
Huntingtin Co-Isolates with Small Extracellular Vesicles from Blood Plasma of TgHD and KI-HD Pig Models of Huntington’s Disease and Human Blood Plasma

 , , , , , , , , , and
, , , , , , , , , and
Abstract
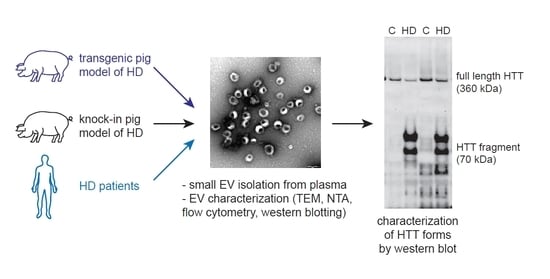
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Isolation and Characterization of the EVs
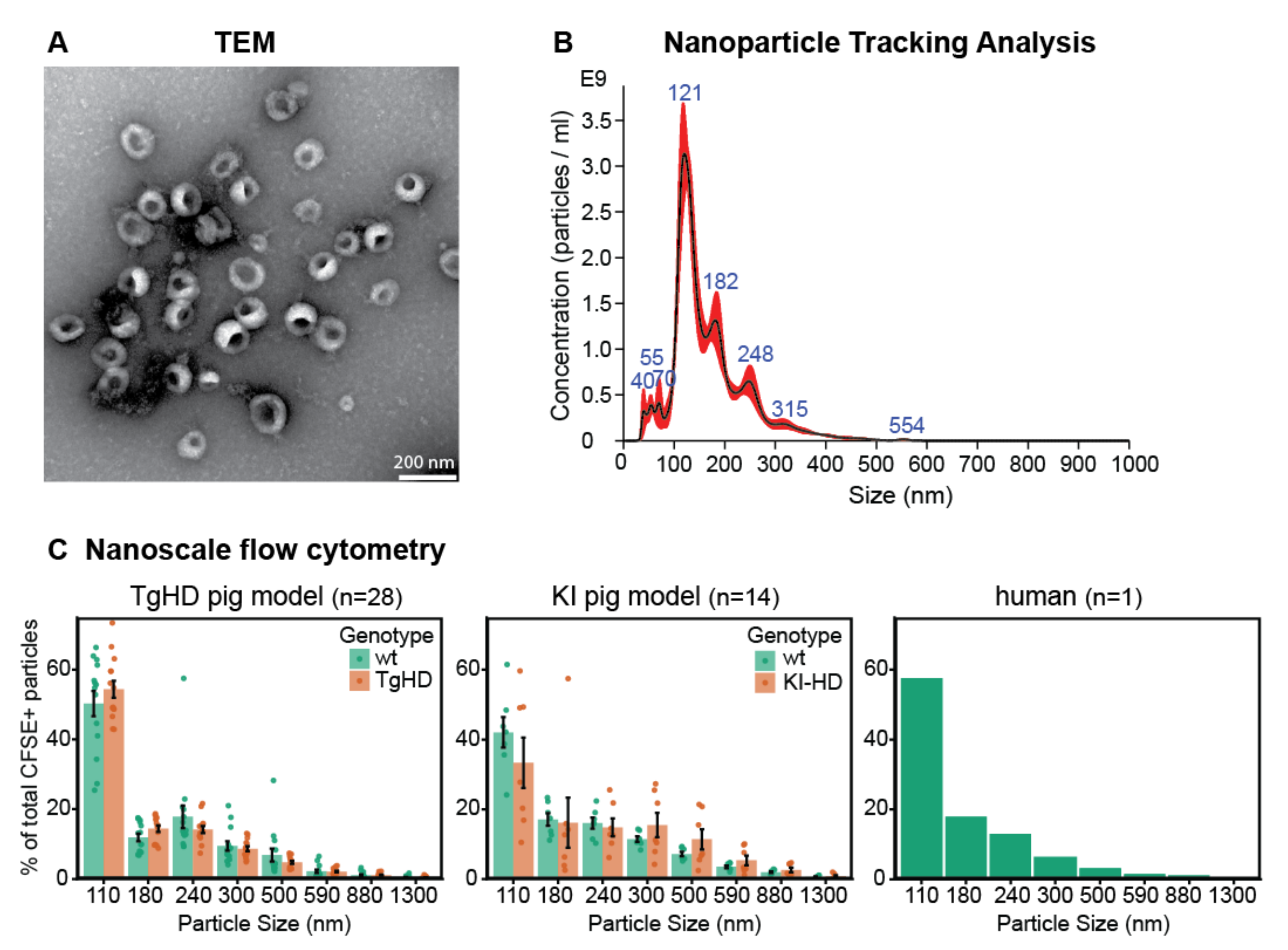
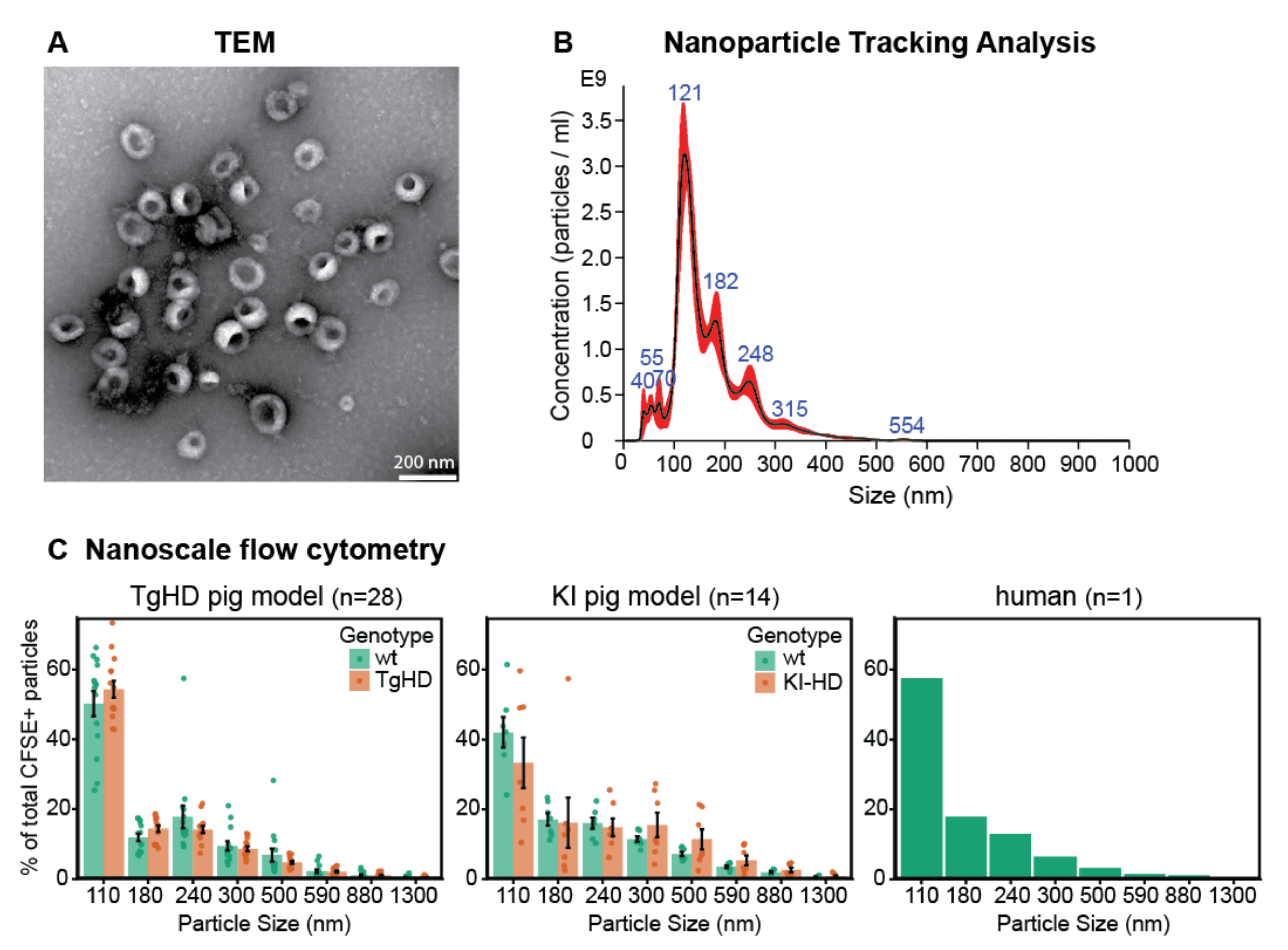
2.1.1. Morphological Characterization of EVs by TEM
2.1.2. Characterization of Particle Size by NTA
2.1.3. Characterization of Particle Size Distribution by Flow Cytometry
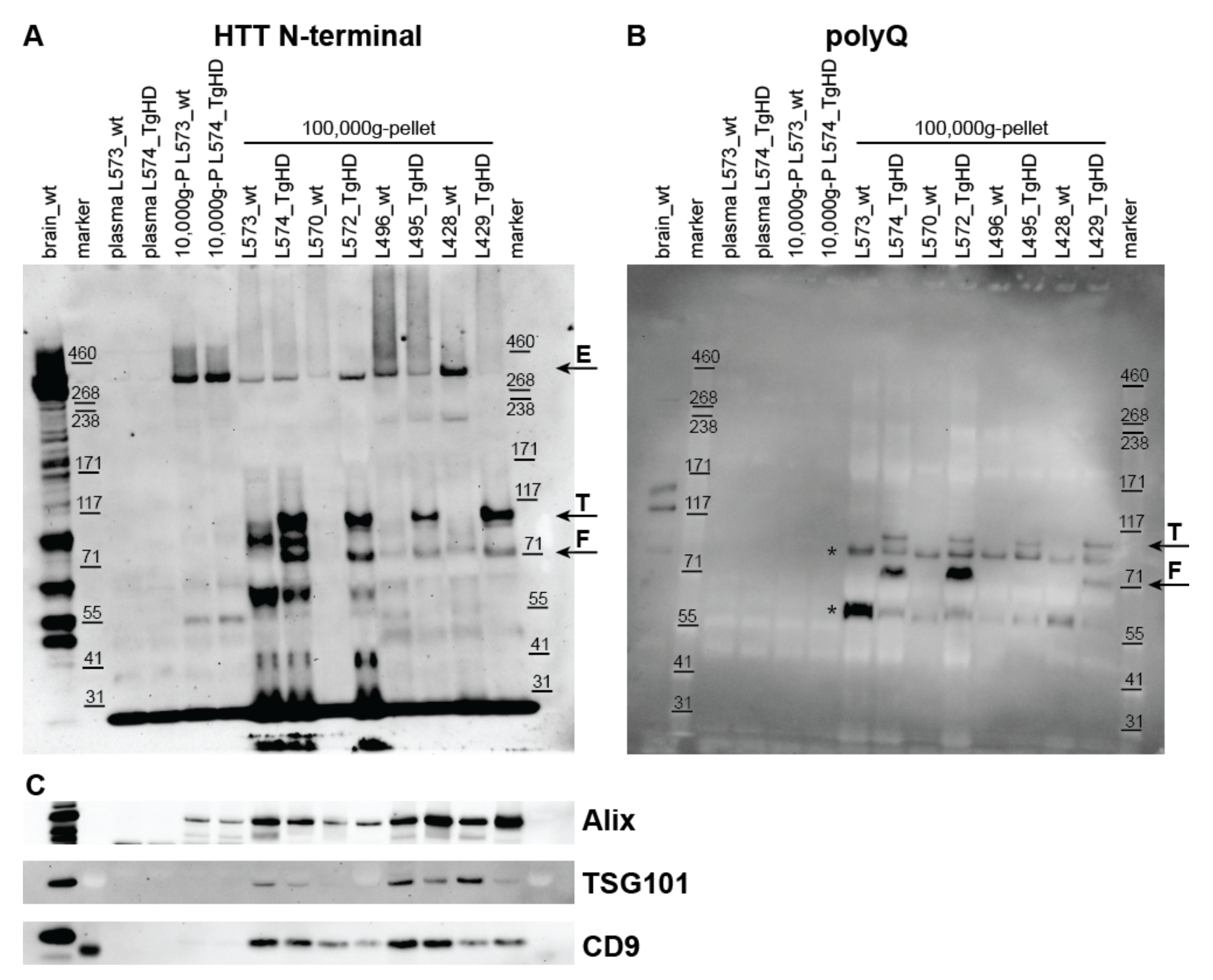
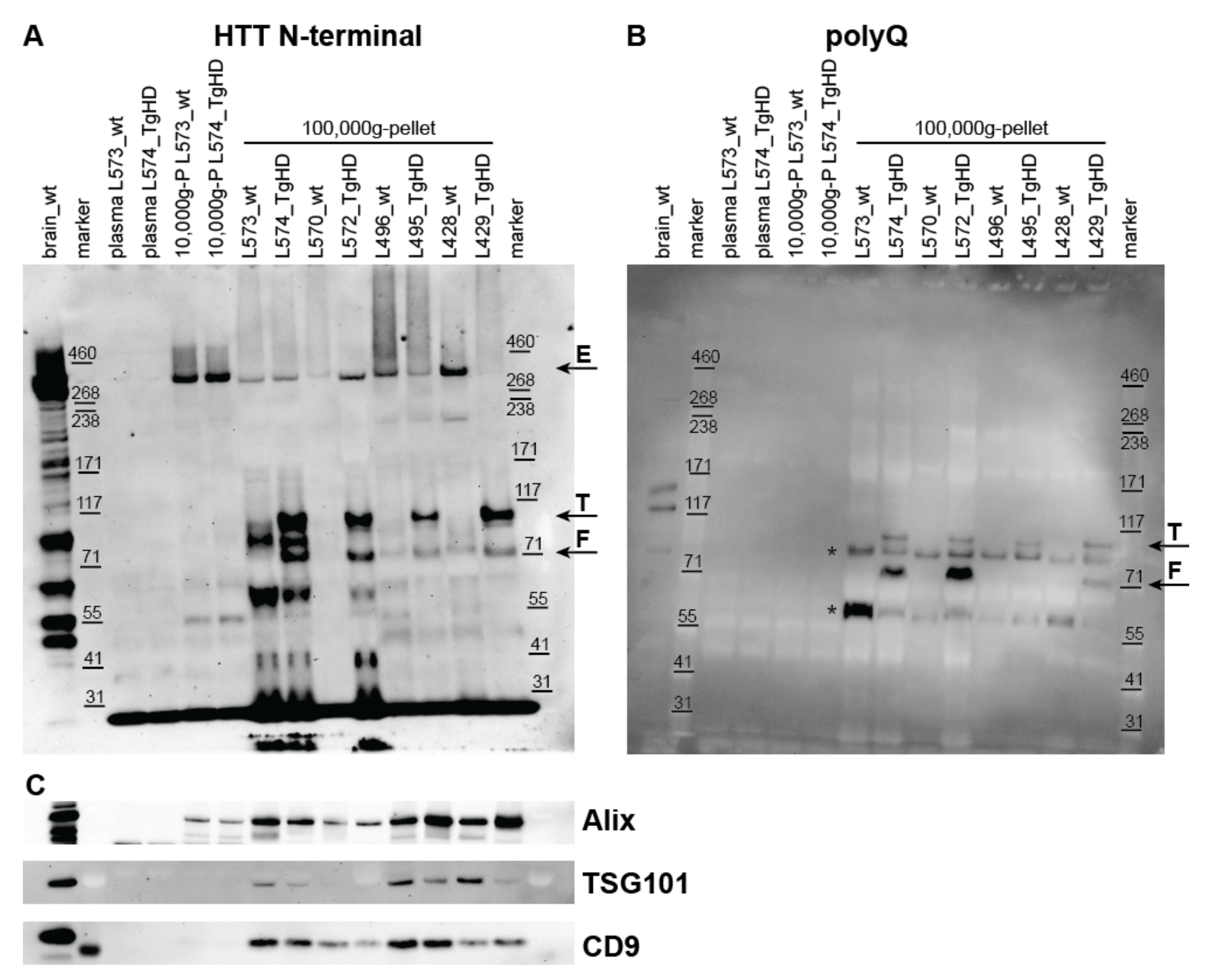
2.2. Detection of HTT, Mutant HTT, and Exosome Markers in UC Pellets
2.2.1. Detection of HTT Forms in UC Pellets from Plasma of the TgHD Model
2.2.2. Detection of HTT Forms in UC Pellets from Plasma of the KI-HD Model
2.2.3. Detection of HTT Forms in UC Pellets from Human Plasma
2.2.4. Detection of Exosome Markers in UC Pellets
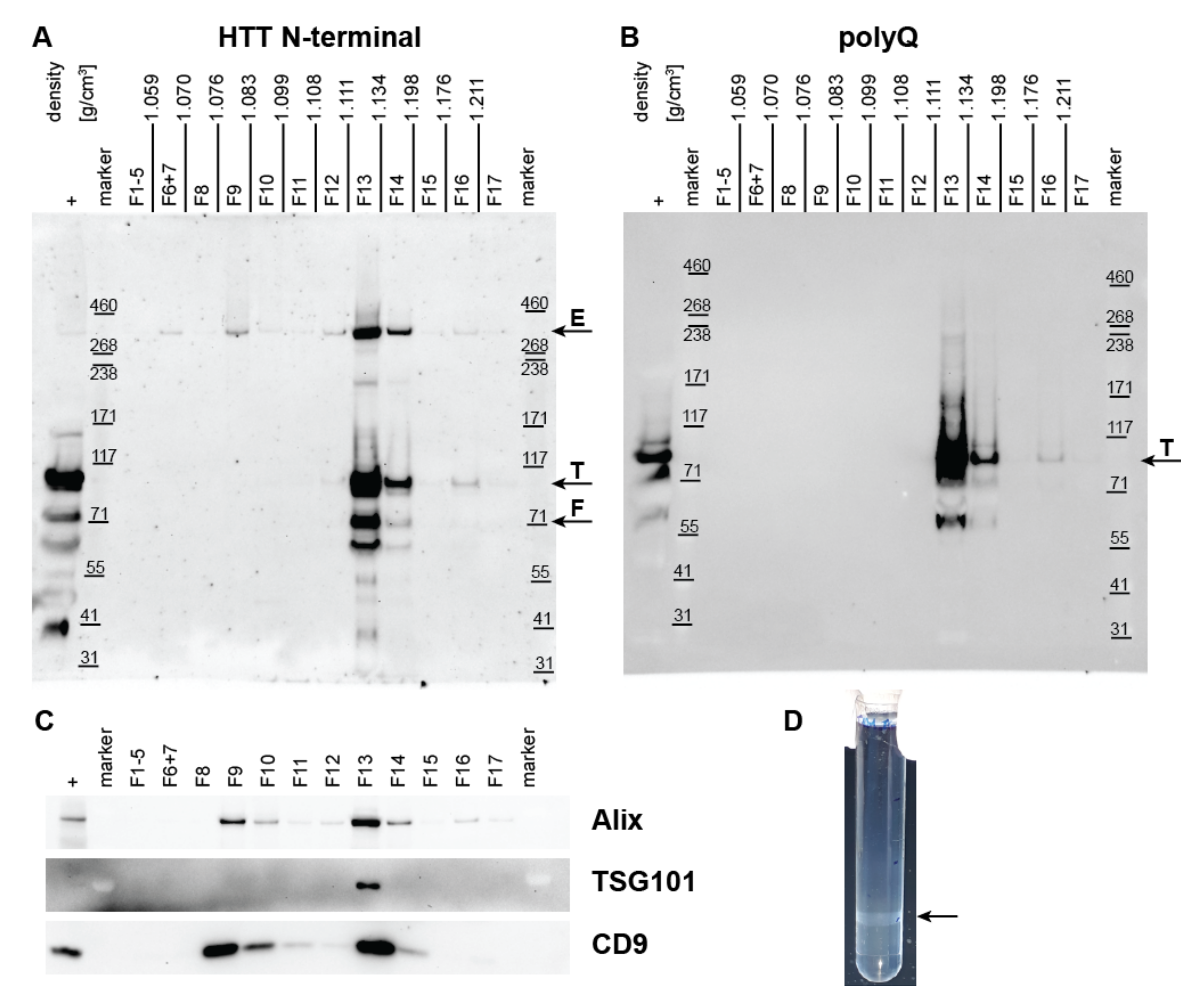
2.3. Density Gradient UC to Confirm Co-Isolation of HTT and mHTT with Exosomes
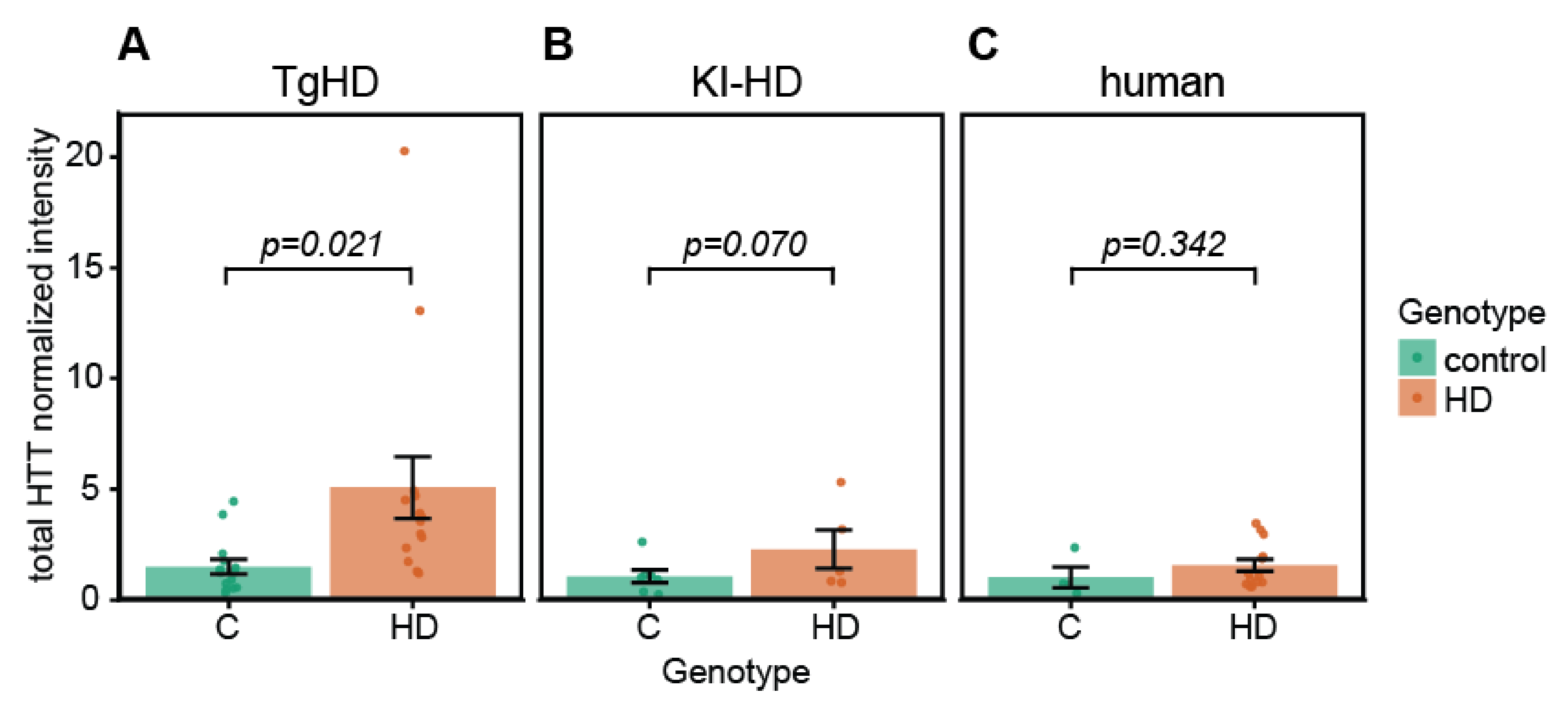
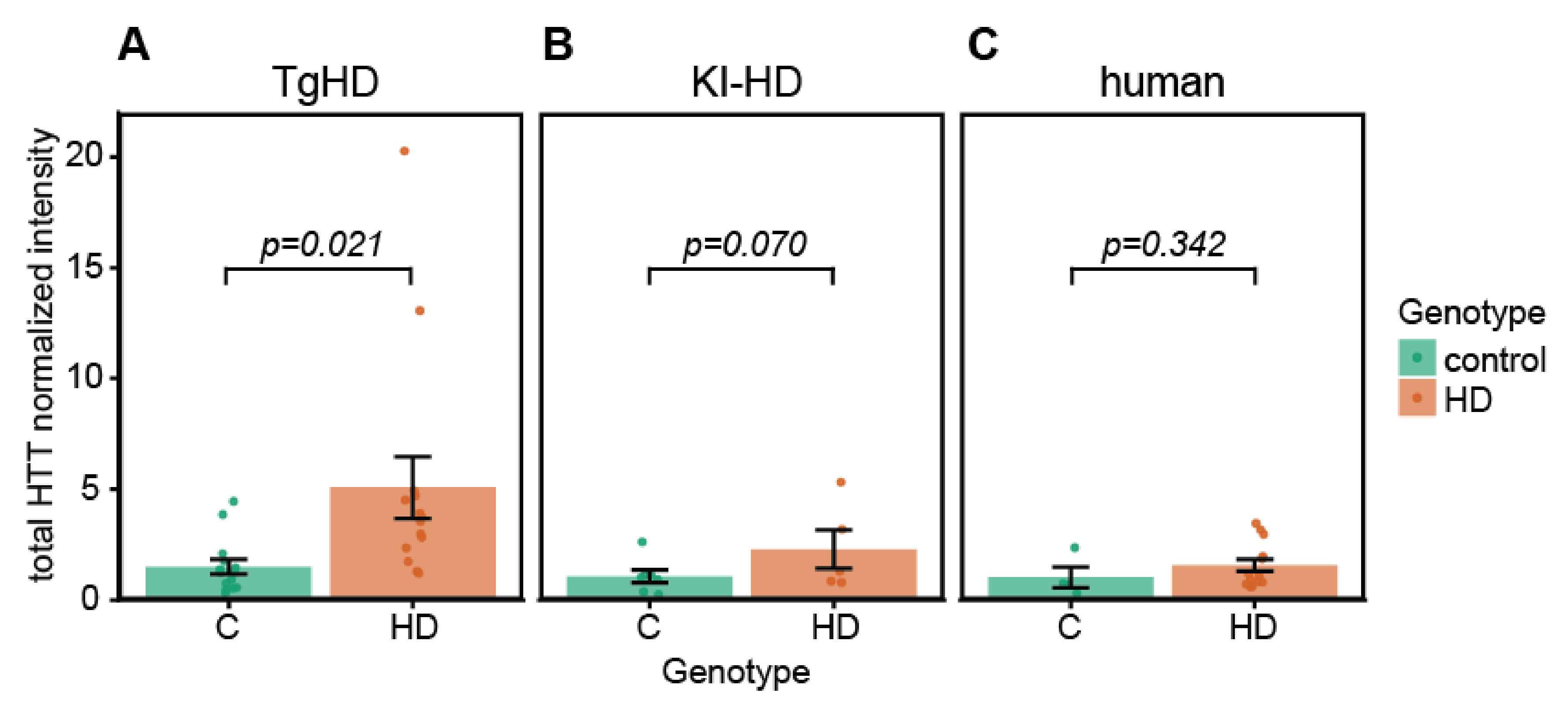
2.4. Quantification of HTT/mHTT Levels in EVs from TgHD and KI-HD Models and HD Patients
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. HD Patients
4.3. Blood Sample Collection and PPP Preparation
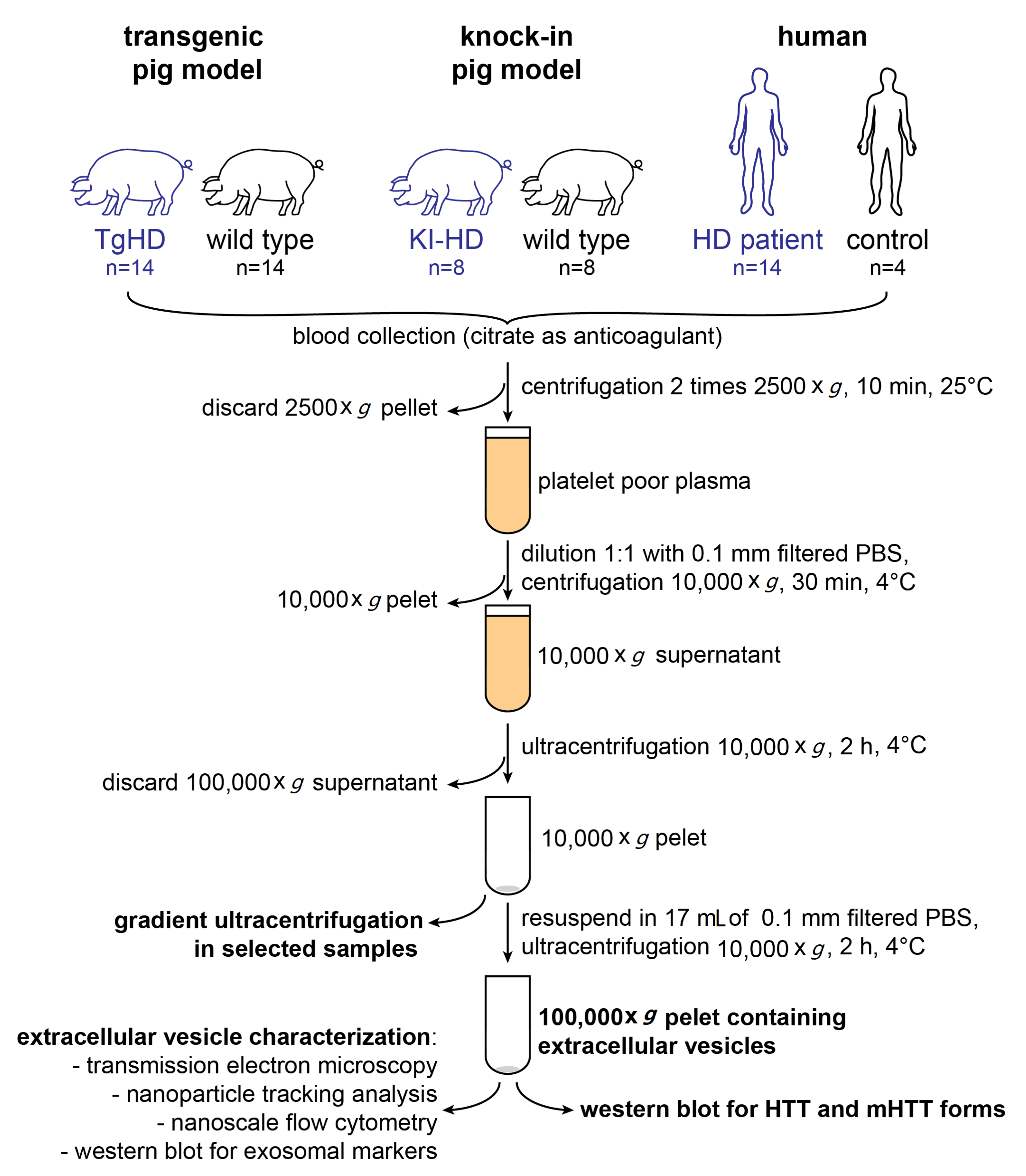
4.4. Isolation of Extracellular Vesicles
4.4.1. UC
4.4.2. OptiPrep™ (Iodixanol) Density Gradient UC
4.5. Extracellular Vesicle Lysate Preparation
4.6. Western Blot Analysis
4.7. TEM
4.8. NTA
4.9. Nanoscale Flow Cytometry
4.10. Data Analysis and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dayalu, P.; Albin, R.L. Huntington Disease. Neurol. Clin. 2015, 33, 101–114. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s Disease: A Clinical Review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Bashir, H. Emerging Therapies in Huntington’s Disease. Expert Rev. Neurother. 2019, 19, 983–995. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington Disease: New Insights into Molecular Pathogenesis and Therapeutic Opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Diamond, M.I. Huntington Disease and the Huntingtin Protein. Prog. Mol. Biol. Transl. Sci. 2012, 107, 189–214. [Google Scholar] [CrossRef]
- Tourette, C.; Li, B.; Bell, R.; O’Hare, S.; Kaltenbach, L.S.; Mooney, S.D.; Hughes, R.E. A Large Scale Huntingtin Protein Interaction Network Implicates Rho GTPase Signaling Pathways in Huntington Disease. J. Biol. Chem. 2014, 289, 6709–6726. [Google Scholar] [CrossRef] [Green Version]
- Wild, E.J.; Tabrizi, S. Therapies Targeting DNA and RNA in Huntington’s Disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Tabrizi, S.J. Huntington’s Disease: From Molecular Pathogenesis to Clinical Treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Nopoulos, P.C. Huntington Disease: A Single-Gene Degenerative Disorder of the Striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar] [CrossRef]
- Van der Burg, J.M.; Björkqvist, M.; Brundin, P. Beyond the Brain: Widespread Pathology in Huntington’s Disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef]
