
Down-Regulating the High Level of 17-Beta-Hydroxysteroid Dehydrogenase 13 Plays a Therapeutic Role for Non-Alcoholic Fatty Liver Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
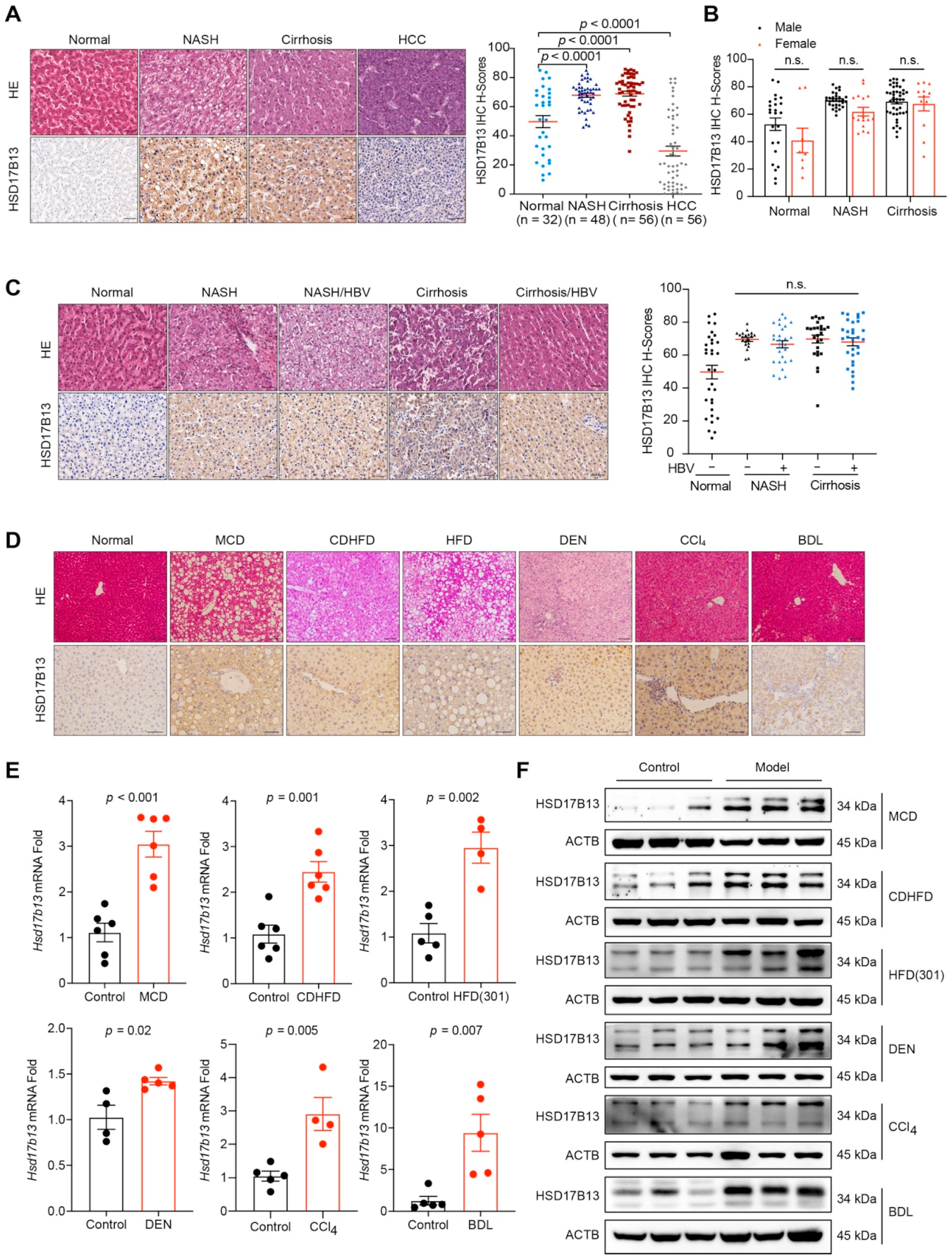
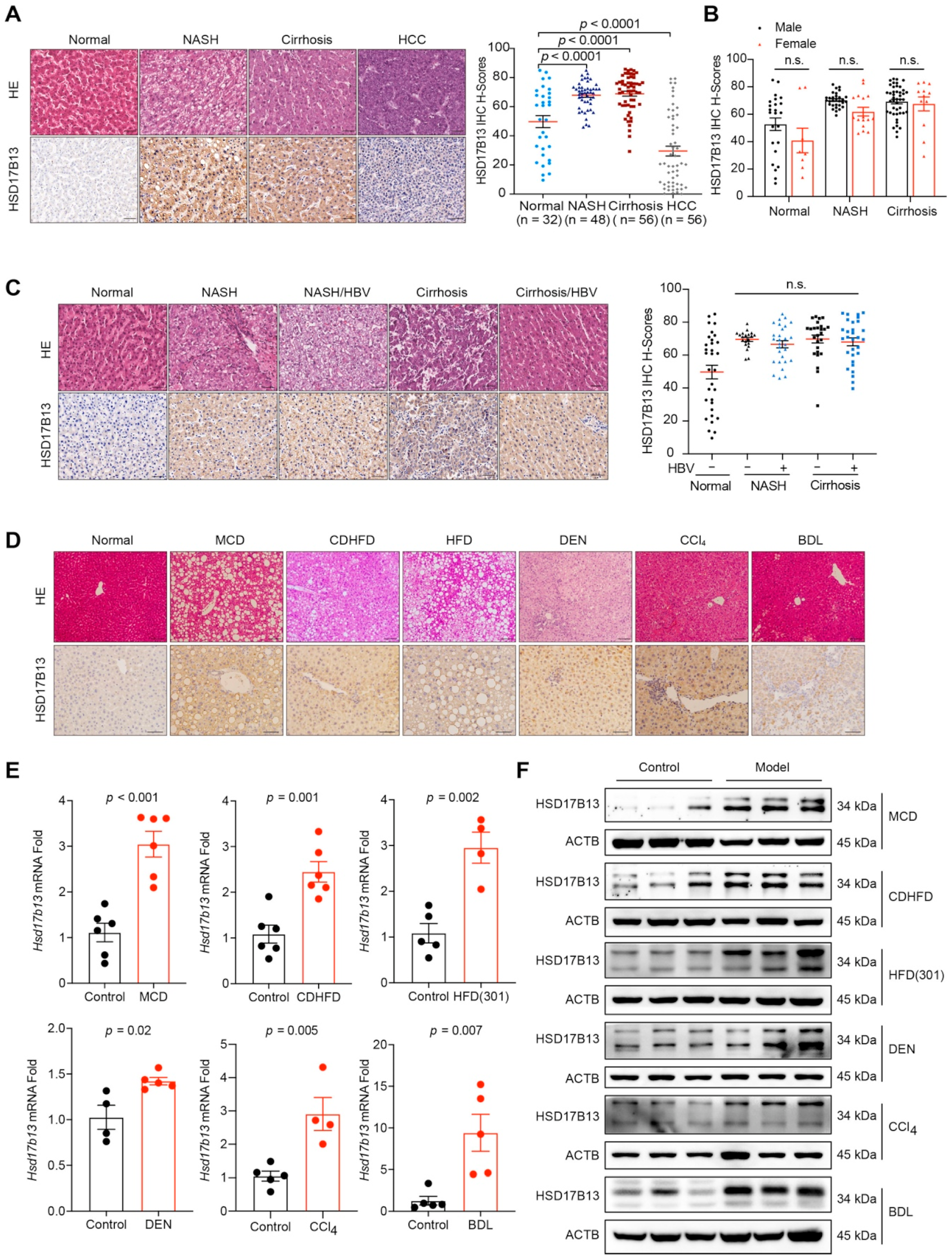
2.1. HSD17B13 Expression Is Increased in the Liver Tissues of Patients and Murine Models with NAFLD
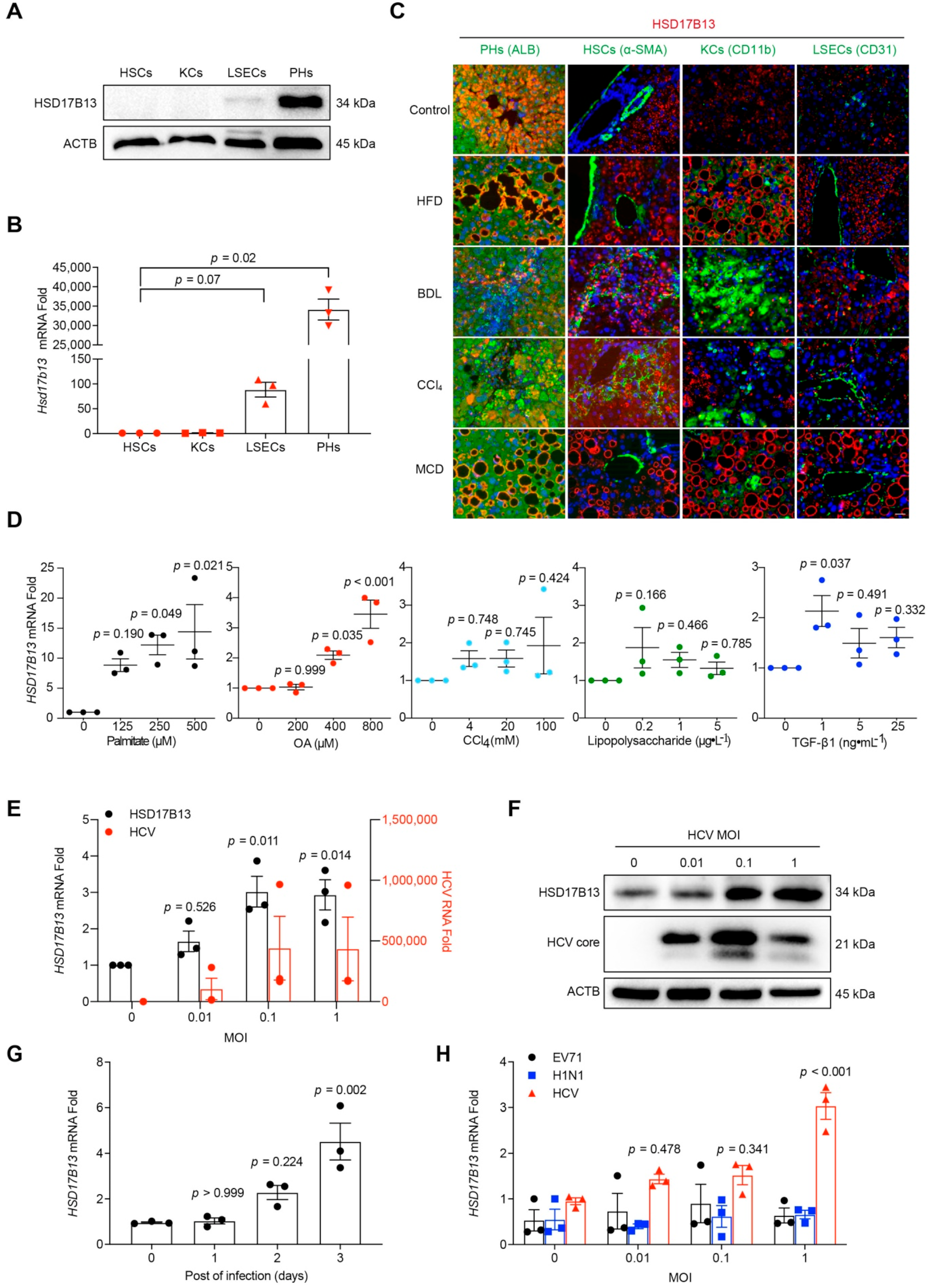
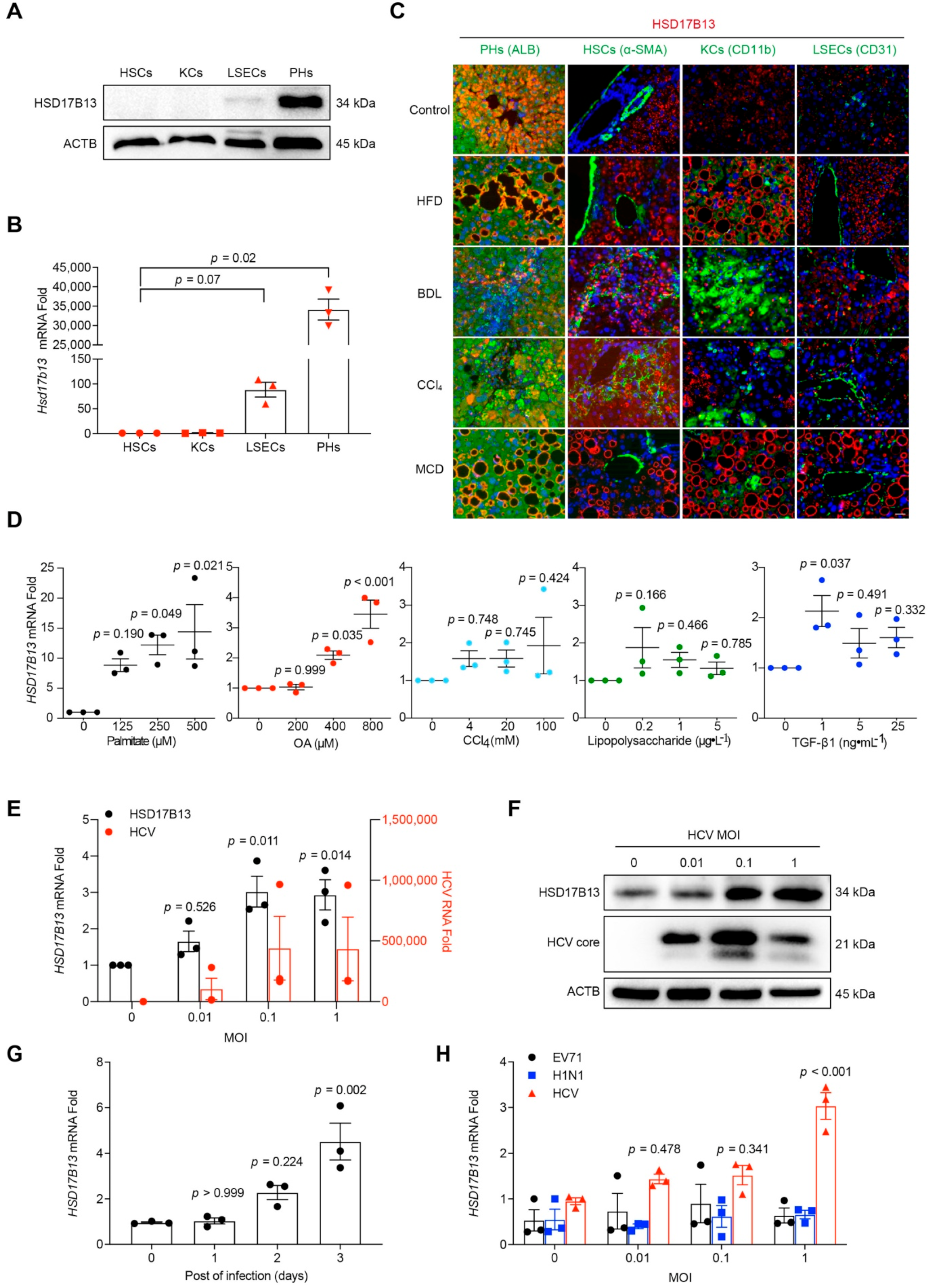
2.2. Higher HSD17B13 Expression Is Mainly in Hepatocytes and Directly Induced by Etiological Agents
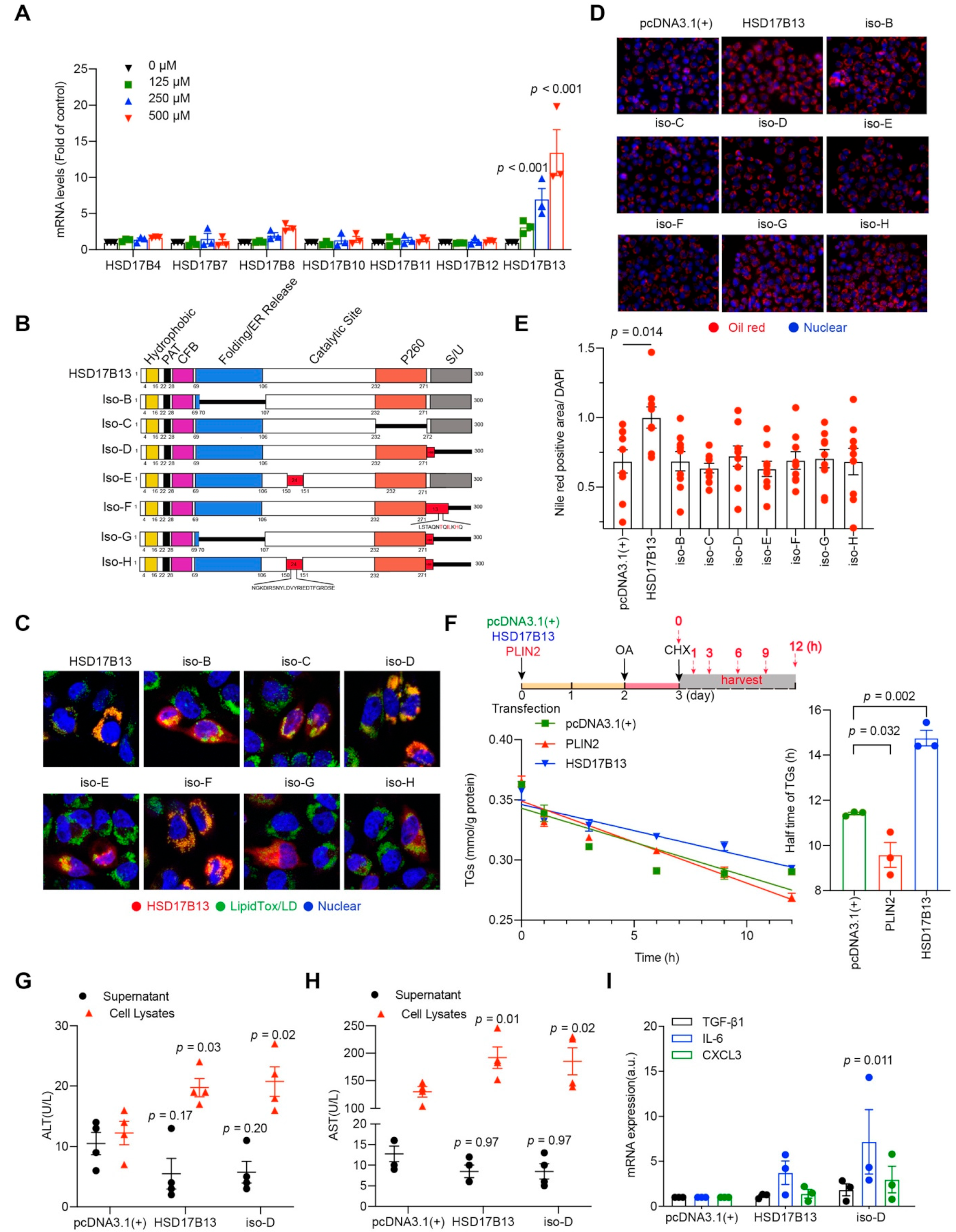
2.3. HSD17B13 Promotes the Accumulation of Intracellular Lipid Droplets and Causes Hepatocyte Injury
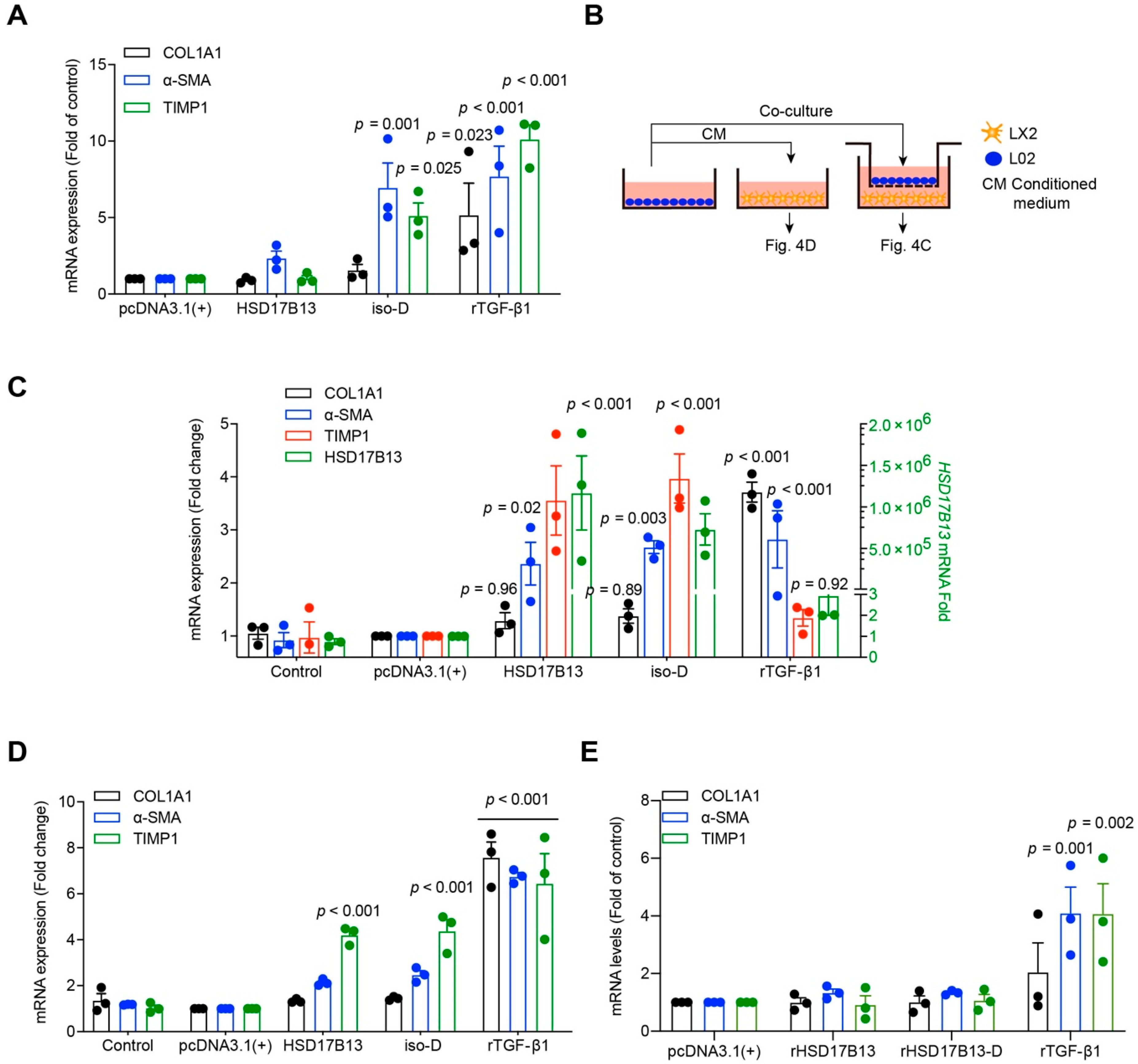
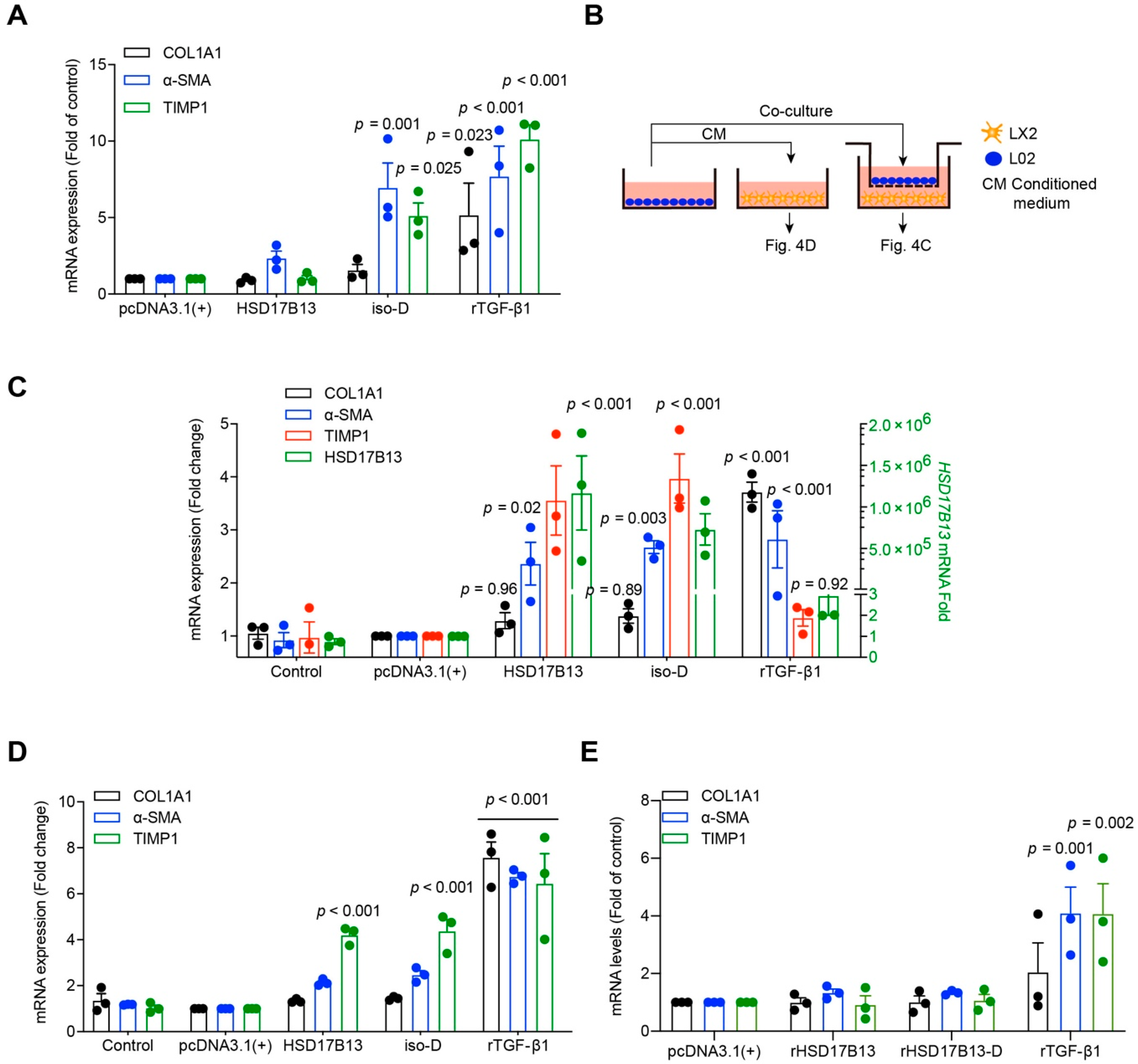
2.4. HSD17B13 in Hepatocytes Indirectly Promotes the Activation of HSCs
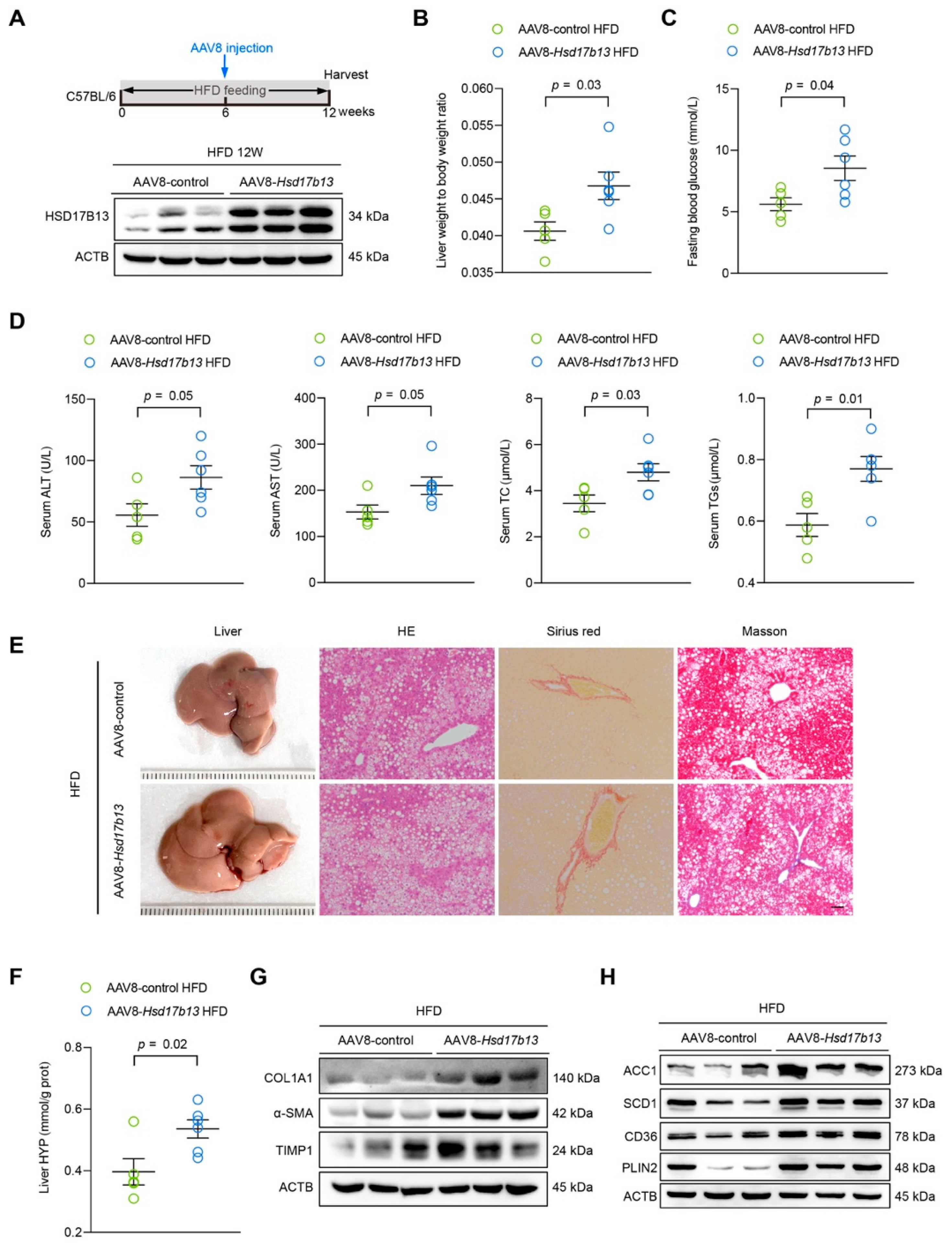
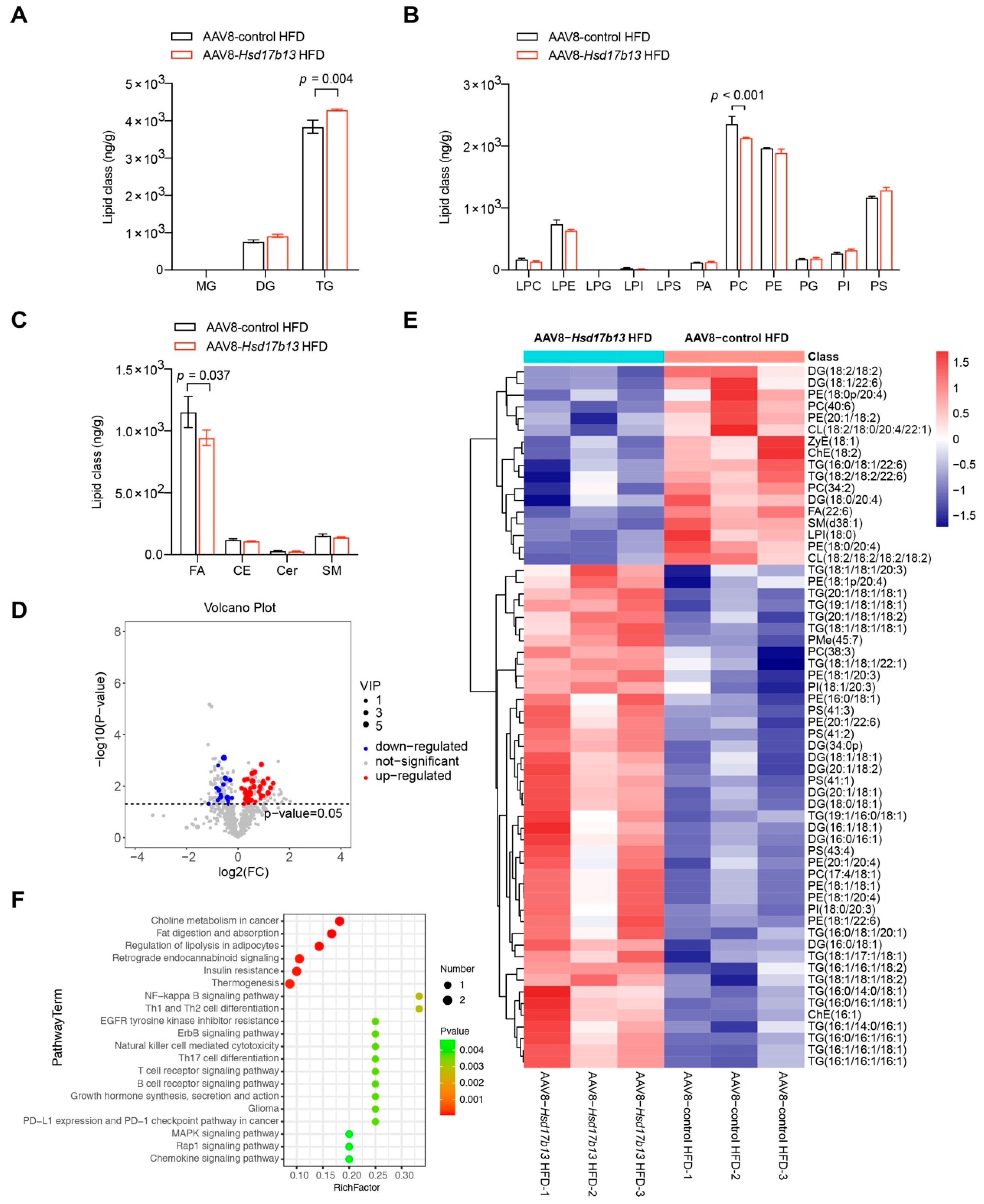
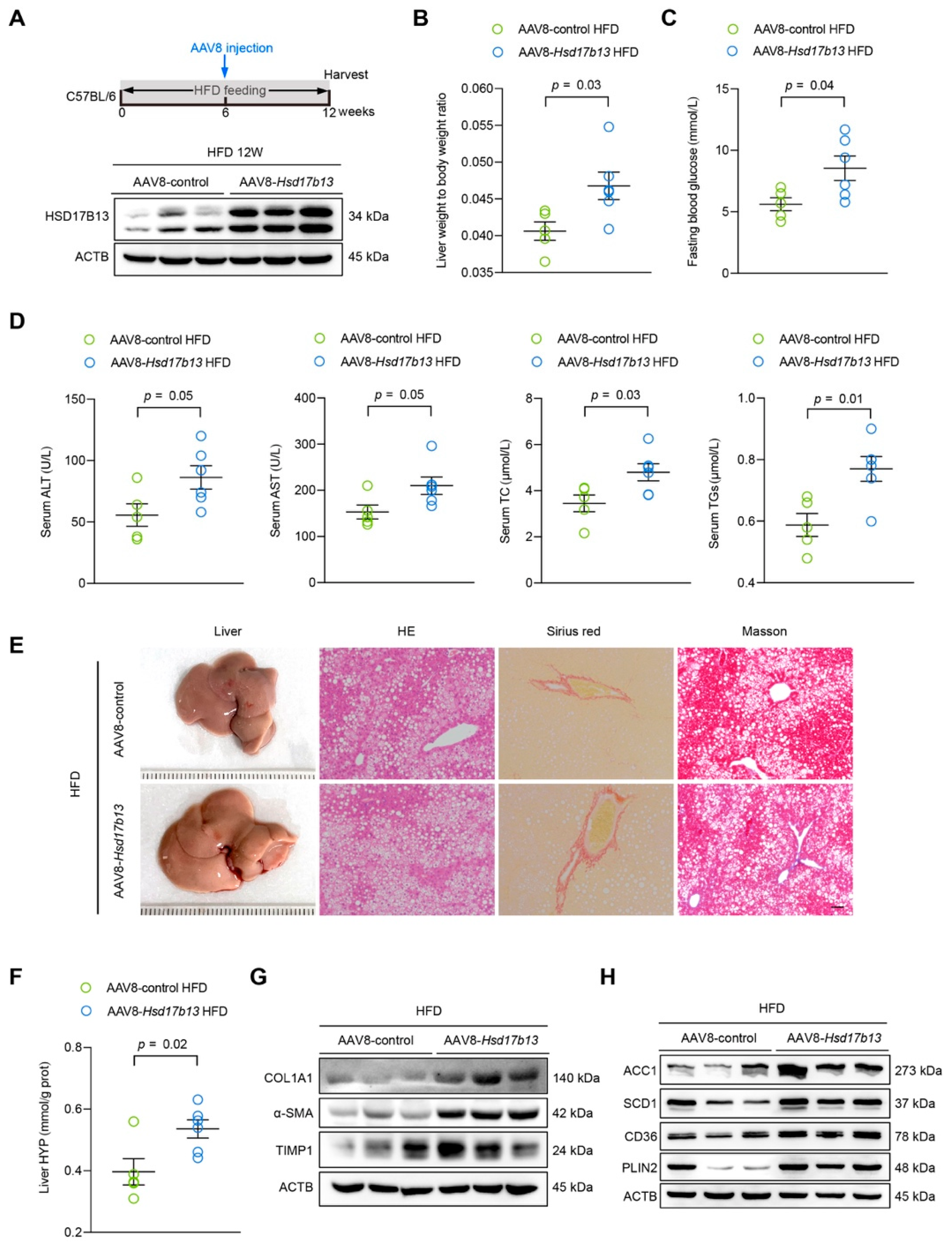
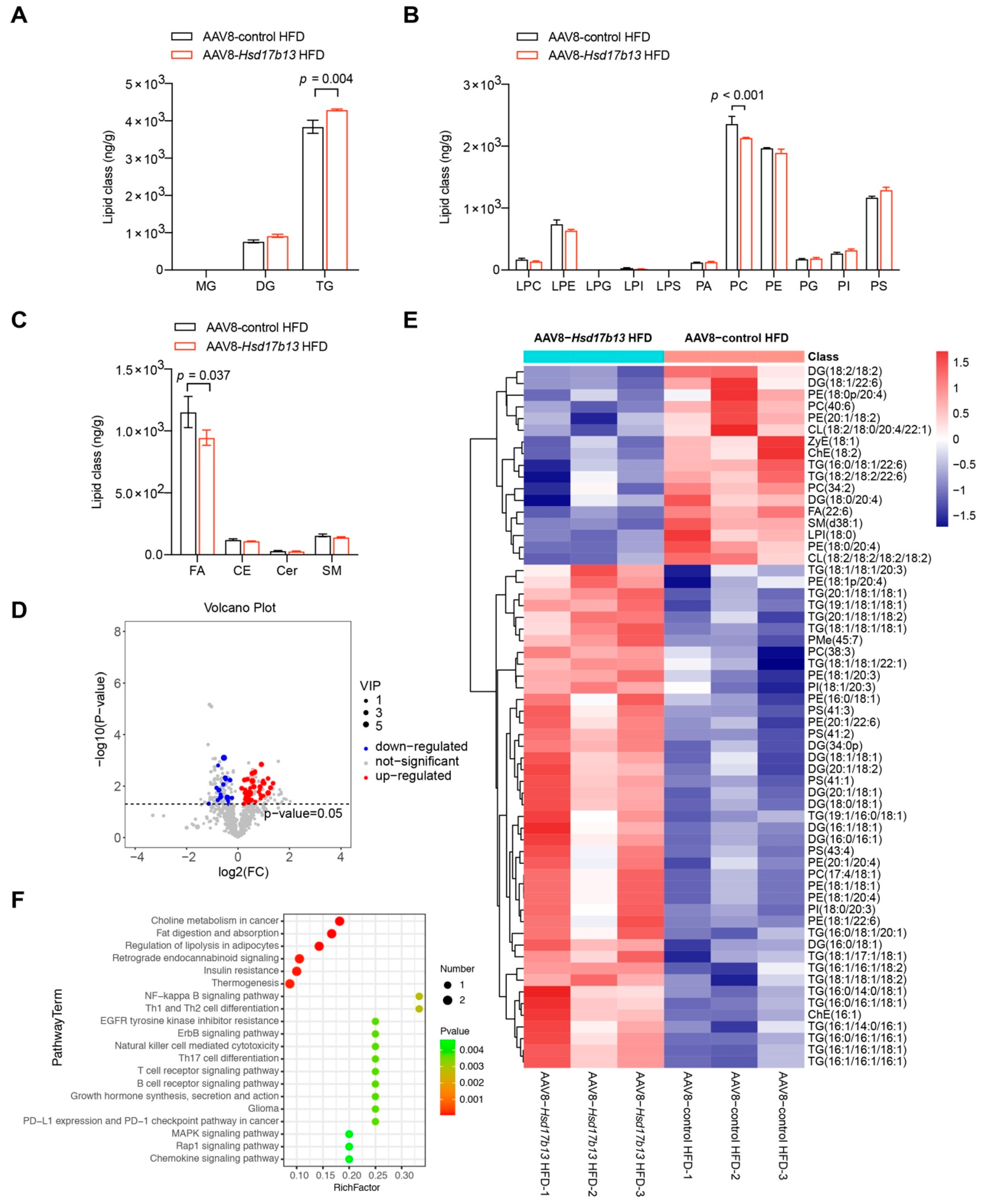
