
Arylquin 1 (Potent Par-4 Secretagogue) Inhibits Tumor Progression and Induces Apoptosis in Colon Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Arylquin-1-Attenuated Cell Viability in CRC Cell Lines
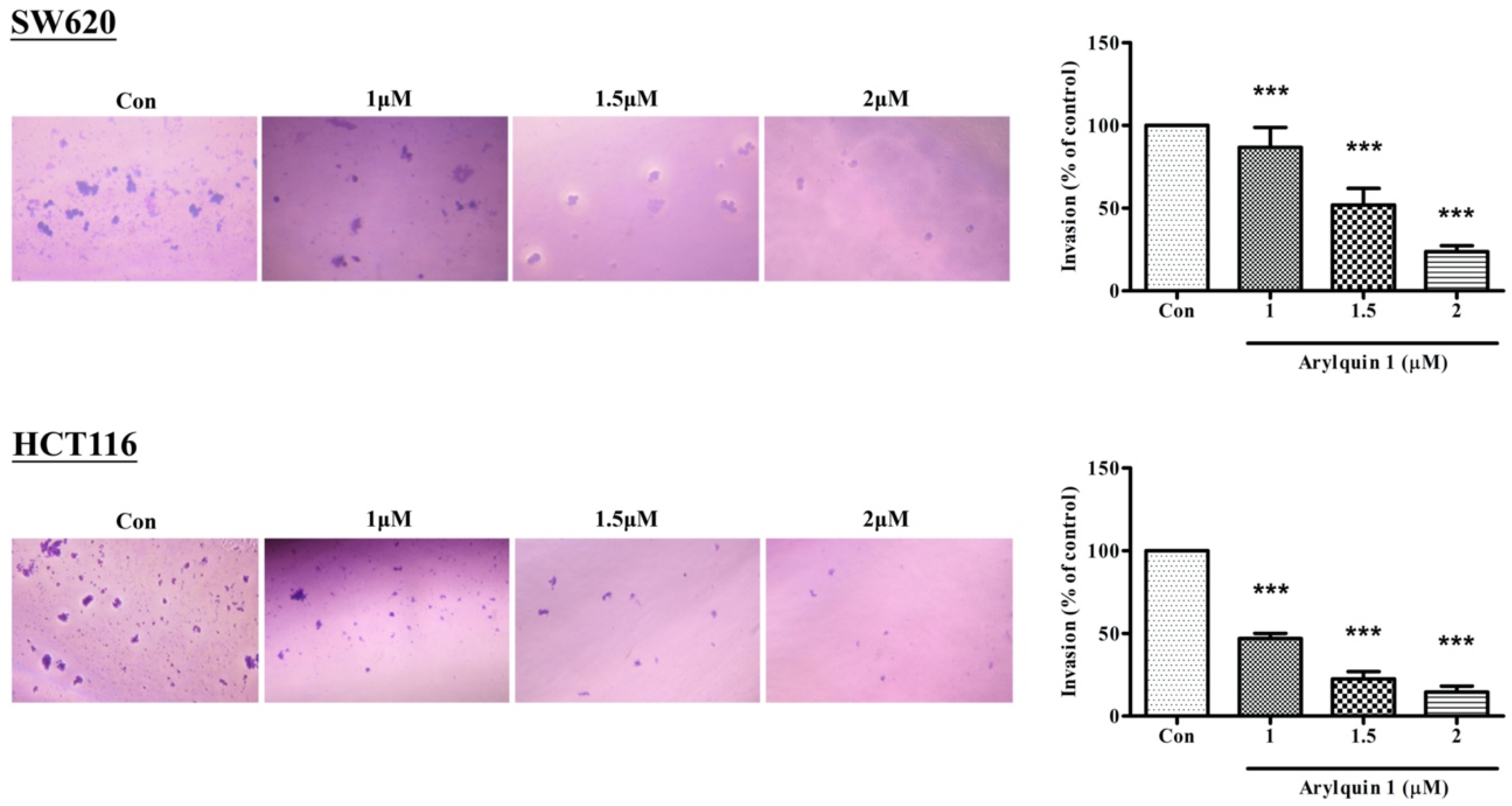
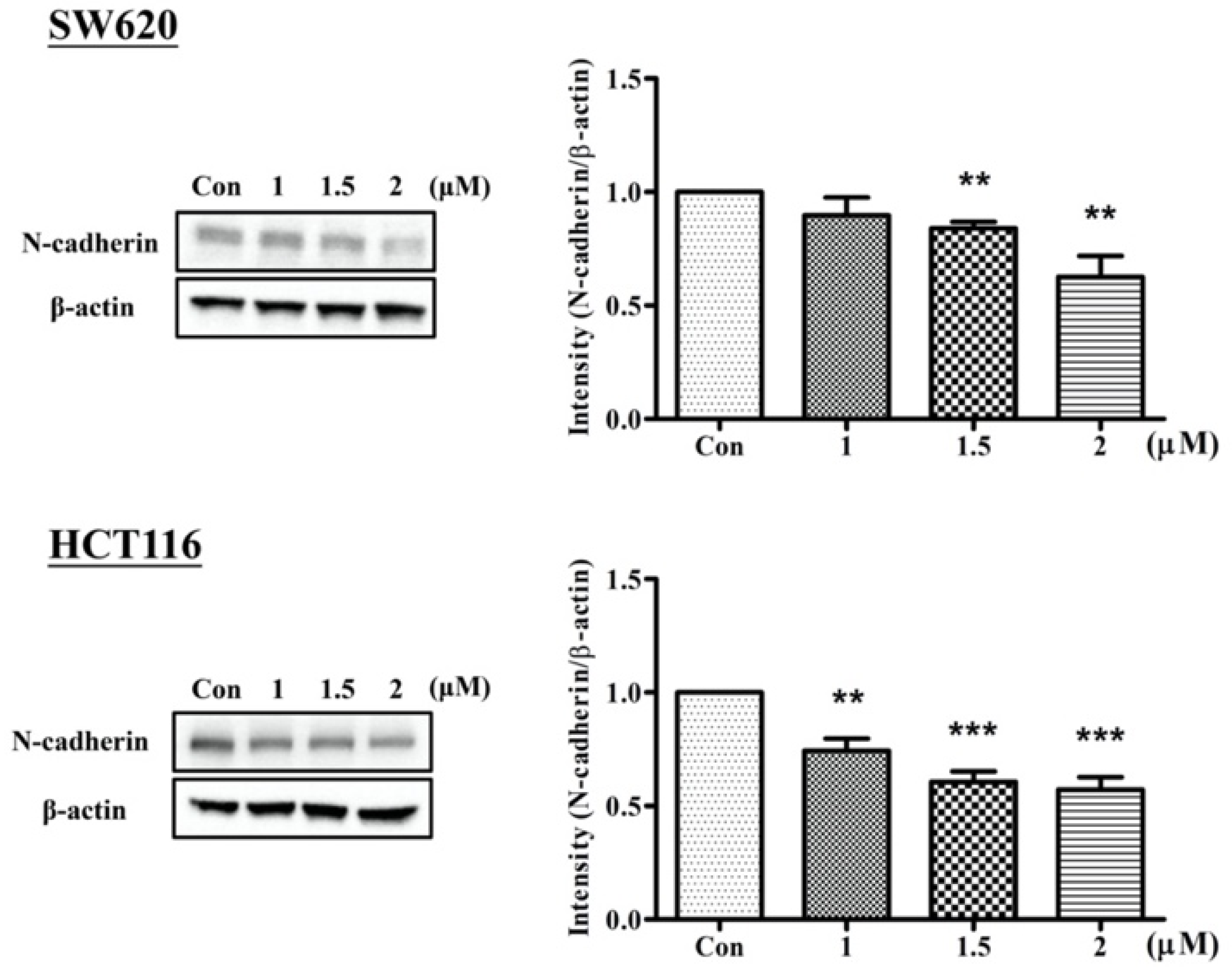
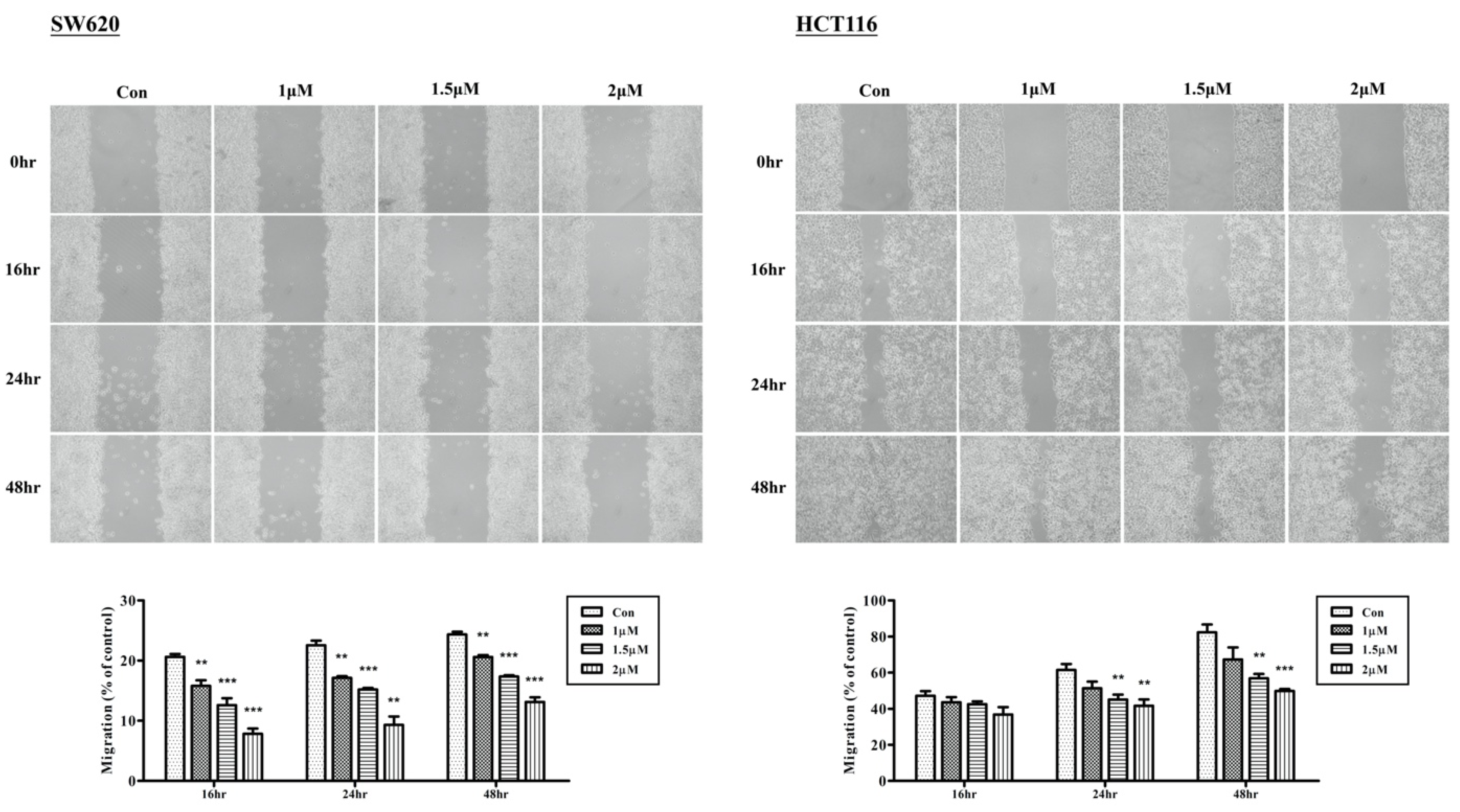
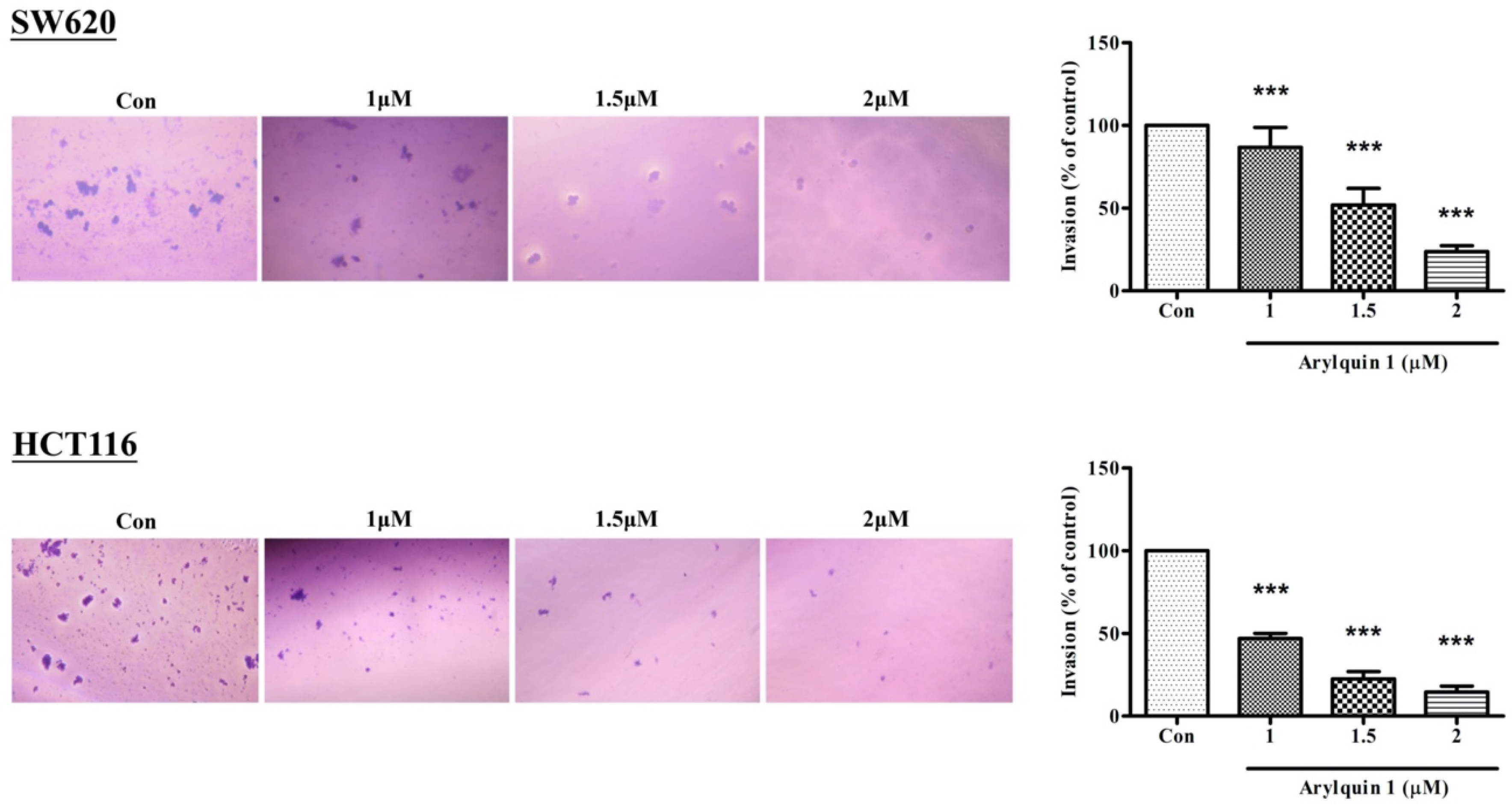
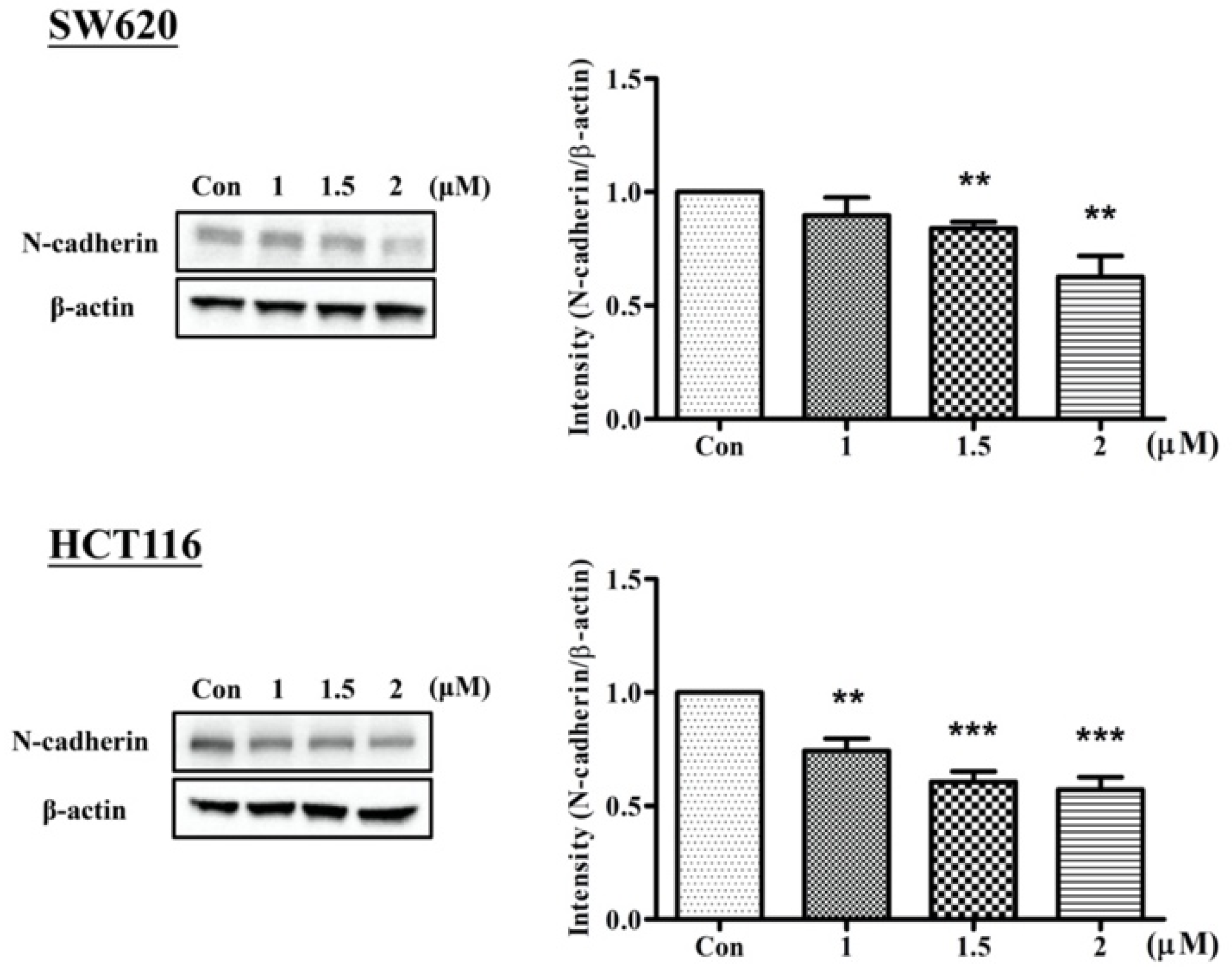
2.2. Arylquin-1-Inhibited Cell Migration, Invasion, and Epithelial–Mesenchymal Transition (EMT) in CRC Cells
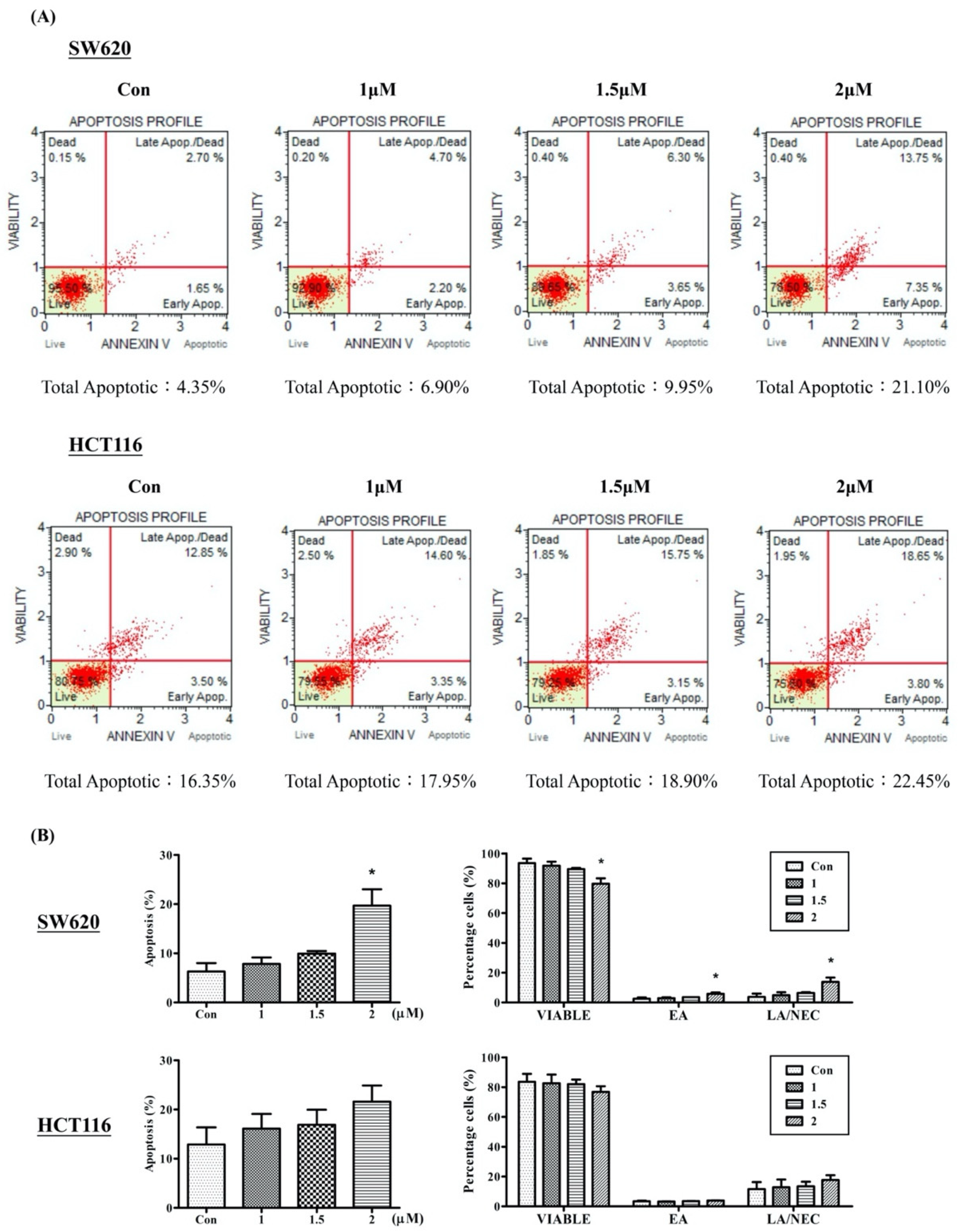
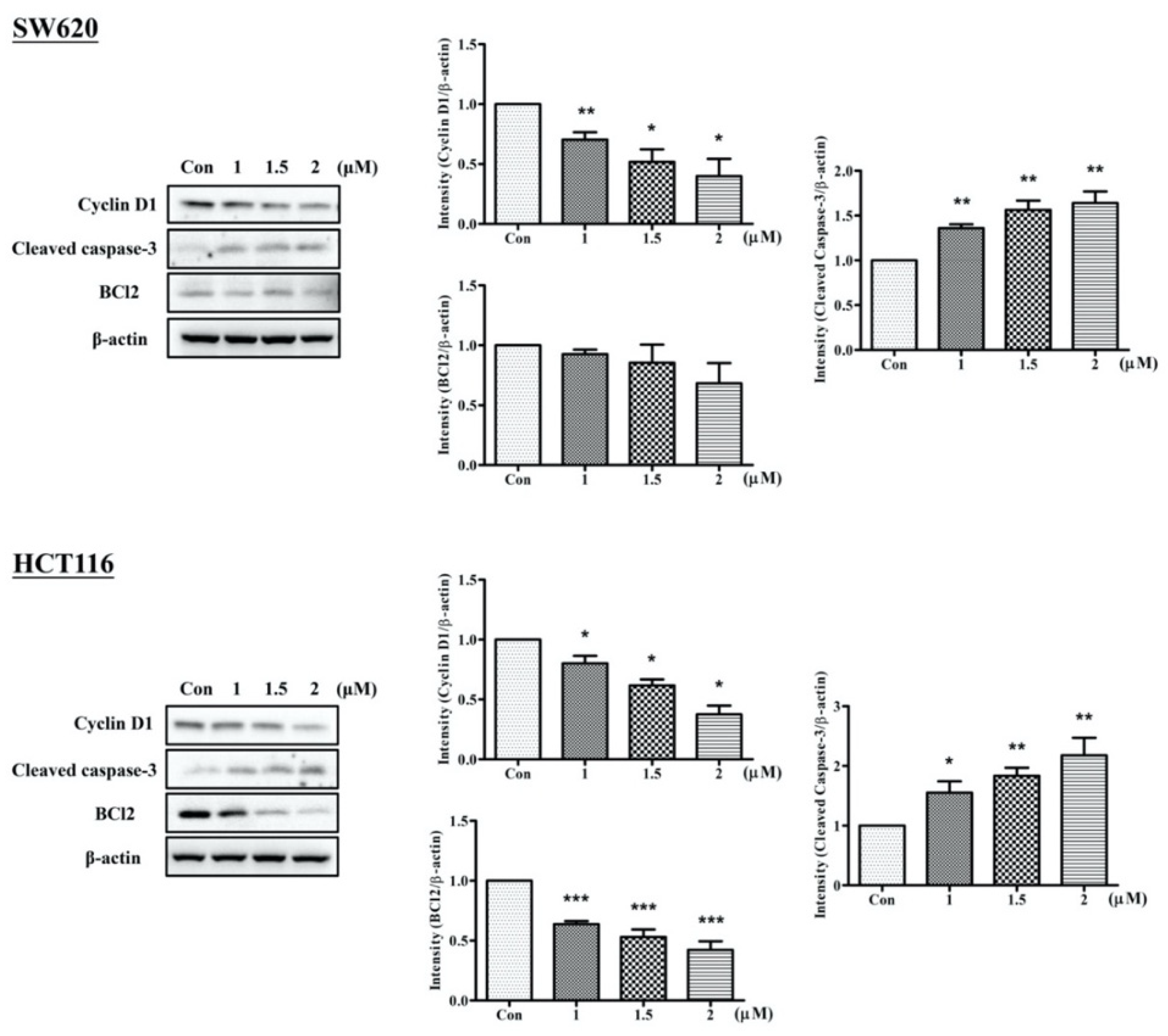
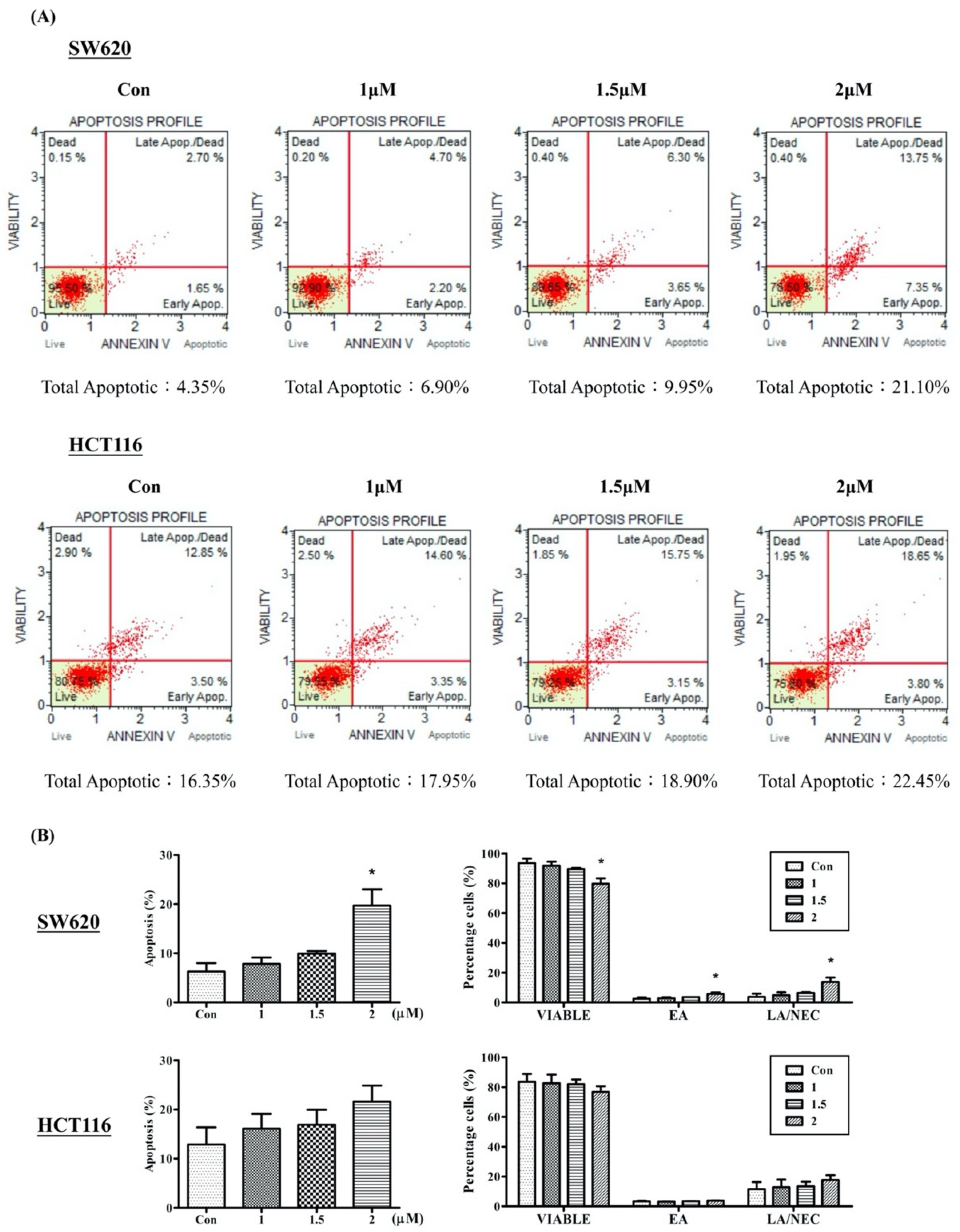
2.3. Arylquin 1 Promotes Apoptosis in Cultured CRC Cells
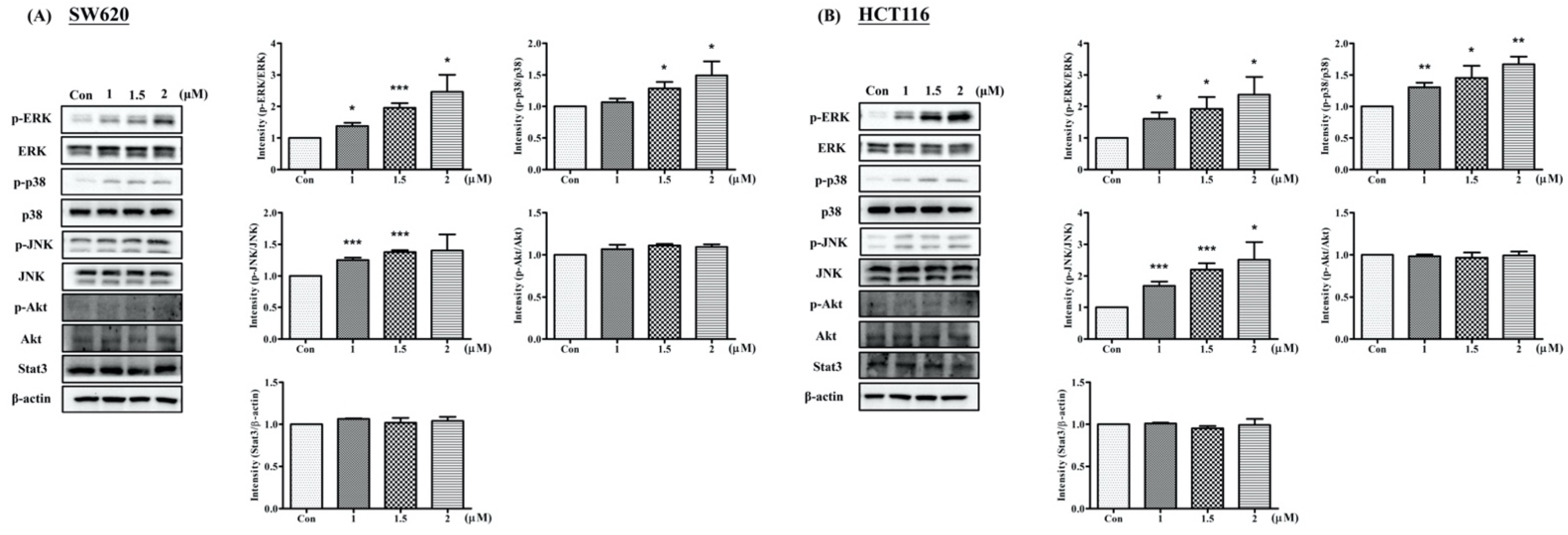
2.4. Arylquin 1 Regulated Apoptosis via the MAPK Pathway in CRC Cells
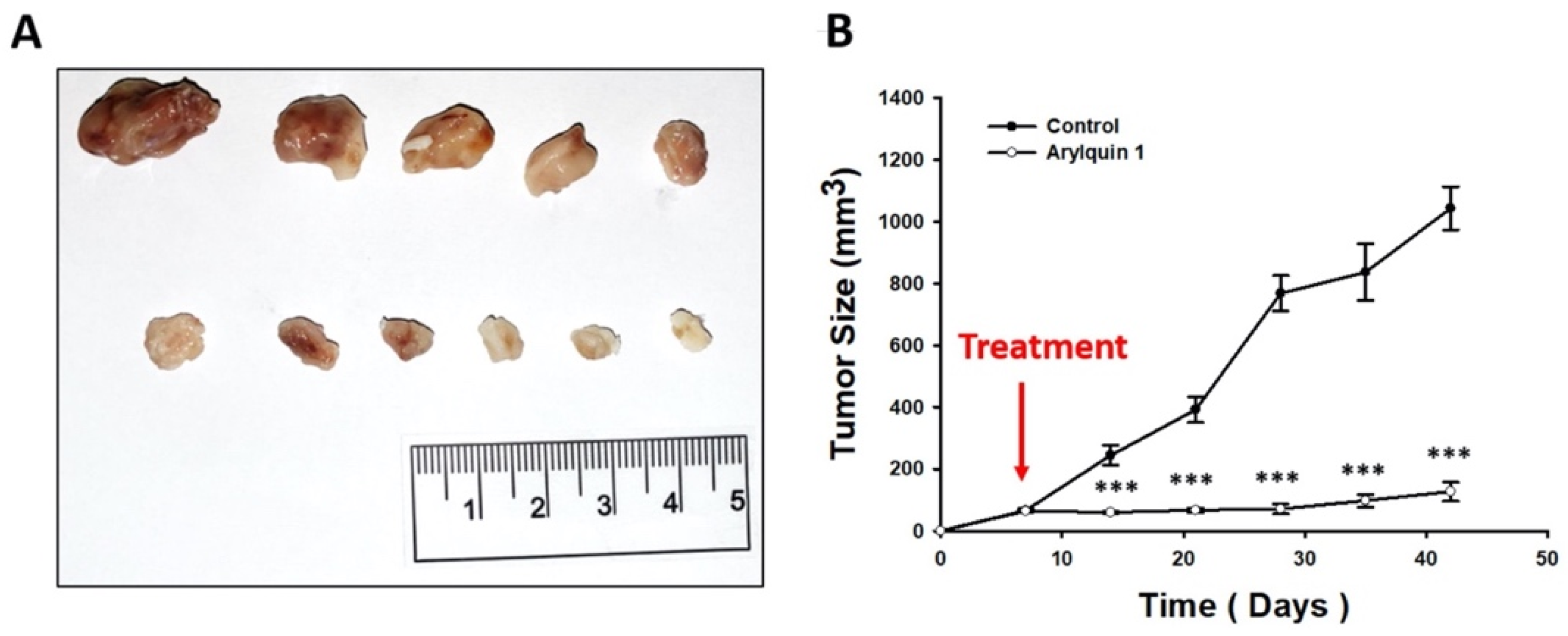
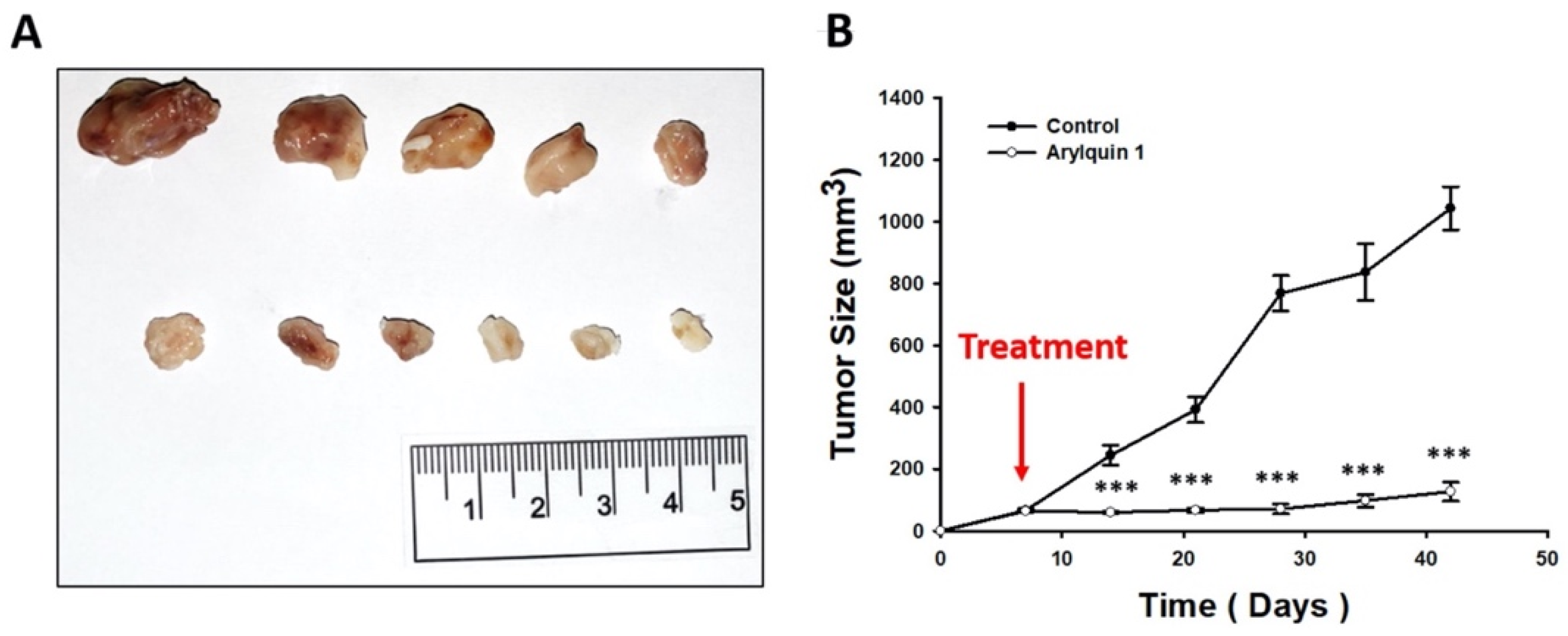
2.5. Arylquin 1 Suppressed the Growth of CRC Cells in Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability
4.3. Migration and Invasion Assays In Vitro
4.4. Protein Extraction and Western Blot
4.5. Flow Cytometry
4.6. Animal Model
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FIAfRoC, L. Global Cancer Observatory: Cancer Today. Available online: https://gcoiarcfr/today/home (accessed on 17 August 2020).