- Bozzi, M.; Sciandra, F. Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease. Int. J. Mol. Sci. 2020, 21, E8314. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Agrawal, N. Metabolism in Huntington’s Disease: A Major Contributor to Pathology. Metab. Brain Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, M.; Glaser, T.; Slominska, E.M.; Ulrich, H.; Smolenski, R.T. Purine Nucleotides Metabolism and Signaling in Huntington’s Disease: Search for a Target for Novel Therapies. Int. J. Mol. Sci. 2021, 22, 6545. [Google Scholar] [CrossRef]
- Tomczyk, M.; Glaser, T.; Ulrich, H.; Slominska, E.M.; Smolenski, R.T. Huntingtin Protein Maintains Balanced Energetics in Mouse Cardiomyocytes. Nucleosides Nucleotides Nucleic Acids 2022, 41, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Ismailoglu, I.; Chen, Q.; Popowski, M.; Yang, L.; Gross, S.S.; Brivanlou, A.H. Huntingtin Protein Is Essential for Mitochondrial Metabolism, Bioenergetics and Structure in Murine Embryonic Stem Cells. Dev. Biol. 2014, 391, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.M.; Miniarikova, J.; Juhas, S.; Vallès, A.; Bohuslavova, B.; Juhasova, J.; Skalnikova, H.K.; Vodicka, P.; Valekova, I.; Brouwers, C.; et al. AAV5-MiHTT Gene Therapy Demonstrates Broad Distribution and Strong Human Mutant Huntingtin Lowering in a Huntington’s Disease Minipig Model. Mol. Ther. 2018, 26, 2163–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling Neurodegeneration in Caenorhabditis Elegans. Dis. Models Mech. 2020, 13, dmm046110. [Google Scholar] [CrossRef]
- Subhan, I.; Siddique, Y.H. Modulation of Huntington’s Disease in Drosophila. CNS Neurol. Disord. Drug Targets 2021, 20, 894–903. [Google Scholar] [CrossRef]
- Wang, J.; Cao, H. Zebrafish and Medaka: Important Animal Models for Human Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 10766. [Google Scholar] [CrossRef]
- Kaye, J.; Reisine, T.; Finkbeiner, S. Huntington’s Disease Mouse Models: Unraveling the Pathology Caused by CAG Repeat Expansion. Fac. Rev. 2021, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Howland, D.; Ellederova, Z.; Aronin, N.; Fernau, D.; Gallagher, J.; Taylor, A.; Hennebold, J.; Weiss, A.R.; Gray-Edwards, H.; McBride, J. Large Animal Models of Huntington’s Disease: What We Have Learned and Where We Need to Go Next. J. Huntingt. Dis. 2020, 9, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Kosior, N.; Leavitt, B.R. Murine Models of Huntington’s Disease for Evaluating Therapeutics. Methods Mol. Biol. 2018, 1780, 179–207. [Google Scholar] [CrossRef] [PubMed]
- Schomberg, D.T.; Tellez, A.; Meudt, J.J.; Brady, D.A.; Dillon, K.N.; Arowolo, F.K.; Wicks, J.; Rousselle, S.D.; Shanmuganayagam, D. Miniature Swine for Preclinical Modeling of Complexities of Human Disease for Translational Scientific Discovery and Accelerated Development of Therapies and Medical Devices. Toxicol. Pathol. 2016, 44, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Baxa, M.; Hruska-Plochan, M.; Juhas, S.; Vodicka, P.; Pavlok, A.; Juhasova, J.; Miyanohara, A.; Nejime, T.; Klima, J.; Macakova, M.; et al. A Transgenic Minipig Model of Huntington’s Disease. J. Huntingt. Dis. 2013, 2, 47–68. [Google Scholar] [CrossRef]
- Yang, W.; Chen, X.; Li, S.; Li, X.-J. Genetically Modified Large Animal Models for Investigating Neurodegenerative Diseases. Cell Biosci. 2021, 11, 218. [Google Scholar] [CrossRef]
