
Therapeutic Options in Neuro-Oncology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
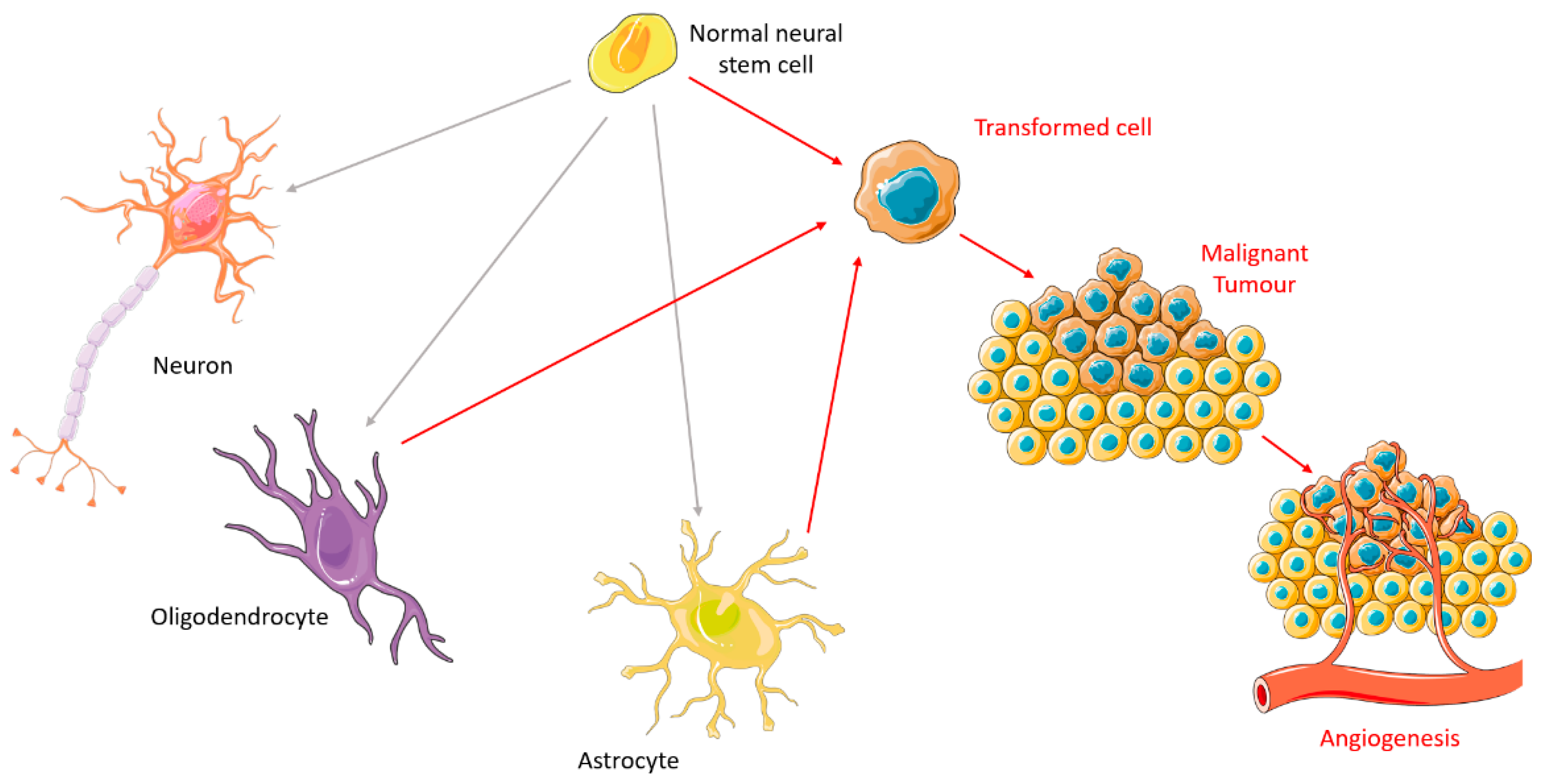
2. Carcinogenesis
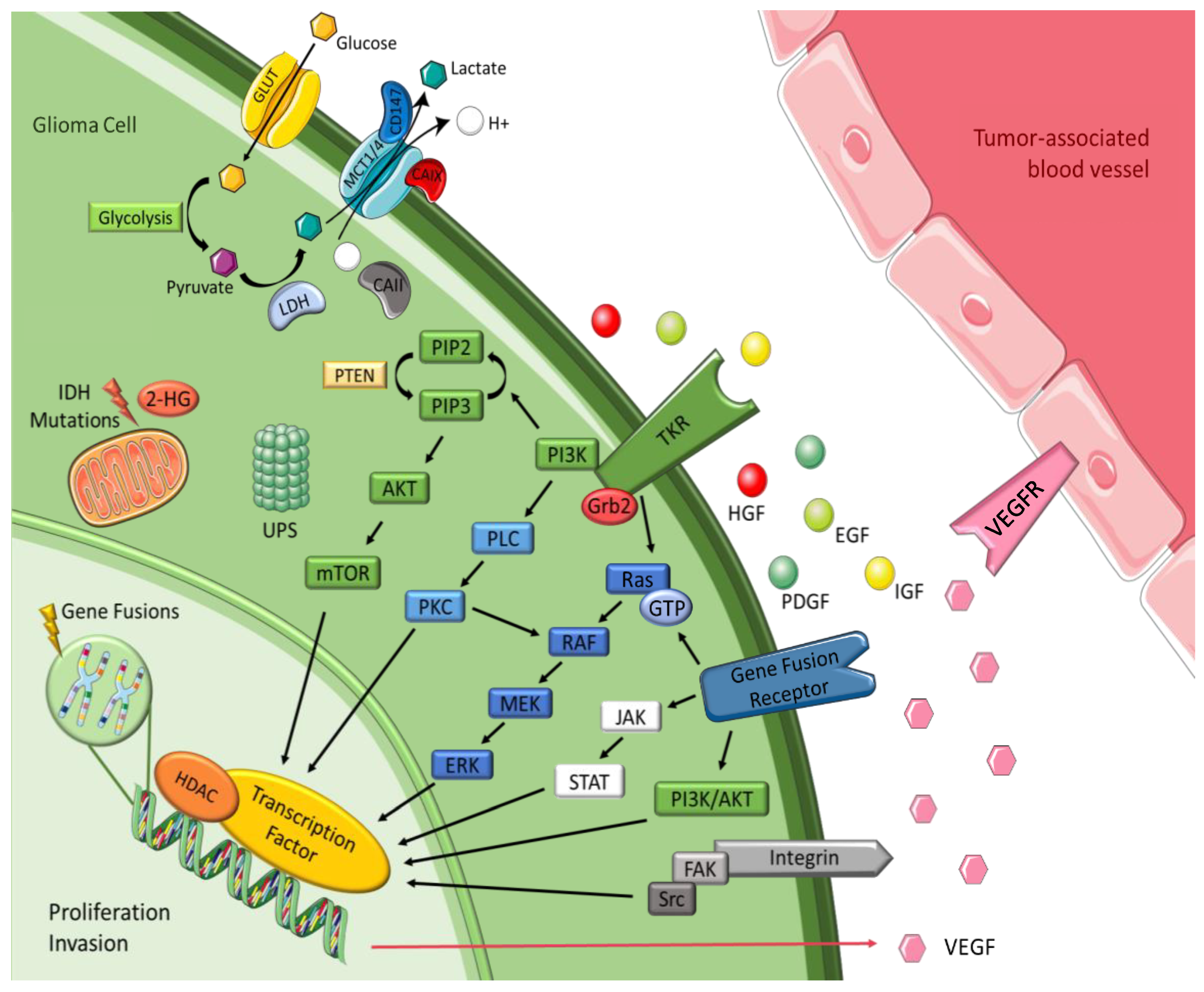
3. Tumor Cell Signaling Pathways
3.1. Tyrosine Kinase Receptor Pathways
3.2. Intracellular Downstream Pathways
3.3. Histones
3.4. Integrins