- Hebbar, N.; Wang, C.; Rangnekar, V.M. Mechanisms of apoptosis by the tumor suppressor Par-4. J. Cell Physiol. 2012, 227, 3715–3721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burikhanov, R.; Zhao, Y.; Goswami, A.; Qiu, S.; Schwarze, S.R.; Rangnekar, V.M. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell 2009, 138, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, M.; Qiu, S.G.; Vasudevan, K.M.; Rangnekar, V.M. Par-4 drives trafficking and activation of Fas and Fasl to induce prostate cancer cell apoptosis and tumor regression. Cancer Res. 2001, 61, 7255–7263. [Google Scholar]
- Cook, J.; Krishnan, S.; Ananth, S.; Sells, S.F.; Shi, Y.; Walther, M.M.; Linehan, W.M.; Sukhatme, V.P.; Weinstein, M.H.; Rangnekar, V.M. Decreased expression of the pro-apoptotic protein Par-4 in renal cell carcinoma. Oncogene 1999, 18, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagtap, J.C.; Parveen, D.; Shah, R.D.; Desai, A.; Bhosale, D.; Chugh, A.; Ranade, D.; Karnik, S.; Khedkar, B.; Mathur, A.; et al. Secretory prostate apoptosis response (Par)-4 sensitizes multicellular spheroids (MCS) of glioblastoma multiforme cells to tamoxifen-induced cell