- Askeland, G.; Rodinova, M.; Štufková, H.; Dosoudilova, Z.; Baxa, M.; Smatlikova, P.; Bohuslavova, B.; Klempir, J.; Nguyen, T.D.; Kuśnierczyk, A.; et al. A Transgenic Minipig Model of Huntington’s Disease Shows Early Signs of Behavioral and Molecular Pathologies. Dis. Models Mech. 2018, 11, dmm035949. [Google Scholar] [CrossRef] [Green Version]
- Baxa, M.; Levinska, B.; Skrivankova, M.; Pokorny, M.; Juhasova, J.; Klima, J.; Klempir, J.; Motlík, J.; Juhas, S.; Ellederova, Z. Longitudinal Study Revealing Motor, Cognitive and Behavioral Decline in a Transgenic Minipig Model of Huntington’s Disease. Dis. Models Mech. 2019, 13, dmm.041293. [Google Scholar] [CrossRef] [Green Version]
- Navarro, R.; Juhas, S.; Keshavarzi, S.; Juhasova, J.; Motlik, J.; Johe, K.; Marsala, S.; Scadeng, M.; Lazar, P.; Tomori, Z.; et al. Chronic Spinal Compression Model in Minipigs: A Systematic Behavioral, Qualitative, and Quantitative Neuropathological Study. J. Neurotrauma 2012, 29, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Borovanský, J.; Horák, V.; Elleder, M.; Fortýn, K.; Smit, N.P.; Kolb, A.M. Biochemical Characterization of a New Melanoma Model—The Minipig MeLiM Strain. Melanoma Res. 2003, 13, 543–548. [Google Scholar] [CrossRef]
- Horak, V.; Palanova, A.; Cizkova, J.; Miltrova, V.; Vodicka, P.; Kupcova Skalnikova, H. Melanoma-Bearing Libechov Minipig (MeLiM): The Unique Swine Model of Hereditary Metastatic Melanoma. Genes 2019, 10, 915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Li, X.; Du, X.; Liu, S.; Xu, Y. Diagnostic and Therapeutic Potential of Exosomes in Neurodegenerative Diseases. Front. Aging Neurosci. 2021, 13, 790863. [Google Scholar] [CrossRef]
- He, M.; Zhang, H.-N.; Tang, Z.-C.; Gao, S.-G. Diagnostic and Therapeutic Potential of Exosomal MicroRNAs for Neurodegenerative Diseases. Neural Plast. 2021, 2021, 8884642. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Mankhong, S.; Kang, J.-H. Extracellular Vesicle as a Source of Alzheimer’s Biomarkers: Opportunities and Challenges. Int. J. Mol. Sci. 2019, 20, 1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Sarko, D.K.; McKinney, C.E. Exosomes: Origins and Therapeutic Potential for Neurodegenerative Disease. Front. Neurosci. 2017, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Yuyama, K.; Igarashi, Y. Physiological and Pathological Roles of Exosomes in the Nervous System. Biomol. Concepts 2016, 7, 53–68. [Google Scholar] [CrossRef]
- Rajendran, L.; Bali, J.; Barr, M.M.; Court, F.A.; Krämer-Albers, E.-M.; Picou, F.; Raposo, G.; van der Vos, K.E.; van Niel, G.; Wang, J.; et al. Emerging Roles of Extracellular Vesicles in the Nervous System. J. Neurosci. 2014, 34, 15482–15489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnatz, A.; Müller, C.; Brahmer, A.; Krämer-Albers, E.-M. Extracellular Vesicles in Neural Cell Interaction and CNS Homeostasis. FASEB BioAdvances 2021, 3, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Gu, J.; Li, Z.; Xu, Z.; Gui, Y. Recent Advances on Extracellular Vesicles in Central Nervous System Diseases. Clin. Interv. Aging 2021, 16, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Busatto, S.; Morad, G.; Guo, P.; Moses, M.A. The Role of Extracellular Vesicles in the Physiological and Pathological Regulation of the Blood-Brain Barrier. FASEB BioAdvances 2021, 3, 665–675. [Google Scholar] [CrossRef]