3.5. Ubiquitin-Proteasome System
3.6. Gene Fusions
3.7. Isocitrate Dehydrogenase
3.8. Transmembrane Monocarboxylate Transporters
4. Therapeutic Options
4.1. General Chemotherapeutic Options
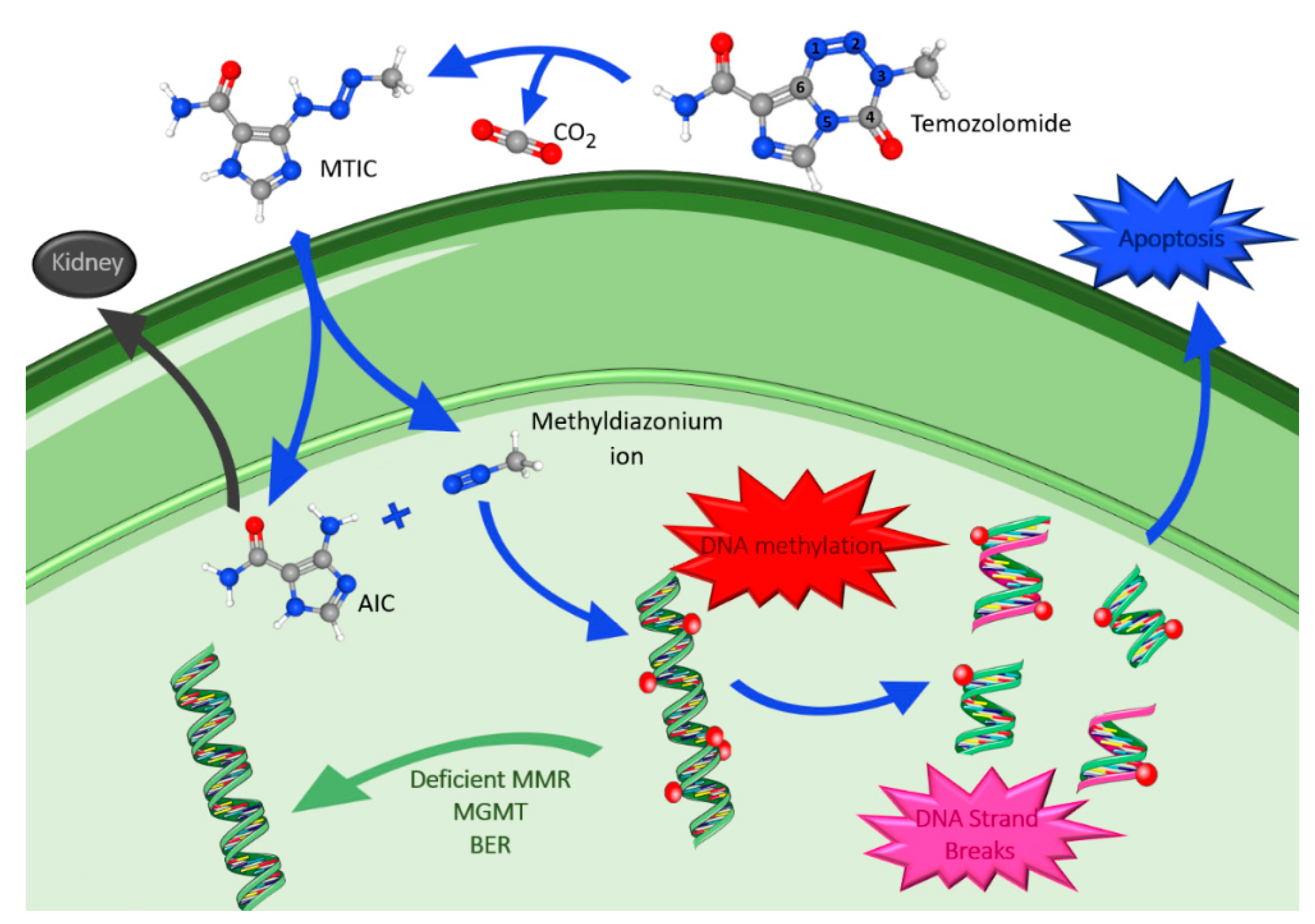
4.1.1. Temozolomide
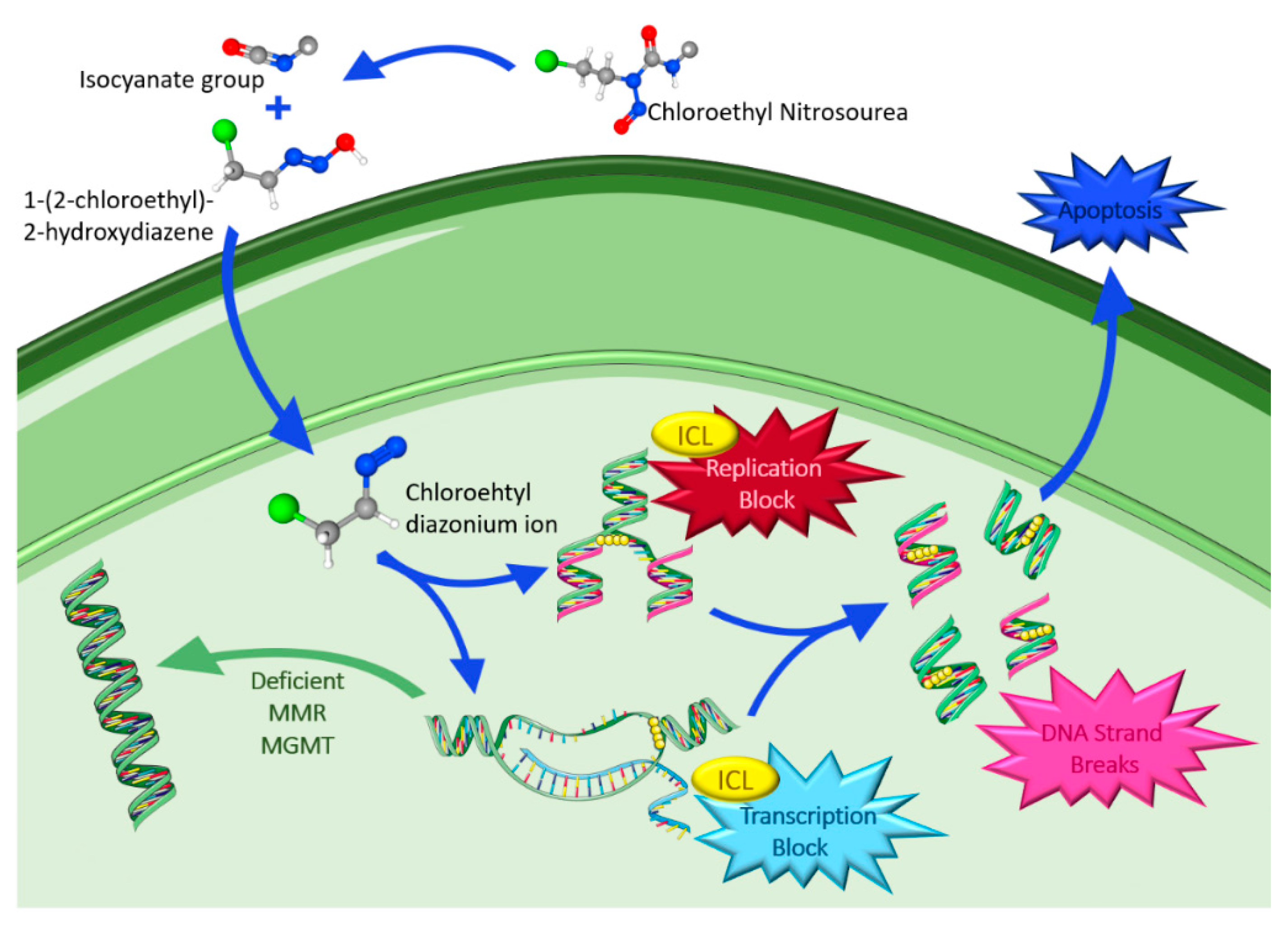
4.1.2. Chloroethyl Nitrosoureas
4.1.3. Polychemotherapy