death. FEBS Open Bio 2015, 5, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.C.; de Bessa-Garcia, S.A.; Burikhanov, R.; Pavanelli, A.C.; Antunes, L.; Rangnekar, V.M.; Nagai, M.A. Prostate apoptosis response-4 is involved in the apoptosis response to docetaxel in MCF-7 breast cancer cells. Int. J. Oncol. 2013, 43, 531–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.J.; Shahriyar, S.A.; Kwon, T.K. Arylquin 1, a potent Par-4 secretagogue, induces lysosomal membrane permeabilization-mediated non-apoptotic cell death in cancer cells. Toxicol. Res. 2020, 36, 167–173. [Google Scholar] [CrossRef]
- Burikhanov, R.; Sviripa, V.M.; Hebbar, N.; Zhang, W.; Layton, W.J.; Hamza, A.; Zhan, C.G.; Watt, D.S.; Liu, C.; Rangnekar, V.M. Arylquins target vimentin to trigger Par-4 secretion for tumor cell apoptosis. Nat. Chem. Biol. 2014, 10, 924–926. [Google Scholar] [CrossRef] [Green Version]
- Sviripa, V.M.; Burikhanov, R.; Obiero, J.M.; Yuan, Y.; Nickell, J.R.; Dwoskin, L.P.; Zhan, C.G.; Liu, C.; Tsodikov, O.V.; Rangnekar, V.M.; et al. Par-4 secretion: Stoichiometry of 3-arylquinoline binding to vimentin. Org. Biomol. Chem. 2016, 14, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Burikhanov, R.; Hebbar, N.; Noothi, S.K.; Shukla, N.; Sledziona, J.; Araujo, N.; Kudrimoti, M.; Wang, Q.J.; Watt, D.S.; Welch, D.R.; et