- Choi, H.; Choi, K.; Kim, D.-H.; Oh, B.-K.; Yim, H.; Jo, S.; Choi, C. Strategies for Targeted Delivery of Exosomes to the Brain: Advantages and Challenges. Pharmaceutics 2022, 14, 672. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of SiRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Properzi, F.; Ferroni, E.; Poleggi, A.; Vinci, R. The Regulation of Exosome Function in the CNS: Implications for Neurodegeneration. Swiss Med. Wkly. 2015, 145, w14204. [Google Scholar] [CrossRef]
- Vella, L.J.; Sharples, R.A.; Nisbet, R.M.; Cappai, R.; Hill, A.F. The Role of Exosomes in the Processing of Proteins Associated with Neurodegenerative Diseases. Eur. Biophys. J. EBJ 2008, 37, 323–332. [Google Scholar] [CrossRef]
- Vandendriessche, C.; Bruggeman, A.; Van Cauwenberghe, C.; Vandenbroucke, R.E. Extracellular Vesicles in Alzheimer’s and Parkinson’s Disease: Small Entities with Large Consequences. Cells 2020, 9, E2485. [Google Scholar] [CrossRef]
- Gagliardi, D.; Bresolin, N.; Comi, G.P.; Corti, S. Extracellular Vesicles and Amyotrophic Lateral Sclerosis: From Misfolded Protein Vehicles to Promising Clinical Biomarkers. Cell. Mol. Life Sci. CMLS 2021, 78, 561–572. [Google Scholar] [CrossRef]
- Ruan, Z.; Pathak, D.; Venkatesan Kalavai, S.; Yoshii-Kitahara, A.; Muraoka, S.; Bhatt, N.; Takamatsu-Yukawa, K.; Hu, J.; Wang, Y.; Hersh, S.; et al. Alzheimer’s Disease Brain-Derived Extracellular Vesicles Spread Tau Pathology in Interneurons. Brain J. Neurol. 2021, 144, 288–309. [Google Scholar] [CrossRef]
- Zhang, X.; Abels, E.R.; Redzic, J.S.; Margulis, J.; Finkbeiner, S.; Breakefield, X.O. Potential Transfer of Polyglutamine and CAG-Repeat RNA in Extracellular Vesicles in Huntington’s Disease: Background and Evaluation in Cell Culture. Cell. Mol. Neurobiol. 2016, 36, 459–470. [Google Scholar] [CrossRef]
- Jeon, I.; Cicchetti, F.; Cisbani, G.; Lee, S.; Li, E.; Bae, J.; Lee, N.; Li, L.; Im, W.; Kim, M.; et al. Human-to-Mouse Prion-like Propagation of Mutant Huntingtin Protein. Acta Neuropathol. 2016, 132, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; McKinney, K.Q.; Pavlopoulos, A.J.; Han, M.H.; Kim, S.-H.; Kim, H.J.; Hwang, S. Exosomal Proteome Analysis of Cerebrospinal Fluid Detects Biosignatures of Neuromyelitis Optica and Multiple Sclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 462, 118–126. [Google Scholar] [CrossRef]
- Hong, Y.; Zhao, T.; Li, X.-J.; Li, S. Mutant Huntingtin Inhibits AB-Crystallin Expression and Impairs Exosome Secretion from Astrocytes. J. Neurosci. 2017, 37, 9550–9563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Koutras, C.; Donnelier, J.; Alshehri, M.; Fotouhi, M.; Girard, M.; Casha, S.; McPherson, P.S.; Robbins, S.M.; Braun, J.E.A. Neurons Export Extracellular Vesicles Enriched in Cysteine String Protein and Misfolded Protein Cargo. Sci. Rep. 2017, 7, 956. [Google Scholar] [CrossRef] [Green Version]