4.2. Targeted Therapies
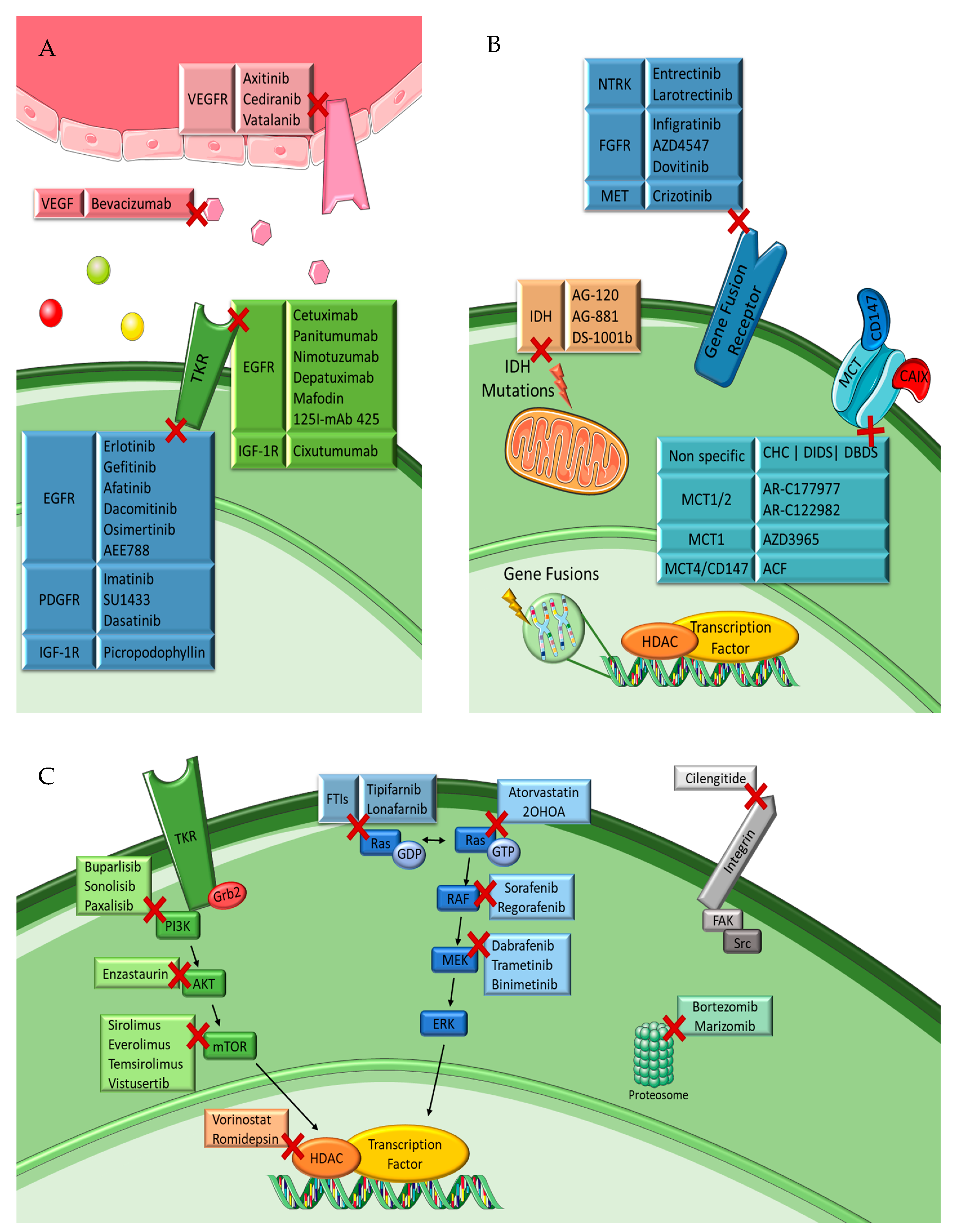
4.2.1. Tyrosine Kinase Receptor Inhibitors
EGFR Inhibitors
VEGFR Inhibitors
VEGF Trap
PDGFR Inhibitors
IGF-1R Inhibitors
4.2.2. Intracellular Downstream Pathway Inhibitors
Ras/RAF/MEK/ERK Pathway Inhibitors
PIP3/AKT/mTOR Pathway Inhibitors
4.2.3. Proteasome Inhibitors