al. Chloroquine-Inducible Par-4 Secretion Is Essential for Tumor Cell Apoptosis and Inhibition of Metastasis. Cell Rep. 2017, 18, 508–519. [Google Scholar] [CrossRef]
- Kang, Y.; Massagué, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef] [PubMed]
- Katoch, A.; Jamwal, V.L.; Faheem, M.M.; Kumar, S.; Senapati, S.; Yadav, G.; Gandhi, S.G.; Goswami, A. Overlapping targets exist between the Par-4 and miR-200c axis which regulate EMT and proliferation of pancreatic cancer cells. Transl. Oncol. 2021, 14, 100879. [Google Scholar] [CrossRef]
- Chaudhry, P.; Fabi, F.; Singh, M.; Parent, S.; Leblanc, V.; Asselin, E. Prostate apoptosis response-4 mediates TGF-beta-induced epithelial-to-mesenchymal transition. Cell Death Dis. 2014, 5, e1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohd Faheem, M.; Rasool, R.U.; Ahmad, S.M.; Jamwal, V.L.; Chakraborty, S.; Katoch, A.; Gandhi, S.G.; Bhagat, M.; Goswami, A. Par-4 mediated Smad4 induction in PDAC cells restores canonical TGF-β/Smad4 axis driving the cells towards lethal EMT. Eur. J. Cell Biol. 2020, 99, 151076. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E. Apoptosis (programmed cell death) and its signals—A review. Braz. J. Biol. 2021, 81, 1133–1143. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Sells, S.F.; Wood, D.P., Jr.; Joshi-Barve, S.S.; Muthukumar, S.; Jacob, R.J.; Crist, S.A.; Humphreys, S.; Rangnekar, V.M. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ. 1994, 5, 457–466. [Google Scholar]
- Wang, B.D.; Kline, C.L.; Pastor, D.M.; Olson, T.L.; Frank, B.; Luu, T.; Sharma, A.K.; Robertson, G.; Weirauch, M.T.; Patierno, S.R.; et al. Prostate apoptosis response protein 4 sensitizes human colon cancer cells to chemotherapeutic 5-FU through mediation of an NF kappaB and microRNA network. Mol. Cancer 2010, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Hardy, B.; Raiter, A.; Yakimov, M.; Vilkin, A.; Niv, Y. Colon cancer cells expressing cell surface GRP78 as a marker for reduced tumorigenicity. Cell Oncol. 2012, 35, 345–354. [Google Scholar] [CrossRef]
- Belfi, C.A.; Chatterjee, S.; Gosky, D.M.; Berger, S.J.; Berger, N.A. Increased sensitivity of human colon cancer cells to DNA cross-linking agents after GRP78 up-regulation. Biochem. Biophys. Res. Commun. 