- Ananbeh, H.; Vodicka, P.; Kupcova Skalnikova, H. Emerging Roles of Exosomes in Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 4085. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of Extracellular Vesicle Biomarkers With Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019, 76, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Sergeant, N.; Buée, L. Potential Contribution of Exosomes to the Prion-Like Propagation of Lesions in Alzheimer’s Disease. Front. Physiol. 2012, 3, 229. [Google Scholar] [CrossRef] [Green Version]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of Pre-Clinical Alzheimer’s Disease by a Profile of Pathogenic Proteins in Neurally-Derived Blood Exosomes: A Case-Control Study. Alzheimers Dement. J. Alzheimers Assoc. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered Lysosomal Proteins in Neural-Derived Plasma Exosomes in Preclinical Alzheimer Disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of Conversion from Mild Cognitive Impairment to Dementia with Neuronally Derived Blood Exosome Protein Profile. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma Exosomal α-Synuclein Is Likely CNS-Derived and Increased in Parkinson’s Disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Tomlinson, P.R.; Zheng, Y.; Fischer, R.; Heidasch, R.; Gardiner, C.; Evetts, S.; Hu, M.; Wade-Martins, R.; Turner, M.R.; Morris, J.; et al. Identification of Distinct Circulating Exosomes in Parkinson’s Disease. Ann. Clin. Transl. Neurol. 2015, 2, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-Y.; Lu, J.-M.; Zhao, Z.-Q.; Li, M.-C.; Lu, T.; An, X.-S.; Xue, L.-J. MicroRNA Biomarkers of Parkinson’s Disease in Serum Exosome-like Microvesicles. Neurosci. Lett. 2017, 644, 94–99. [Google Scholar] [CrossRef]
- You, Y.; Ikezu, T. Emerging Roles of Extracellular Vesicles in Neurodegenerative Disorders. Neurobiol. Dis. 2019, 130, 104512. [Google Scholar] [CrossRef]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular Propagated Misfolding of Wild-Type Cu/Zn Superoxide Dismutase Occurs via Exosome-Dependent and -Independent Mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, N.; Doi, H.; Kurata, Y.; Kagawa, H.; Atobe, Y.; Funakoshi, K.; Tada, M.; Katsumoto, A.; Tanaka, K.; Kunii, M.; et al. Proteomic Analysis of Exosome-Enriched Fractions Derived from Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients. Neurosci. Res. 2020, 160, 43–49. [Google Scholar] [CrossRef]
- Wang, J.K.T.; Langfelder, P.; Horvath, S.; Palazzolo, M.J. Exosomes and Homeostatic Synaptic Plasticity Are Linked to Each Other and to Huntington’s, Parkinson’s, and Other Neurodegenerative Diseases by Database-Enabled Analyses of Comprehensively Curated Datasets. Front. Neurosci. 2017, 11, 149. [Google Scholar] [CrossRef]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.-M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-Derived Extracellular Vesicles from Superoxide Dismutase 1 (SOD1)G93A ALS Mice Originate from Astrocytes and Neurons and Carry Misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.-C.; Chan, L.; Chen, J.-H.; Bamodu, O.A.; Chiu, H.-W.; Hong, C.-T. Plasma Extracellular Vesicles Tau and β-Amyloid as Biomarkers of Cognitive Dysfunction of Parkinson’s Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21895. [Google Scholar] [CrossRef]