2.5. Overexpression of HSD17B13 in the Liver Aggravates Liver Steatosis and Fibrosis in HFD-Fed Mice
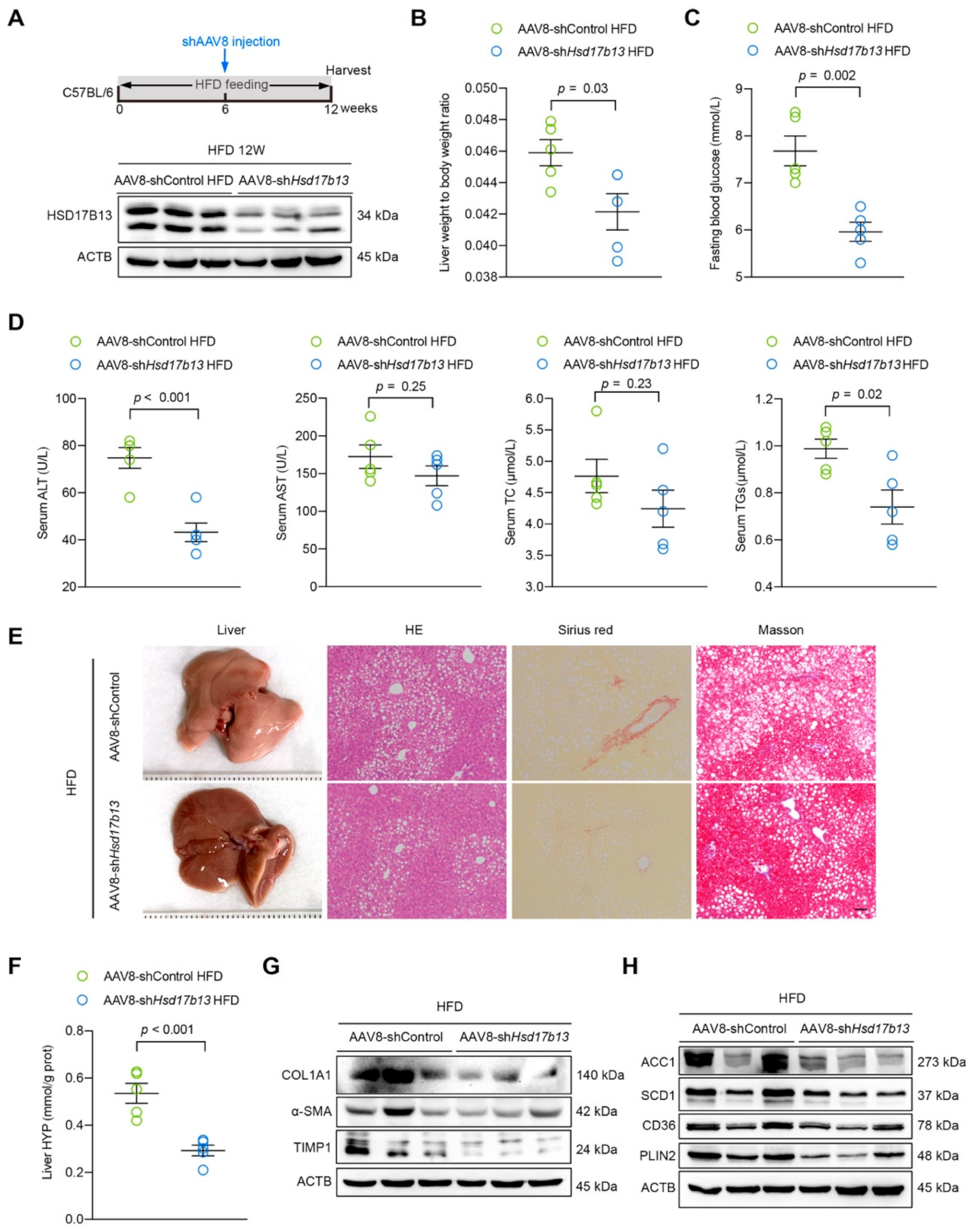
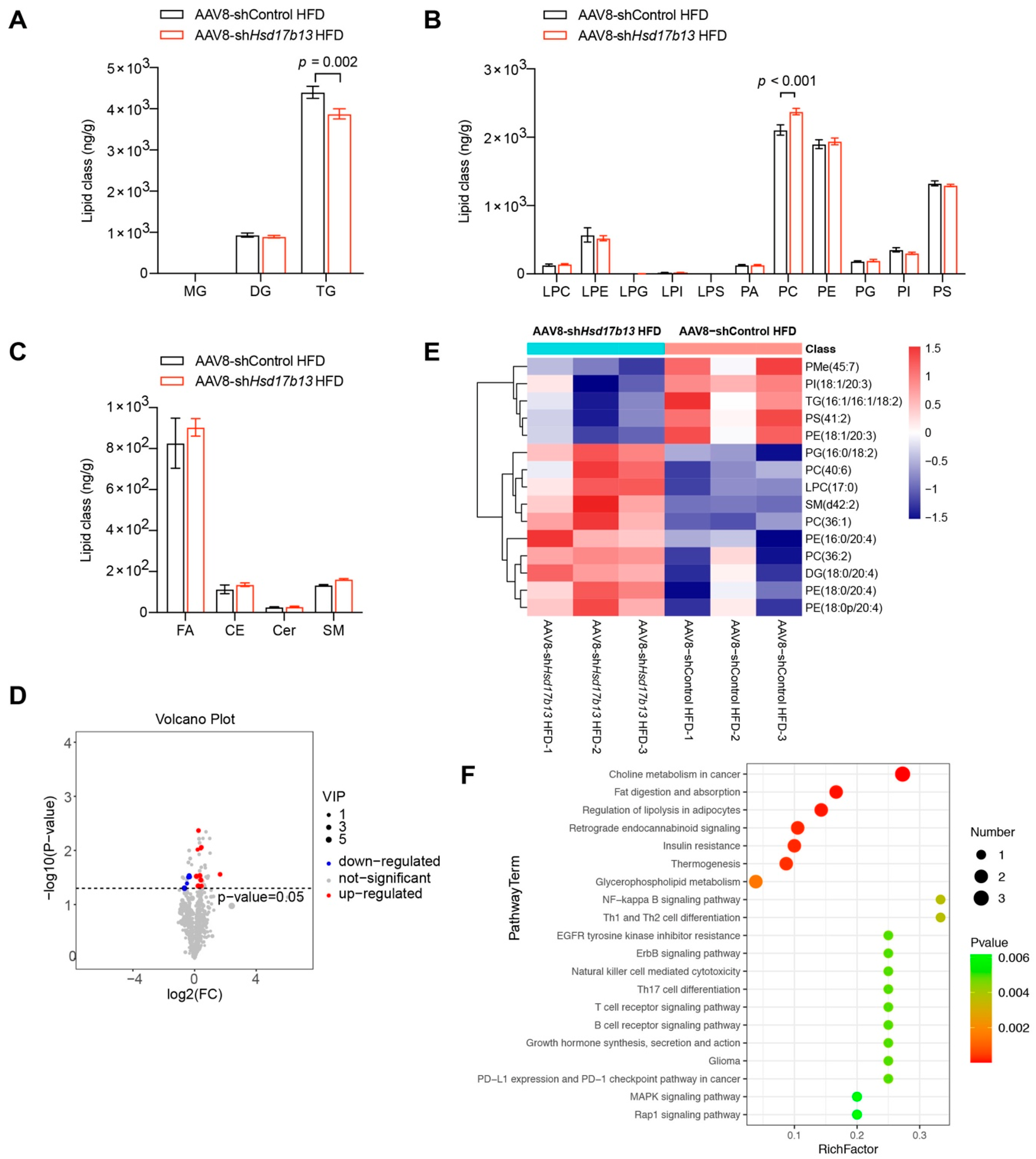
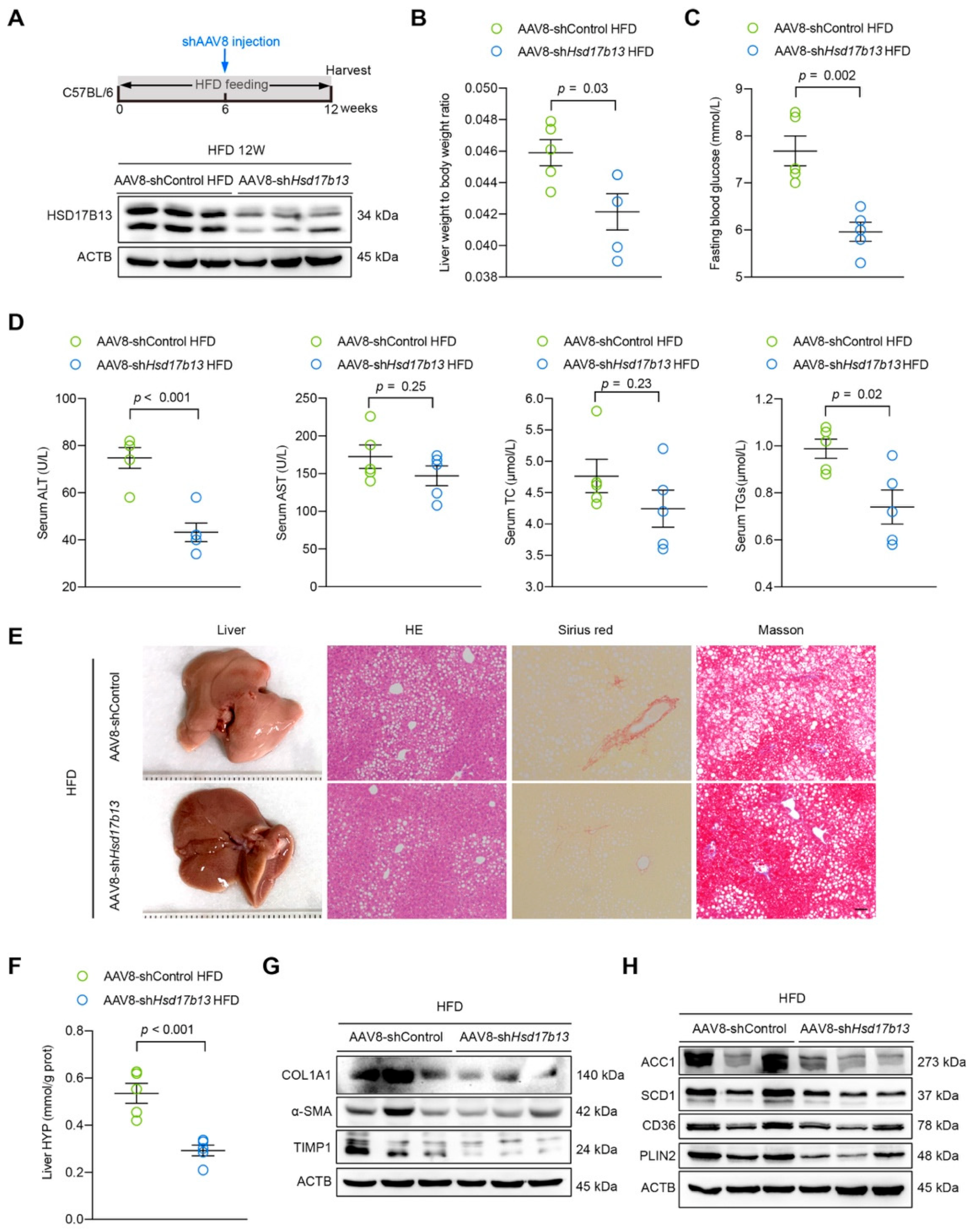
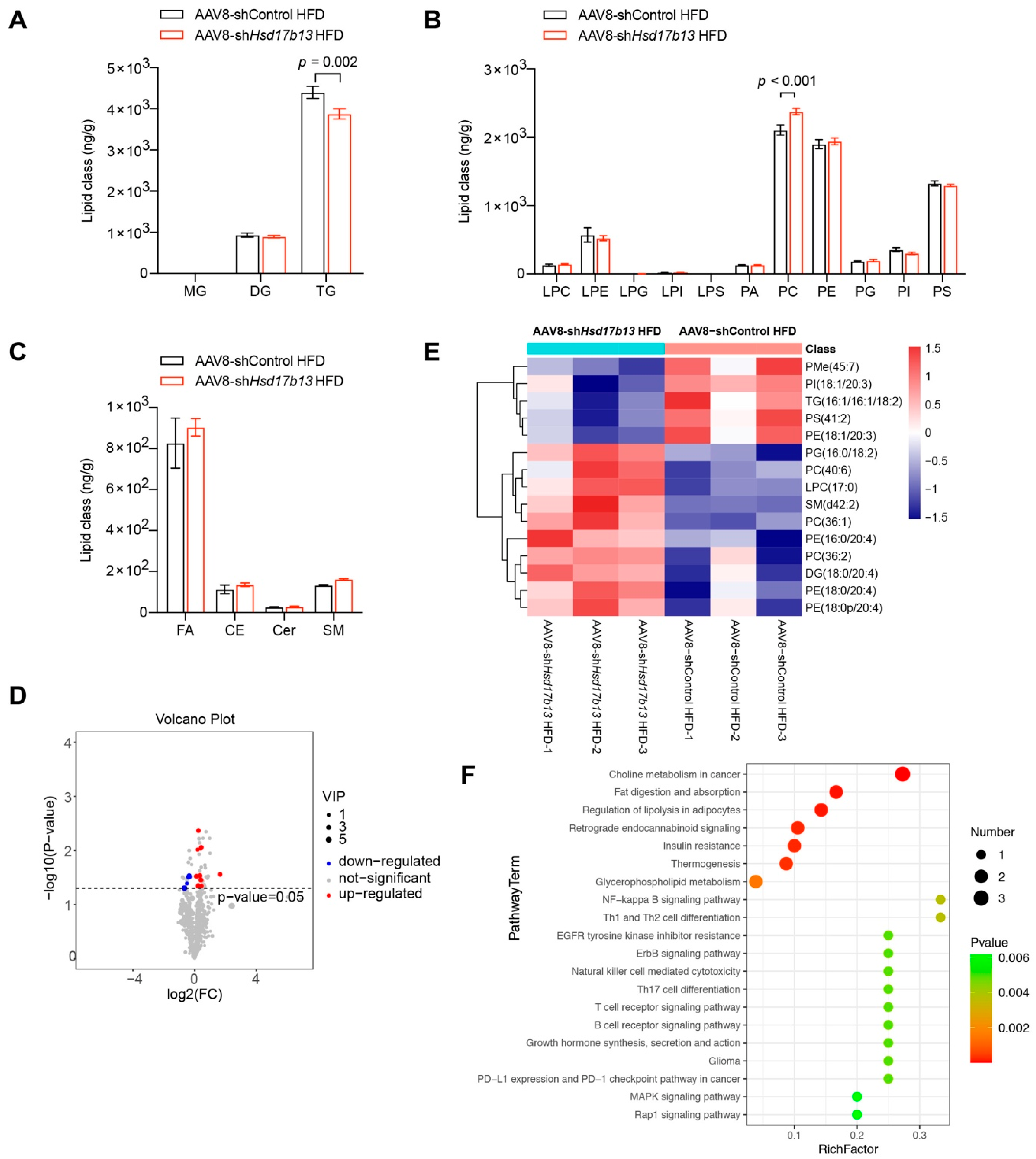
2.6. Down-Regulation of HSD17B13 Plays a Therapeutic Role in NAFLD Mice
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Cell Culture
4.3. Cell Treatment
4.4. Virus Infection
4.5. Isolation of Primary Hepatocytes
4.6. Nile Red Staining Assay
4.7. Confocal Assays
4.8. TG Turnover Study
4.9. Co-Culture of Hepatocytes and HSCs
4.10. Intracellular Transaminase Analyses
4.11. Western Blot Analyses and Antibodies
4.12. Plasmid Construction
4.13. Expression and Purification of Recombinant Proteins
4.14. Quantitative Real-Time PCR Analyses
4.15. Quantification of HCV RNA
4.16. Histopathologic Analysis
4.17. Immunofluorescence Staining
4.18. Fasting Blood Glucose Test
4.19. Liver Function and Biochemical Test
4.20. Adenovirus Vector Construction and Infection
4.21. Lipid Metabolomics Analysis
4.22. Immunohistochemical Staining