4.2.4. Histone Deacetylase Inhibitors
4.2.5. Integrin Inhibitor
4.2.6. Gene Fusions Inhibitors
4.2.7. Mutant IDH Enzymes Inhibitors
4.2.8. Transmembrane Monocarboxylate Transporters Inhibitors
4.3. Immune Therapies
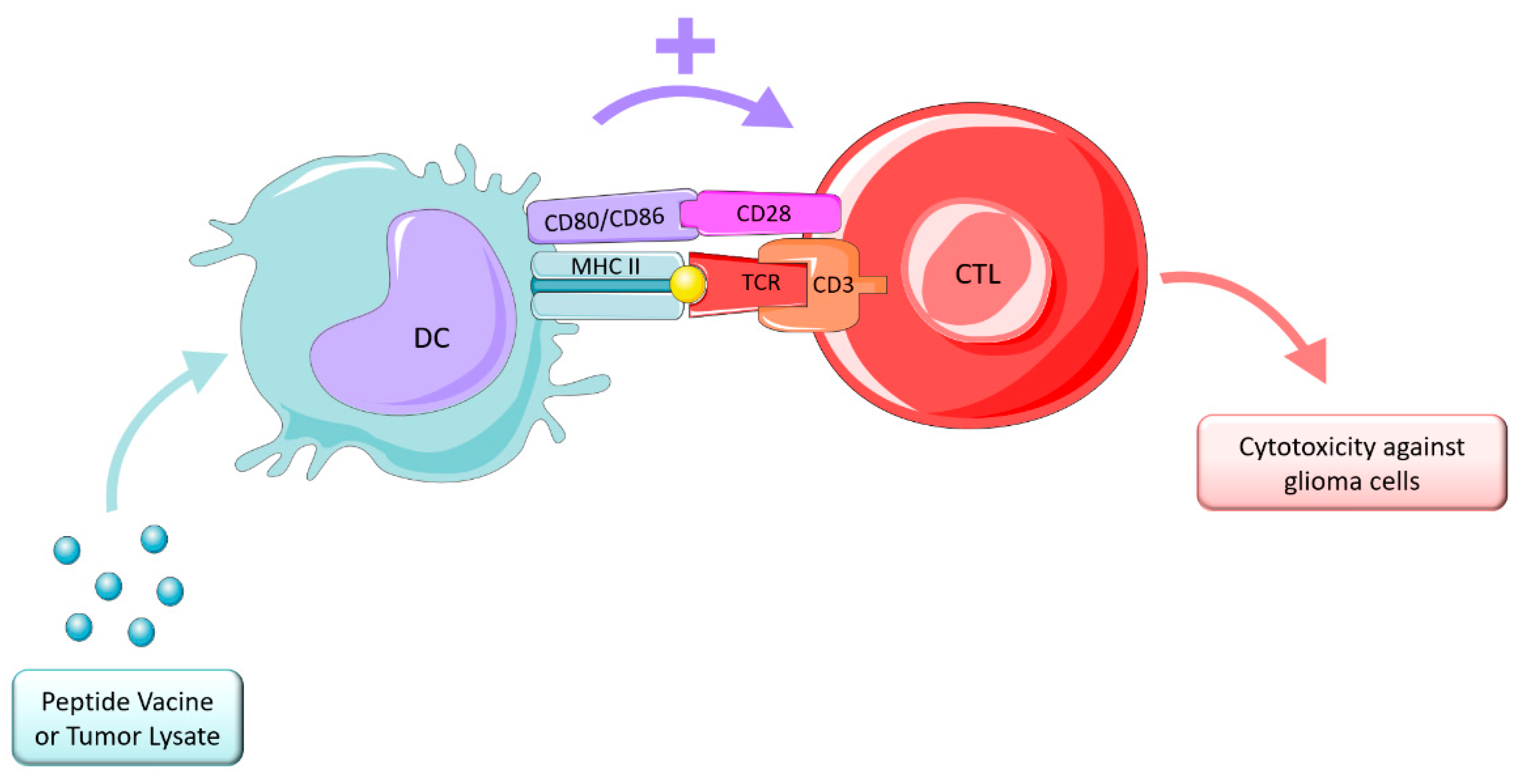
4.3.1. Vaccine Therapy
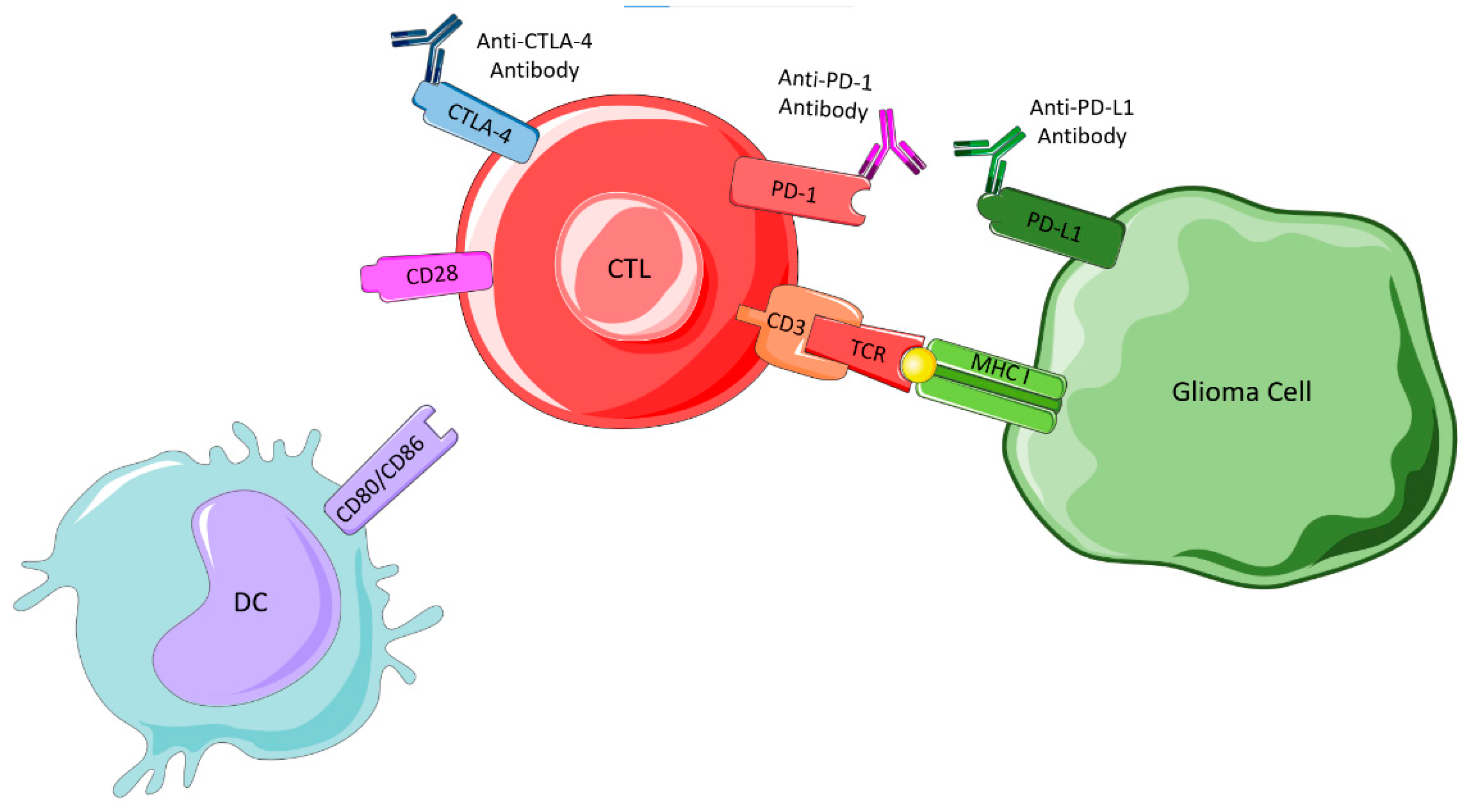
4.3.2. Immune Checkpoint Blockade