- Eren, E.; Leoutsakos, J.-M.; Troncoso, J.; Lyketsos, C.G.; Oh, E.S.; Kapogiannis, D. Neuronal-Derived EV Biomarkers Track Cognitive Decline in Alzheimer’s Disease. Cells 2022, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Wang, X.; Ma, F.; Li, F.; Li, H.; Li, F.; Zhang, A.; Gao, Y. Association between T-Tau Protein and Aβ42 in Plasma Neuronal-Derived Exosomes and Cognitive Impairment in Patients with Permanent Atrial Fibrillation and the Role of Anticoagulant Therapy and Inflammatory Mechanisms. J. Card. Surg. 2022, 37, 909–918. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Wong, D.K.; Hong, K.Y.; Raffai, R.L. Cushioned-Density Gradient Ultracentrifugation (C-DGUC): A Refined and High Performance Method for the Isolation, Characterization, and Use of Exosomes. Methods Mol. Biol. 2018, 1740, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein Misfolding, Aggregation, and Conformational Strains in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Weiss, A.; Abramowski, D.; Bibel, M.; Bodner, R.; Chopra, V.; DiFiglia, M.; Fox, J.; Kegel, K.; Klein, C.; Grueninger, S.; et al. Single-Step Detection of Mutant Huntingtin in Animal and Human Tissues: A Bioassay for Huntington’s Disease. Anal. Biochem. 2009, 395, 8–15. [Google Scholar] [CrossRef]
- Wild, E.J.; Boggio, R.; Langbehn, D.; Robertson, N.; Haider, S.; Miller, J.R.C.; Zetterberg, H.; Leavitt, B.R.; Kuhn, R.; Tabrizi, S.J.; et al. Quantification of Mutant Huntingtin Protein in Cerebrospinal Fluid from Huntington’s Disease Patients. J. Clin. Investig. 2015, 125, 1979–1986. [Google Scholar] [CrossRef]
- Macdonald, D.; Tessari, M.A.; Boogaard, I.; Smith, M.; Pulli, K.; Szynol, A.; Albertus, F.; Lamers, M.B.A.C.; Dijkstra, S.; Kordt, D.; et al. Quantification Assays for Total and Polyglutamine-Expanded Huntingtin Proteins. PLoS ONE 2014, 9, e96854. [Google Scholar] [CrossRef]
- Baldo, B.; Sajjad, M.U.; Cheong, R.Y.; Bigarreau, J.; Vijayvargia, R.; McLean, C.; Perrier, A.L.; Seong, I.S.; Halliday, G.; Petersén, Å.; et al. Quantification of Total and Mutant Huntingtin Protein Levels in Biospecimens Using a Novel AlphaLISA Assay. eNeuro 2018, 5, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallès, A.; Evers, M.M.; Stam, A.; Sogorb-Gonzalez, M.; Brouwers, C.; Vendrell-Tornero, C.; Acar-Broekmans, S.; Paerels, L.; Klima, J.; Bohuslavova, B.; et al. Widespread and Sustained Target Engagement in Huntington’s Disease Minipigs upon Intrastriatal MicroRNA-Based Gene Therapy. Sci. Transl. Med. 2021, 13, eabb8920. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-L.; Demontis, F. Systemic Manifestation and Contribution of Peripheral Tissues to Huntington’s Disease Pathogenesis. Ageing Res. Rev. 2021, 69, 101358. [Google Scholar] [CrossRef] [PubMed]
- Macakova, M.; Bohuslavova, B.; Vochozkova, P.; Pavlok, A.; Sedlackova, M.; Vidinska, D.; Vochyanova, K.; Liskova, I.; Valekova, I.; Baxa, M.; et al. Mutated Huntingtin Causes Testicular Pathology in Transgenic Minipig Boars. Neurodegener. Dis. 2016, 16, 245–259. [Google Scholar] [CrossRef]
- Palviainen, M.; Saraswat, M.; Varga, Z.; Kitka, D.; Neuvonen, M.; Puhka, M.; Joenväärä, S.; Renkonen, R.; Nieuwland, R.; Takatalo, M.; et al. Extracellular Vesicles from Human Plasma and Serum Are Carriers of Extravesicular Cargo-Implications for Biomarker Discovery. PLoS ONE 2020, 15, e0236439. [Google Scholar] [CrossRef]