4.23. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.-S.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlitina, J. Genetic Risk Factors and Disease Modifiers of Nonalcoholic Steatohepatitis. Gastroenterol. Clin. N. Am. 2020, 49, 25–44. [Google Scholar] [CrossRef]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Abdelmalek, M.F. Nonalcoholic fatty liver disease: Another leap forward. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 85–86. [Google Scholar] [CrossRef]
- Tacke, F.; Weiskirchen, R. An update on the recent advances in antifibrotic therapy. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 1143–1152. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Therapeutic Landscape for NAFLD in 2020. Gastroenterology 2020, 158, 1984–1998.e3. [Google Scholar] [CrossRef]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; De Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Kozlitina, J.; Stender, S.; Hobbs, H.H.; Cohen, J.C. HSD17B13 and Chronic Liver Disease in Blacks and Hispanics. N. Engl. J. Med. 2018, 379, 1876–1877. [Google Scholar] [CrossRef]
- Thomas, H. An HSD17B13 variant reduces cirrhosis risk. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 328. [Google Scholar] [CrossRef]
- Basyte-Bacevice, V.; Skieceviciene, J.; Valantiene, I.; Sumskiene, J.; Petrenkiene, V.; Kondrackiene, J.; Petrauskas, D.; Lammert, F.; Kupcinskas, J. SERPINA1 and HSD17B13 Gene Variants in Patients with Liver Fibrosis and Cirrhosis. J. Gastrointestin Liver Dis. 2019, 28, 297–302. [Google Scholar] [CrossRef]
- Stickel, F.; Lutz, P.; Buch, S.; Nischalke, H.D.; Silva, I.; Rausch, V.; Fischer, J.; Weiss, K.H.; Gotthardt, D.; Rosendahl, J.; et al. Genetic Variation in HSD17B13 Reduces the Risk of Developing Cirrhosis and Hepatocellular Carcinoma in Alcohol Misusers. Hepatology 2020, 72, 88–102. [Google Scholar] [CrossRef]
- Liu, S.; Huang, C.; Li, D.; Ren, W.; Zhang, H.; Qi, M.; Li, X.; Yu, L. Molecular cloning and expression analysis of a new gene for short-chain dehydrogenase/reductase 9. Acta Biochim. Pol. 2007, 54, 213–218. [Google Scholar] [CrossRef]