CTLA-4 Inhibitors
PD-1/PD-L1 Interaction Inhibitors
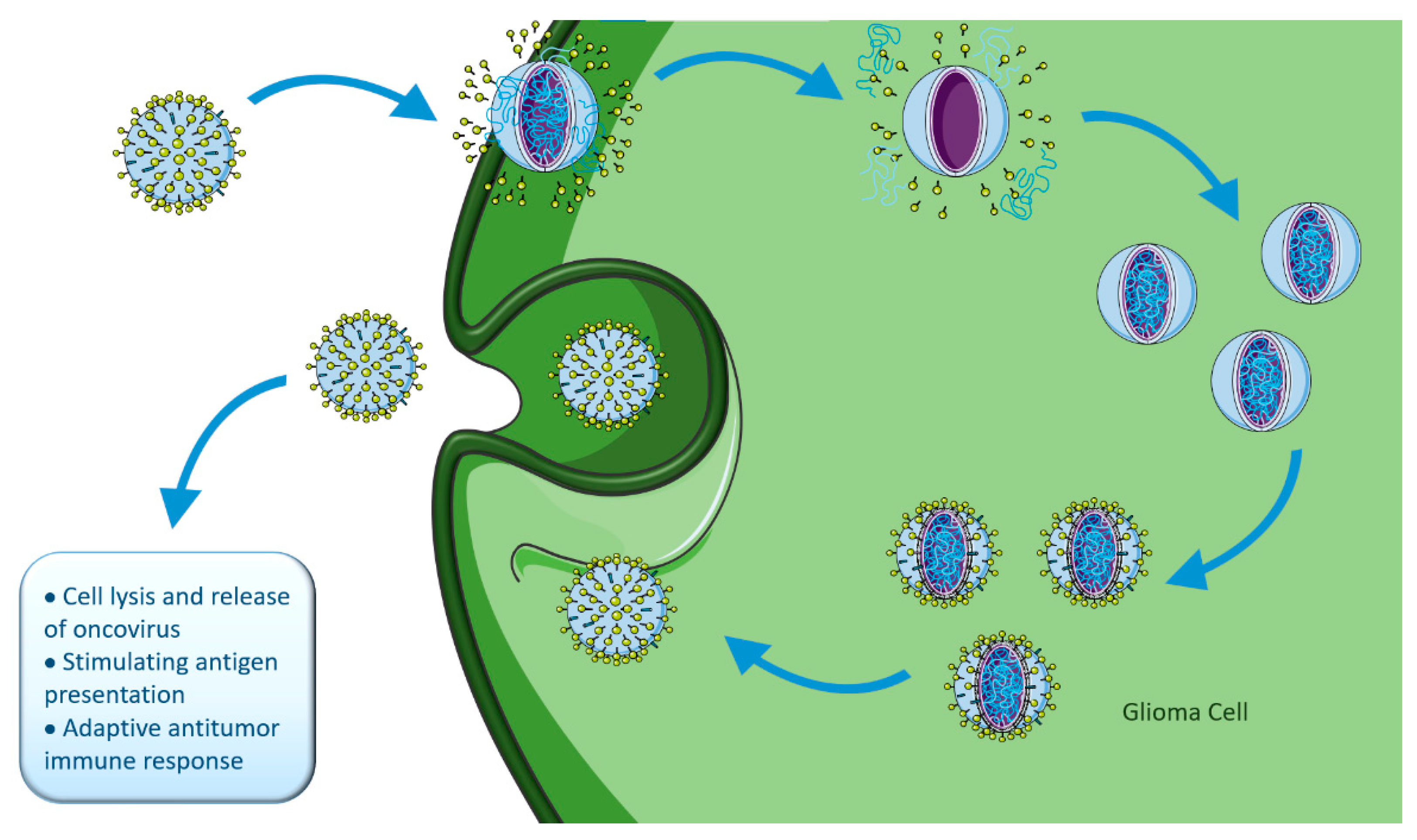
4.3.3. Oncolytic Viral Therapy
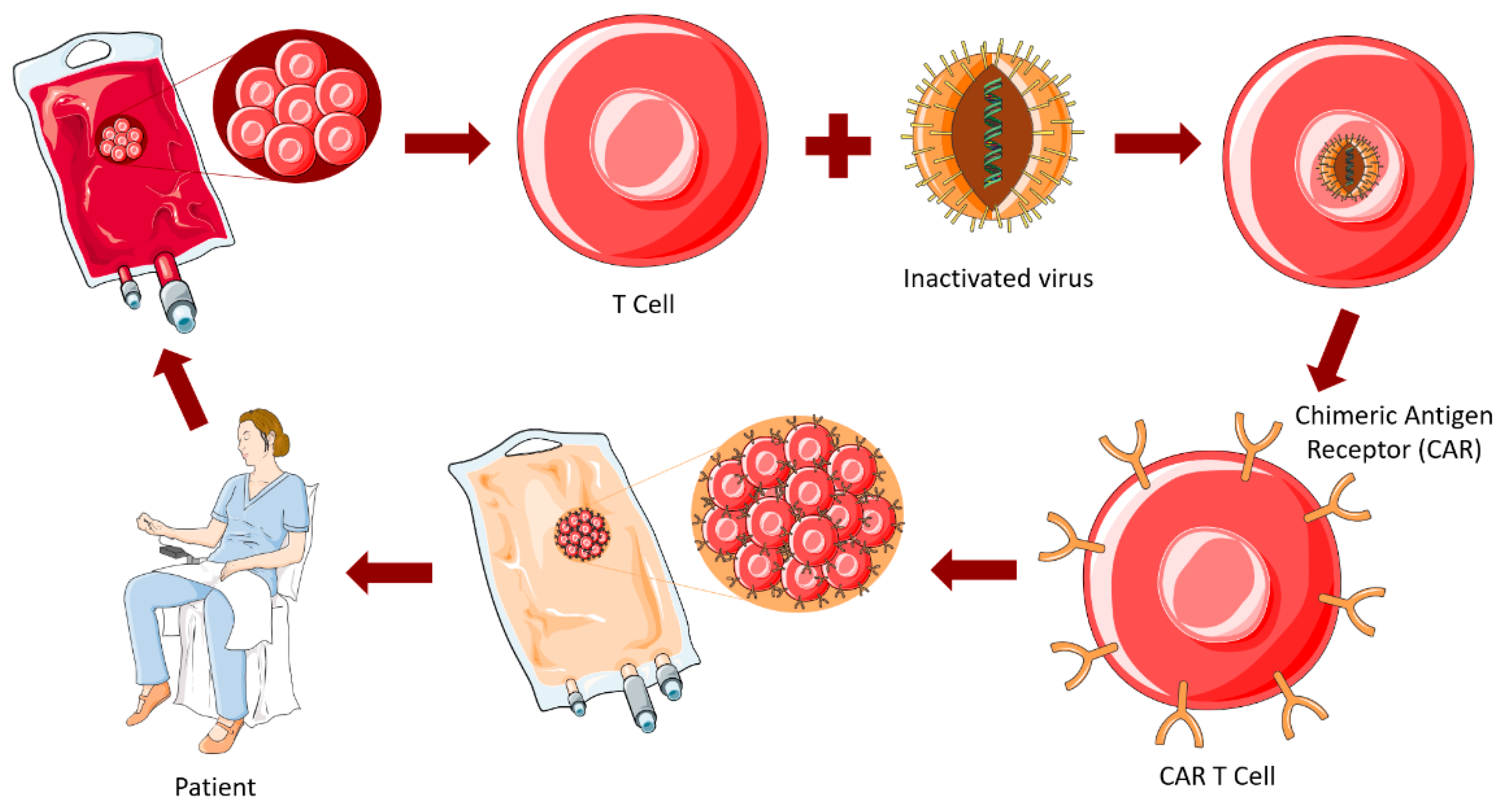
4.3.4. CAR-T Cell Therapy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chamberlain, M.C. International Neurology-Neuro-Oncology Overview, 2nd ed.; Lisak, R.P., Truong, D., Carroll, W.M., Bhidayasiri, R., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Weller, M. Next generation neuro-oncology. Eur. J. Cancer 2018, 96, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brem, S.S.; Bierman, P.J.; Black, P.; Blumenthal, D.T.; Brem, H.; Chamberlain, M.C.; Chiocca, E.A.; DeAngelis, L.M.; Fenstermaker, R.A.; Fine, H.A.; et al. Central nervous system cancers clinical practice guidelines in oncology. JNCCN J. Natl. Compr. Cancer Netw. 2013, 3, 1114–1151. [Google Scholar]
- Acoustic Neuroma. Available online: https://www.mayoclinic.org/diseases-conditions/acoustic-neuroma/symptoms-causes/syc-20356127 (accessed on 28 February 2021).