- Denis, H.L.; Lamontagne-Proulx, J.; St-Amour, I.; Mason, S.L.; Rowley, J.W.; Cloutier, N.; Tremblay, M.-È.; Vincent, A.T.; Gould, P.V.; Chouinard, S.; et al. Platelet Abnormalities in Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry 2019, 90, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Denis, H.L.; Lamontagne-Proulx, J.; St-Amour, I.; Mason, S.L.; Weiss, A.; Chouinard, S.; Barker, R.A.; Boilard, E.; Cicchetti, F. Platelet-Derived Extracellular Vesicles in Huntington’s Disease. J. Neurol. 2018, 265, 2704–2712. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A Comparison of Methods for the Isolation and Separation of Extracellular Vesicles from Protein and Lipid Particles in Human Serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef] [Green Version]
- Roux, Q.; Van Deun, J.; Dedeyne, S.; Hendrix, A. The EV-TRACK Summary Add-on: Integration of Experimental Information in Databases to Ensure Comprehensive Interpretation of Biological Knowledge on Extracellular Vesicles. J. Extracell. Vesicles 2020, 9, 1699367. [Google Scholar] [CrossRef]
- Van Deun, J.; Mestdagh, P.; Sormunen, R.; Cocquyt, V.; Vermaelen, K.; Vandesompele, J.; Bracke, M.; De Wever, O.; Hendrix, A. The Impact of Disparate Isolation Methods for Extracellular Vesicles on Downstream RNA Profiling. J. Extracell. Vesicles 2014, 3, 24858. [Google Scholar] [CrossRef] [Green Version]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.F.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized Exosome Isolation Protocol for Cell Culture Supernatant and Human Plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, T.; Herczeg, K.; Onódi, Z.; Voszka, I.; Módos, K.; Marton, N.; Nagy, G.; Mäger, I.; Wood, M.J.; El Andaloussi, S.; et al. Isolation of Exosomes from Blood Plasma: Qualitative and Quantitative Comparison of Ultracentrifugation and Size Exclusion Chromatography Methods. PLoS ONE 2015, 10, e0145686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupcova Skalnikova, H.; Bohuslavova, B.; Turnovcova, K.; Juhasova, J.; Juhas, S.; Rodinova, M.; Vodicka, P. Isolation and Characterization of Small Extracellular Vesicles from Porcine Blood Plasma, Cerebrospinal Fluid, and Seminal Plasma. Proteomes 2019, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, T.; Tartakoff, A.M. Nuclear Relocation of Normal Huntingtin. Traffic Cph. Den. 2001, 2, 385–394. [Google Scholar] [CrossRef]
- Karimi, N.; Cvjetkovic, A.; Jang, S.C.; Crescitelli, R.; Hosseinpour Feizi, M.A.; Nieuwland, R.; Lötvall, J.; Lässer, C. Detailed Analysis of the Plasma Extracellular Vesicle Proteome after Separation from Lipoproteins. Cell. Mol. Life Sci. CMLS 2018, 75, 2873–2886. [Google Scholar] [CrossRef] [Green Version]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Ji, H.; Mathivanan, S.; Scott, A.M.; Simpson, R.J. Comparison of Ultracentrifugation, Density Gradient Separation, and Immunoaffinity Capture Methods for Isolating Human Colon Cancer Cell Line LIM1863-Derived Exosomes. Methods 2012, 56, 293–304. [Google Scholar] [CrossRef]
- Fauré, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes Are Released by Cultured Cortical Neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef]
- Onódi, Z.; Pelyhe, C.; Terézia Nagy, C.; Brenner, G.B.; Almási, L.; Kittel, Á.; Manček-Keber, M.; Ferdinandy, P.; Buzás, E.I.; Giricz, Z. Isolation of High-Purity Extracellular Vesicles by the Combination of Iodixanol Density Gradient Ultracentrifugation and Bind-Elute Chromatography From Blood Plasma. Front. Physiol. 2018, 9, 1479. [Google Scholar] [CrossRef] [Green Version]