- Su, W.; Mao, Z.; Liu, Y.; Zhang, X.; Zhang, W.; Gustafsson, J.-A.; Guan, Y. Role of HSD17B13 in the liver physiology and pathophysiology. Mol. Cell. Endocrinol. 2018, 489, 119–125. [Google Scholar] [CrossRef]
- Hiltunen, J.K.; Kastaniotis, A.J.; Autio, K.J.; Jiang, G.; Chen, Z.; Glumoff, T. 17B-hydroxysteroid dehydrogenases as acyl thioester metabolizing enzymes. Mol. Cell. Endocrinol. 2018, 489, 107–118. [Google Scholar] [CrossRef]
- Horiguchi, Y.; Araki, M.; Motojima, K. 17beta-Hydroxysteroid dehydrogenase type 13 is a liver-specific lipid droplet-associated protein. Biochem. Biophys. Res. Commun. 2008, 370, 235–238. [Google Scholar] [CrossRef]
- Su, W.; Wang, Y.; Jia, X.; Wu, W.; Li, L.; Tian, X.; Li, S.; Wang, C.; Xu, H.; Cao, J.; et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11437–11442. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Peng, J.; Li, S.; Dai, Y.-B.; Wang, C.-J.; Xu, H.; Gao, M.; Ruan, X.-Z.; Gustafsson, J.; Guan, Y.-F.; et al. Liver X receptor α induces 17β-hydroxysteroid dehydrogenase-13 expression through SREBP-1c. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E357–E367. [Google Scholar] [CrossRef]