1999, 257, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Gifford, J.B.; Hill, R. GRP78 Influences Chemoresistance and Prognosis in Cancer. Curr. Drug Targets 2018, 19, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.I. Growth arrest signaling of the Raf/MEK/ERK pathway in cancer. Front. Biol. 2014, 9, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.S.; Wu, J.R.; Hu, C.T. Signal cross talks for sustained MAPK activation and cell migration: The potential role of reactive oxygen species. Cancer Metastasis Rev. 2008, 27, 303–314. [Google Scholar] [CrossRef]
- Kim, B.; Seo, J.H.; Lee, K.Y.; Park, B. Icariin sensitizes human colon cancer cells to TRAIL-induced apoptosis via ERK-mediated upregulation of death receptors. Int. J. Oncol. 2020, 56, 821–834. [Google Scholar] [CrossRef]
- Zhao, Y.; Xia, S.; Cao, C.; Du, X. Effect of TGF-β1 on Apoptosis of Colon Cancer Cells Via the ERK Signaling Pathway. J. BUON 2019, 24, 449–455. [Google Scholar]
- Tournier, C. The 2 Faces of JNK Signaling in Cancer. Genes Cancer 2013, 4, 397–400. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohebali, N.; Pandurangan, A.K.; Mustafa, M.R.; Anandasadagopan, S.K.; Alagumuthu, T. Vernodalin induces apoptosis through the activation of ROS/JNK pathway in human colon cancer cells. J. Biochem. Mol. Toxicol. 2020, 34, e22587. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Meng, Y.; Sheng, X.; Guan, Y.; Zhang, F.; Han, Z.; Kang, Y.; Tai, G.; Zhou, Y.; Cheng, H. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anticancer Drugs 2017, 28, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Lee, M.W.; Park, S.C.; Yang, Y.G.; Yim, S.O.; Chae, H.S.; Bach, J.H.; Lee, H.J.; Kim, K.Y.; Lee, W.B.; Kim, S.S. The involvement of reactive oxygen species (ROS) and p38 mitogen-activated protein (MAP) kinase in TRAIL/Apo2L-induced apoptosis. FEBS Lett. 2002, 512, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Li, W.Y.; Xie, J.; Wang, Z.L.; Wen, Y.L.; Zhao, C.C.; Tao, L.; Li, L.F.; Tian, Y.; Sheng, J. Astragalin Inhibits the Proliferation and Migration of Human Colon Cancer HCT116 Cells by Regulating the NF-κB Signaling Pathway. Front. Pharmacol. 2021, 12, 639256. [Google Scholar] [CrossRef]
- Rajput, A.; San Martin, I.D.; Rose, R.; Beko, A.; Levea, C.; Sharratt, E.; Mazurchuk, R.; Hoffman, R.M.; Brattain, M.G.; Wang, J. Characterization of HCT116 human colon cancer cells in an orthotopic model. J. Surg. Res. 2008, 147, 276–281. [Google Scholar] [CrossRef]