- Vidinská, D.; Vochozková, P.; Šmatlíková, P.; Ardan, T.; Klíma, J.; Juhás, Š.; Juhásová, J.; Bohuslavová, B.; Baxa, M.; Valeková, I.; et al. Gradual Phenotype Development in Huntington Disease Transgenic Minipig Model at 24 Months of Age. Neurodegener. Dis. 2018, 18, 107–119. [Google Scholar] [CrossRef]
- Jozefovicova, M.; Herynek, V.; Jiru, F.; Dezortova, M.; Juhasova, J.; Juhas, S.; Motlik, J.; Hajek, M. Minipig Model of Huntington’s Disease: 1H Magnetic Resonance Spectroscopy of the Brain. Physiol. Res. 2016, 65, 155–163. [Google Scholar] [CrossRef]
- Ardan, T.; Baxa, M.; Levinská, B.; Sedláčková, M.; Nguyen, T.D.; Klíma, J.; Juhás, Š.; Juhásová, J.; Šmatlíková, P.; Vochozková, P.; et al. Transgenic Minipig Model of Huntington’s Disease Exhibiting Gradually Progressing Neurodegeneration. Dis. Models Mech. 2019, 13, dmm041319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Červenka, J.; Tylečková, J.; Kupcová Skalníková, H.; Vodičková Kepková, K.; Poliakh, I.; Valeková, I.; Pfeiferová, L.; Kolář, M.; Vaškovičová, M.; Pánková, T.; et al. Proteomic Characterization of Human Neural Stem Cells and Their Secretome during in Vitro Differentiation. Front. Cell. Neurosci. 2021, 14, 612560. [Google Scholar] [CrossRef] [PubMed]
- Pospichalova, V.; Svoboda, J.; Dave, Z.; Kotrbova, A.; Kaiser, K.; Klemova, D.; Ilkovics, L.; Hampl, A.; Crha, I.; Jandakova, E.; et al. Simplified Protocol for Flow Cytometry Analysis of Fluorescently Labeled Exosomes and Microvesicles Using Dedicated Flow Cytometer. J. Extracell. Vesicles 2015, 4, 25530. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ananbeh, H.; Novak, J.; Juhas, S.; Juhasova, J.; Klempir, J.; Doleckova, K.; Rysankova, I.; Turnovcova, K.; Hanus, J.; Hansikova, H.; et al. Huntingtin Co-Isolates with Small Extracellular Vesicles from Blood Plasma of TgHD and KI-HD Pig Models of Huntington’s Disease and Human Blood Plasma. Int. J. Mol. Sci. 2022, 23, 5598. https://doi.org/10.3390/ijms23105598
Ananbeh H, Novak J, Juhas S, Juhasova J, Klempir J, Doleckova K, Rysankova I, Turnovcova K, Hanus J, Hansikova H, et al. Huntingtin Co-Isolates with Small Extracellular Vesicles from Blood Plasma of TgHD and KI-HD Pig Models of Huntington’s Disease and Human Blood Plasma. International Journal of Molecular Sciences. 2022; 23(10):5598. https://doi.org/10.3390/ijms23105598
Chicago/Turabian StyleAnanbeh, Hanadi, Jaromir Novak, Stefan Juhas, Jana Juhasova, Jiri Klempir, Kristyna Doleckova, Irena Rysankova, Karolina Turnovcova, Jaroslav Hanus, Hana Hansikova, and et al. 2022. "Huntingtin Co-Isolates with Small Extracellular Vesicles from Blood Plasma of TgHD and KI-HD Pig Models of Huntington’s Disease and Human Blood Plasma" International Journal of Molecular Sciences 23, no. 10: 5598. https://doi.org/10.3390/ijms23105598
APA StyleAnanbeh, H., Novak, J., Juhas, S., Juhasova, J., Klempir, J., Doleckova, K., Rysankova, I., Turnovcova, K., Hanus, J., Hansikova, H., Vodicka, P., & Kupcova Skalnikova, H. (2022). Huntingtin Co-Isolates with Small Extracellular Vesicles from Blood Plasma of TgHD and KI-HD Pig Models of Huntington’s Disease and Human Blood Plasma. International Journal of Molecular Sciences, 23(10), 5598. https://doi.org/10.3390/ijms23105598