- Di Sessa, A.; Umano, G.R.; Cirillo, G.; Marzuillo, P.; Arienzo, M.R.; Pedullà, M.; del Giudice, E.M. The rs72613567: TA Variant in the Hydroxysteroid 17-beta Dehydrogenase 13 Gene Reduces Liver Damage in Obese Children. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 371–374. [Google Scholar] [CrossRef]
- Kallwitz, E.; Tayo, B.O.; Kuniholm, M.H.; Daviglus, M.; Zeng, D.; Isasi, C.R.; Cotler, S.J. Association of HSD17B13 rs72613567:TA with non-alcoholic fatty liver disease in Hispanics/Latinos. Liver Int. 2020, 40, 889–893. [Google Scholar] [CrossRef]
- Wang, P.; Wu, C.X.; Li, Y.; Shen, N. HSD17B13 rs72613567 protects against liver diseases and histological progression of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8997–9007. [Google Scholar]
- Adam, M.; Heikelä, H.; Sobolewski, C.; Portius, D.; Mäki-Jouppila, J.; Mehmood, A.; Adhikari, P.; Esposito, I.; Elo, L.L.; Zhang, F.-P.; et al. Hydroxysteroid (17β) dehydrogenase 13 deficiency triggers hepatic steatosis and inflammation in mice. FASEB J. 2018, 32, 3434–3447. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhuo, J.-Y.; Yang, F.; Liu, Z.-K.; Zhou, L.; Xie, H.-Y.; Xu, X.; Zheng, S.-S. 17-beta-hydroxysteroid dehydrogenase 13 inhibits the progression and recurrence of hepatocellular carcinoma. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 220–226. [Google Scholar] [CrossRef]
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases From 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616. [Google Scholar] [CrossRef]
- Wong, M.; Huang, J.L.W.; George, J.; Huang, J.; Leung, C.; Eslam, M.; Chan, H.L.Y.; Ng, S.C. The changing epidemiology of liver diseases in the Asia-Pacific region. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 57–73. [Google Scholar] [CrossRef]
- Available online: http://biogps.org/#goto=genereport&id=243168 (accessed on 12 January 2022).
- Available online: http://biogps.org/#goto=genereport&id=345275 (accessed on 12 January 2022).