- Phang, C.W.; Abd Malek, S.N.; Karsani, S.A. Flavokawain C exhibits antitumor effects on in vivo HCT 116 xenograft and identification of its apoptosis-linked serum biomarkers via proteomic analysis. Biomed. Pharmacother. 2021, 137, 110846. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-T.; Tseng, T.-T.; Tsai, H.-P.; Huang, M.-Y. Arylquin 1 (Potent Par-4 Secretagogue) Inhibits Tumor Progression and Induces Apoptosis in Colon Cancer Cells. Int. J. Mol. Sci. 2022, 23, 5645. https://doi.org/10.3390/ijms23105645
Chen Y-T, Tseng T-T, Tsai H-P, Huang M-Y. Arylquin 1 (Potent Par-4 Secretagogue) Inhibits Tumor Progression and Induces Apoptosis in Colon Cancer Cells. International Journal of Molecular Sciences. 2022; 23(10):5645. https://doi.org/10.3390/ijms23105645
Chicago/Turabian StyleChen, Yi-Ting, Tzu-Ting Tseng, Hung-Pei Tsai, and Ming-Yii Huang. 2022. "Arylquin 1 (Potent Par-4 Secretagogue) Inhibits Tumor Progression and Induces Apoptosis in Colon Cancer Cells" International Journal of Molecular Sciences 23, no. 10: 5645. https://doi.org/10.3390/ijms23105645
APA StyleChen, Y.-T., Tseng, T.-T., Tsai, H.-P., & Huang, M.-Y. (2022). Arylquin 1 (Potent Par-4 Secretagogue) Inhibits Tumor Progression and Induces Apoptosis in Colon Cancer Cells. International Journal of Molecular Sciences, 23(10), 5645. https://doi.org/10.3390/ijms23105645