307–312. [Google Scholar] [CrossRef]
- Miller, A.J.; Levy, C.; Davis, I.J.; Razin, E.; Fisher, D.E. Sumoylation of MITF and Its Related Family Members TFE3 and TFEB. J. Biol. Chem. 2005, 280, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Rota, R.; Ciarapica, R.; Miele, L.; Locatelli, F. Notch Signaling in Pediatric Soft Tissue Sarcomas. BMC Med. 2012, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Kolligs, F.T.; Bommer, G.; Göke, B. Wnt/Beta-Catenin/Tcf Signaling: A Critical Pathway in Gastrointestinal Tumorigenesis. Digestion 2002, 66, 131–144. [Google Scholar] [CrossRef]
- Alshareef, A.; Gupta, N.; Zhang, H.-F.; Wu, C.; Haque, M.; Lai, R. High Expression of β-Catenin Contributes to the Crizotinib Resistant Phenotype in the Stem-like Cell Population in Neuroblastoma. Sci. Rep. 2017, 7, 16863. [Google Scholar] [CrossRef]
- Lee, C.-J.; Wozniak, A.; Van Cann, T.; Timmermans, I.; Wellens, J.; Vanleeuw, U.; Briaire-de Bruijn, I.H.; Britschgi, C.; Bovée, J.V.M.G.; Zlobec, I.; et al. Establishment of an Academic Tissue Microarray Platform as a Tool for Soft Tissue Sarcoma Research. Sarcoma 2021, 2021, e6675260. [Google Scholar] [CrossRef]
- Cattoretti, G.; Bosisio, F.M.; Marcelis, L.; Bolognesi, M.M. Multiple Iterative Labeling by Antibody Neodeposition (MILAN). Protoc. Exch. 2019. [Google Scholar] [CrossRef]
- Bolognesi, M.M.; Manzoni, M.; Scalia, C.R.; Zannella, S.; Bosisio, F.M.; Faretta, M.; Cattoretti, G. Multiplex Staining by Sequential Immunostaining and Antibody Removal on Routine Tissue Sections. J. Histochem. Cytochem. 2017, 65, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Loo, P.V.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-Specific Copy Number Analysis of Tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 16910–16915. [Google Scholar] [PubMed]
- Scheinin, I.; Sie, D.; Bengtsson, H.; van de Wiel, M.A.; Olshen, A.B.; van Thuijl, H.F.; van Essen, H.F.; Eijk, P.P.; Rustenburg, F.; Meijer, G.A.; et al. DNA Copy Number Analysis of Fresh and Formalin-Fixed Specimens by Shallow Whole-Genome Sequencing with Identification and Exclusion of Problematic Regions in the Genome Assembly. Genome Res. 2014, 24, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing Genetic Dysfunction across All Human Cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with Open Crowdsource Efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. G:Profiler—a Web-Based Toolset for Functional Profiling of Gene Lists from Large-Scale Experiments. Nucleic Acids Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef]
- Sidiropoulos, K.; Viteri, G.; Sevilla, C.; Jupe, S.; Webber, M.; Orlic-Milacic, M.; Jassal, B.; May, B.; Shamovsky, V.; Duenas, C.; et al. Reactome Enhanced Pathway Visualization. Bioinformatics 2017, 33, 3461–3467. [Google Scholar] [CrossRef]
- Reimand, J.; Isser, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway Enrichment Analysis and Visualization of Omics Data Using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study SeqID | Gender/Age | Tissue Source/ Type of Lesion | MET Status (EORTC 90,101 Protocol) | Best Response (RECIST) | Progression Status on Crizotinib | PFS (Months) | Survival Status | OS (Months) | Exploratory Study | |