- Ma, Y.; Brown, P.; Lin, D.D.; Ma, J.; Feng, D.; Belyaeva, O.V.; Podszun, M.C.; Roszik, J.; Allen, J.N.; Umarova, R.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Deficiency Does Not Protect Mice From Obesogenic Diet Injury. Hepatology 2020, 73, 1701–1716. [Google Scholar] [CrossRef]
- Khatun, M.; Ray, R.B. Mechanisms Underlying Hepatitis C Virus-Associated Hepatic Fibrosis. Cells 2019, 8, 1249. [Google Scholar] [CrossRef] [Green Version]
- About, F.; Abel, L.; Cobat, A. HCV-Associated Liver Fibrosis and HSD17B13. N. Engl. J. Med. 2018, 379, 1875–1876. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, M.; Yamaji, T.; Saito, K.; Shirasago, Y.; Satomura, K.; Endo, T.; Fukasawa, M.; Hanada, K.; Osada, N. Identification of Characteristic Genomic Markers in Human Hepatoma HuH-7 and Huh7.5.1-8 Cell Lines. Front. Genet. 2020, 11, 546106. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; McKeating, J.; Rice, C.M. Highly Permissive Cell Lines for Subgenomic and Genomic Hepatitis C Virus RNA Replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardani, I.; Dalen, K.T.; Drevinge, C.; Miljanovic, A.; Ståhlman, M.; Klevstig, M.; Täng, M.S.; Fogelstrand, P.; Levin, M.; Ekstrand, M.; et al. Plin2-deficiency reduces lipophagy and results in increased lipid accumulation in the heart. Sci. Rep. 2019, 9, 6909. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Gellert-Kristensen, H.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; Stender, S. High Risk of Fatty Liver Disease Amplifies the Alanine Transaminase–Lowering Effect of a HSD17B13 Variant. Hepatology 2019, 71, 56–66. [Google Scholar] [CrossRef]
- Moriles, K.E.; Azer, S.A. Alanine Amino Transferase. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 145–159. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Motomura, T.; Amirneni, S.; Diaz-Aragon, R.; Faccioli, L.; Malizio, M.; Coard, M.; Kocas-Kilicarslan, Z.; Frau, C.; Haep, N.; Ostrowska, A.; et al. Is HSD17B13 Genetic Variant a Protector for Liver Dysfunction? Future Perspective as a Potential Therapeutic Target. J. Pers. Med. 2021, 11, 619. [Google Scholar] [CrossRef]
- Ma, Y.; Karki, S.; Brown, P.M.; Lin, D.D.; Podszun, M.C.; Zhou, W.; Belyaeva, O.V.; Kedishvili, N.Y.; Rotman, Y. Characterization of essential domains in HSD17B13 for cellular localization and enzymatic activity. J. Lipid Res. 2020, 61, 1400–1409. [Google Scholar] [CrossRef]
- Mato, J.M.; Alonso, C.; Noureddin, M.; Lu, S.C. Biomarkers and subtypes of deranged lipid metabolism in non-alcoholic fatty liver disease. World J. Gastroenterol. 2019, 25, 3009–3020. [Google Scholar] [CrossRef]
- Goh, V.J.; Silver, D.L. The Lipid Droplet as a Potential Therapeutic Target in NAFLD. Semin. Liver Dis. 2013, 33, 312–320. [Google Scholar] [CrossRef]
- Pereira-Dutra, F.S.; Teixeira, L.; Costa, M.F.D.S.; Bozza, P.T. Fat, fight, and beyond: The multiple roles of lipid droplets in infections and inflammation. J. Leukoc. Biol. 2019, 106, 563–580. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Li, H.-Y.; Peng, Z.-G. Targeting lipophagy as a potential therapeutic strategy for nonalcoholic fatty liver disease. Biochem. Pharmacol. 2022, 197, 114933. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Tukiainen, T.; Juuti, A.; Sammalkorpi, H.; Haridas, P.N.; Niemelä, O.; Arola, J.; Orho-Melander, M.; Hakkarainen, A.; Kovanen, P.T.; et al. Hydroxysteroid 17-β dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI Insight 2020, 5, e132158. [Google Scholar] [CrossRef]
- Komatsu, G.; Nonomura, T.; Sasaki, M.; Ishida, Y.; Arai, S.; Miyazaki, T. AIM-deficient mouse fed a high-trans fat, high-cholesterol diet: A new animal model for nonalcoholic fatty liver disease. Exp. Anim. 2019, 68, 147–158. [Google Scholar] [CrossRef]
- Márquez-Quiroga, L.V.; Arellanes-Robledo, J.; Vásquez-Garzón, V.R.; Villa-Treviño, S.; Muriel, P. Models of nonalcoholic steatohepatitis potentiated by chemical inducers leading to hepatocellular carcinoma. Biochem. Pharmacol. 2021, 195, 114845. [Google Scholar] [CrossRef]
- Qu, C.; Zheng, D.; Li, S.; Liu, Y.; Lidofsky, A.; Holmes, J.A.; Chen, J.; He, L.; Wei, L.; Liao, Y.; et al. Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis. Hepatology 2018, 68, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2018, 69, 2241–2257. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Mennone, A.; Boyer, J.L.; Cai, S.-Y. Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells. Hepatology 2010, 53, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.G.; Zhao, Z.Y.; Li, Y.P.; Wang, Y.P.; Hao, L.H.; Fan, B.; Li, Y.H.; Wang, Y.M.; Shan, Y.Q.; Han, Y.X.; et al. Host apolipoprotein B messenger RNA-editing enzyme catalytic polypeptide-like 3G is an innate defensive factor and drug target against hepatitis C virus. Hepatology 2011, 53, 1080–1089. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, M.; Li, Y.; Li, K.; Wu, S.; Guo, T.; Cen, S.; Jiang, J.; Li, Z.; Li, Y. APOBEC3G is a restriction factor of EV71 and mediator of IMB-Z antiviral activity. Antivir. Res. 2019, 165, 23–33. [Google Scholar] [CrossRef]
- Ma, L.-L.; Zhang, P.; Wang, H.-Q.; Li, Y.-F.; Hu, J.; Jiang, J.-D.; Li, Y.-H. heme oxygenase-1 agonist CoPP suppresses influenza virus replication through IRF3-mediated generation of IFN-α/β. Virology 2018, 528, 80–88. [Google Scholar] [CrossRef]
- Yan, H.-Y.; Wang, H.-Q.; Zhong, M.; Wu, S.; Yang, L.; Li, K.; Li, Y.-H. PML Suppresses Influenza Virus Replication by Promoting FBXW7 Expression. Virol. Sin. 2021, 36, 1154–1164. [Google Scholar] [CrossRef]
- Tsai, T.-H.; Chen, E.; Li, L.; Saha, P.; Lee, H.-J.; Huang, L.-S.; Shelness, G.S.; Chan, L.; Chang, B.H.-J. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy 2017, 13, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wen, H.; Fu, J.; Cai, L.; Li, P.; Zhao, C.; Dong, Z.; Ma, J.P.; Wang, X.; Tian, H.; et al. Hepatocyte TNF Receptor–Associated Factor 6 Aggravates Hepatic Inflammation and Fibrosis by Promoting Lysine 6–Linked Polyubiquitination of Apoptosis Signal-Regulating Kinase 1. Hepatology 2019, 71, 93–111. [Google Scholar] [CrossRef]
- Zou, L.L.; Li, J.R.; Li, H.; Tan, J.L.; Wang, M.X.; Liu, N.N.; Gao, R.M.; Yan, H.Y.; Wang, X.K.; Dong, B.; et al. TGF-β isoforms inhibit hepatitis C virus propagation in transforming growth factor beta/SMAD protein signalling pathway dependent and independent manners. J. Cell. Mol. Med. 2021, 25, 3498–3510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, P.X.; Zhao, L.P.; Zhang, X.; Ji, Y.X.; Zhang, X.J.; Fang, C.; Lu, Y.X.; Yang, X.; Gao, M.M.; et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat. Med. 2018, 24, 84–94. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Li, J.; Li, H.; Dong, B.; Jiang, J.; Liu, N.; Tan, J.; Wang, X.; Lei, L.; Li, H.; et al. Down-Regulating the High Level of 17-Beta-Hydroxysteroid Dehydrogenase 13 Plays a Therapeutic Role for Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 5544. https://doi.org/10.3390/ijms23105544
Wang M, Li J, Li H, Dong B, Jiang J, Liu N, Tan J, Wang X, Lei L, Li H, et al. Down-Regulating the High Level of 17-Beta-Hydroxysteroid Dehydrogenase 13 Plays a Therapeutic Role for Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2022; 23(10):5544. https://doi.org/10.3390/ijms23105544
Chicago/Turabian StyleWang, Meixi, Jianrui Li, Hu Li, Biao Dong, Jing Jiang, Nannan Liu, Jiali Tan, Xuekai Wang, Lei Lei, Hongying Li, and et al. 2022. "Down-Regulating the High Level of 17-Beta-Hydroxysteroid Dehydrogenase 13 Plays a Therapeutic Role for Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 23, no. 10: 5544. https://doi.org/10.3390/ijms23105544
APA StyleWang, M., Li, J., Li, H., Dong, B., Jiang, J., Liu, N., Tan, J., Wang, X., Lei, L., Li, H., Sun, H., Tang, M., Wang, H., Yan, H., Li, Y., Jiang, J., & Peng, Z. (2022). Down-Regulating the High Level of 17-Beta-Hydroxysteroid Dehydrogenase 13 Plays a Therapeutic Role for Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 23(10), 5544. https://doi.org/10.3390/ijms23105544