|---|---|---|---|---|---|---|---|---|---|---|
| Status | % Cells Positive for FISH | |||||||||
| 4 | M/51 | Primary | MET + | nd | PD | Progression | 1.5 | Death | 14.7 | TMA+Sequencing |
| 7 | M/25 | Metastatic | MET + | nd | SD | Progression | 2.5 | Death | 39.4 | TMA |
| 8 | F/28 | Primary | MET + | nd | PD | Progression | 1.3 | Alive | 3.3 | TMA |
| 14 | M/35 | Primary | nd | nd | SD | Progression | 10.3 | Death | 27.5 | TMA+Sequencing |
| 16 | M/30 | Metastatic | MET + | 60 | SD | Progression | 12.8 | Alive | 39.0 | TMA |
| 17 | F/21 | Primary | MET + | 85 | PD | Progression | 1.5 | Death | 15.7 | TMA+Sequencing |
| 19 | M/23 | Metastatic | MET + | 91 | SD | Progression | 3.0 | Death | 13.4 | TMA+Sequencing |
| 24 | F/31 | Primary | MET + | 80 | SD | Progression | 4.2 | Death | 40.1 | TMA |
| 28 | M/33 | Primary | MET + | 61 | SD | Progression | 6.0 | Death | 11.2 | TMA+Sequencing |
| 39 | M/51 | Primary | MET + | nd | PD | Progression | 0.8 | Alive | 35.3 | TMA+Sequencing |
| 48 | M/30 | Primary | MET + | 75 | SD | Progression | 7.6 | Alive | 39.7 | TMA+Sequencing |
| 53 | M/69 | Metastatic | MET − * | 0 | PR | Progression | 34.4 | Alive | 41.9 | TMA+Sequencing |
| 54 | M/54 | Primary | MET + | 61 | SD | Progression | 2.8 | Alive | 39.5 | TMA+Sequencing |
| 61 | M/43 | Primary | MET + | 31 | SD | Progression | 4.2 | Alive | 39.1 | TMA |
| 65 | F/42 | Primary | MET + | 61 | SD | Progression | 38.8 | Alive | 41.9 | TMA |
| 67 | M/34 | Primary | MET + | 31 | SD | No progression | 33.5 | Alive | 33.5 | TMA |
| 70 | M/33 | Primary | MET + | 55 | PD | Progression | 1.0 | Death | 26.4 | TMA+Sequencing |
| 73 | F/18 | Primary | MET + | 67 | SD | Progression | 4.1 | Death | 23.0 | TMA+Sequencing |
| 74 | F/20 | Primary | MET + | 21 | SD | Progression | 2.8 | Alive | 35.4 | TMA+Sequencing |
| 76 | M/24 | Primary | MET + | 76 | SD | No progression | 37.4 | Alive | 37.4 | TMA+Sequencing |
| 77 | F/40 | Primary | MET + | 69 | SD | Progression | 11.6 | Death | 20.3 | TMA |
| 78 | M/37 | Primary | MET − | 0 | SD | Progression | 2.8 | Alive | 31.3 | TMA+Sequencing |
| 79 | M/45 | Primary | MET − | 0 | SD | Progression | 4.2 | Death | 10.1 | TMA+Sequencing |
| 83 | F/19 | Metastatic | MET + | 74 | SD | Progression | 18.3 | Alive | 33.3 | TMA+Sequencing |
| 89 | F/33 | Primary | MET + | 43 | SD | Progression | 5.7 | Alive | 24.6 | TMA |
| 90 | F/22 | Metastatic | MET + | 31 | SD | Progression | 4.1 | Alive | 33.3 | TMA |
| 92 | M/29 | Primary | MET + | 24 | SD | Progression | 8.3 | Death | 25.2 | TMA+Sequencing |
| 93 | M/24 | Metastatic | MET + | 43 | SD | Progression | 4.2 | Death | 10.8 | TMA |
| 96 | F/28 | Primary | MET + | 82 | SD | Progression | 8.1 | Death | 19.0 | TMA+Sequencing |
| 97 | F/37 | Primary | MET + | 81 | SD | Progression | 1.8 | Alive | 30.0 | TMA |
| 98 | M/33 | Primary | MET + | 51 | SD | Progression | 15.2 | Alive | 34.6 | TMA |
| 99 | M/17 | Primary | MET + | 67 | nd | nd | nd | Alive | nd | TMA+Sequencing |
| 105 | M/37 | Primary | MET + | 60 | SD | Progression | 21.6 | Alive | 33.5 | TMA+Sequencing |
| 109 | M/39 | Primary | MET + | 80 | SD | Progression | 7.9 | Alive | 26.1 | TMA+Sequencing |
| 114 | F/28 | Primary | MET + | 91 | PD | Progression | 1.4 | Alive | 26.0 | TMA+Sequencing |
| 120 | M/31 | Primary | MET + | 37 | PR | Progression | 10.3 | Alive | 28.5 | TMA+Sequencing |
| 125 | F/32 | Metastatic | MET + | 26 | nd | nd | nd | Alive | nd | TMA+Sequencing |
| 129 | F/16 | Primary | MET + | 65 | SD | Progression | 13.7 | Alive | 29.6 | TMA+Sequencing |
| 131 | M/33 | Primary | MET + | 47 | nd | nd | nd | Alive | nd | TMA+Sequencing |
| 133 | F/45 | Primary | MET + | 49 | SD | Progression | 10.1 | Alive | 27.1 | TMA+Sequencing |
| 143 | F/33 | Primary | MET + | 36 | SD | No progression | 26.6 | Alive | 26.6 | TMA+Sequencing |
| 149 | F/18 | Metastatic | MET + | 40 | SD | No progression | 26.2 | Alive | 26.2 | TMA+Sequencing |
| 150 | M/25 | Primary | MET + | 19 | nd | nd | nd | Alive | nd | TMA+Sequencing |
| 153 | M/24 | Metastatic | MET + | 35 | SD | Progression | 2.8 | Alive | 2.8 | TMA+Sequencing |
| 155 | F/26 | Primary | MET + | 46 | SD | Progression | 2.8 | Alive | 25.0 | TMA+Sequencing |
| 158 | F/30 | Primary | MET + | 65 | SD | Progression | 15.0 | Alive | 21.4 | TMA+Sequencing |
| 168 | F/34 | Primary | MET + | 71 | SD | No progression | 21.4 | Alive | 21.4 | TMA+Sequencing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-J.; Modave, E.; Boeckx, B.; Kasper, B.; Aamdal, S.; Leahy, M.G.; Rutkowski, P.; Bauer, S.; Debiec-Rychter, M.; Sciot, R.; et al. Correlation of Immunological and Molecular Profiles with Response to Crizotinib in Alveolar Soft Part Sarcoma: An Exploratory Study Related to the EORTC 90101 “CREATE” Trial. Int. J. Mol. Sci. 2022, 23, 5689. https://doi.org/10.3390/ijms23105689
Lee C-J, Modave E, Boeckx B, Kasper B, Aamdal S, Leahy MG, Rutkowski P, Bauer S, Debiec-Rychter M, Sciot R, et al. Correlation of Immunological and Molecular Profiles with Response to Crizotinib in Alveolar Soft Part Sarcoma: An Exploratory Study Related to the EORTC 90101 “CREATE” Trial. International Journal of Molecular Sciences. 2022; 23(10):5689. https://doi.org/10.3390/ijms23105689
Chicago/Turabian StyleLee, Che-Jui, Elodie Modave, Bram Boeckx, Bernd Kasper, Steinar Aamdal, Michael G. Leahy, Piotr Rutkowski, Sebastian Bauer, Maria Debiec-Rychter, Raf Sciot, and et al. 2022. "Correlation of Immunological and Molecular Profiles with Response to Crizotinib in Alveolar Soft Part Sarcoma: An Exploratory Study Related to the EORTC 90101 “CREATE” Trial" International Journal of Molecular Sciences 23, no. 10: 5689. https://doi.org/10.3390/ijms23105689
APA StyleLee, C.-J., Modave, E., Boeckx, B., Kasper, B., Aamdal, S., Leahy, M. G., Rutkowski, P., Bauer, S., Debiec-Rychter, M., Sciot, R., Lambrechts, D., Wozniak, A., & Schöffski, P. (2022). Correlation of Immunological and Molecular Profiles with Response to Crizotinib in Alveolar Soft Part Sarcoma: An Exploratory Study Related to the EORTC 90101 “CREATE” Trial. International Journal of Molecular Sciences, 23(10), 5689. https://doi.org/10.3390/ijms23105689