- Bauchet, L.; Ostrom, Q.T. Epidemiology and Molecular Epidemiology. Neurosurg. Clin. N. Am. 2019, 30, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of Gliomas. Neuropathol. Appl. Neurobiol. 2017, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Janjua, T.I.; Rewatkar, P.; Ahmed-cox, A.; Saeed, I.; Mansfeld, F.M.; Kulshreshtha, R.; Kumeria, T.; Ziegler, D.S.; Kavallaris, M.; Mazzieri, R.; et al. Frontiers in the treatment of glioblastoma: Past, present and emerging. Adv. Drug Deliv. Rev. 2021, 171, 108–138. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Komori, T. The 2016 WHO classification of tumours of the central nervous system: The major points of revision. Neurol. Med.-Chir. 2017, 57, 301–311. [Google Scholar] [CrossRef]
- Wen, P.Y.; Packe, R.J. The 2021 WHO Classification of Tumors of the Central Nervous System: Clinical implications. Neuro-Oncol. 2021, 23, 1215–1217. [Google Scholar] [CrossRef]
- Nabors, B.; Laterra, J. Neuro-Oncology: Current Concepts and Emerging Therapeutics. Neurotherapeutics 2017, 14, 253–255. [Google Scholar] [CrossRef]
- Villa, C.; Miquel, C.; Mosses, D.; Bernier, M.; Luisa, A.; Stefano, D.; Bernier, M.; Stefano, A. Di Quarterly Medical Review The 2016 World Health Organization classification of tumours of the central nervous system. La Presse Méd. 2018, 47, e187–e200. [Google Scholar] [CrossRef]
- Wen, P.Y.; Huse, J.T. 2016 World Health Organization Classification of Central Nervous System Tumors. CONTINUUM Lifelong Learn. Neurol. 2017, 23, 1531–1547. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, A.V. Carcinogenesis: Evolution of Concepts. Biochemistry 2009, 74, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Cattley, R.; Radinsky, R. Cancer Therapeutics: Understanding the Mechanism of Action. Toxicol. Pathol. 2004, 32, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Gutmann, D.H. Cancer stem cells and brain tumors: Uprooting the bad seeds. Expert Rev. Anticancer Ther. 2007, 7, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, T.; Magnusson, M.K. Stem cell biology and the cellular pathways of carcinogenesis. Apmis 2005, 113, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef]
- Kim, W.; Lee, H. Brain angiogenesis: Mechanism and Therapeutic Intervention in Brain Tumors. FEBS J. 2009, 276, 4653–4664. [Google Scholar] [CrossRef]
- Thakur, C. Nanotechnology-Based Targeted Drug Delivery Systems for Brain Tumors: Angiogenesis in Brain Tumors; Academic Press: Cambridge, MA, USA, 2018; ISBN 9780128122181. [Google Scholar]
- Clark, P.A.; Treisman, D.M.; Ebben, J.; Kuo, J.S. Developmental Signaling Pathways in Brain Tumor-Derived Stem-Like Cells. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 3297–3308. [Google Scholar] [CrossRef]
- Newton, H.B. Molecular neuro-oncology and development of targeted therapeutic strategies for brain tumors. Part 1: Growth factor and Ras signaling pathways. Expert Rev. Anticancer. Ther. 2003, 3, 595–614. [Google Scholar] [CrossRef]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J. Aberrant Signaling Pathways in Glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [PubMed]
- Sathornsumetee, S.; Desjardins, A.; Quinn, J.A.; Vredenburgh, J.J.; Rich, J.N. Molecularly Targeted Therapy for Malignant Glioma. Cancer 2007, 110, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Shahpar, S.; Mhatre, P.V.; Huang, M.E. Update on Brain Tumors: New Developments in Neuro-oncologic Diagnosis and Treatment, and Impact on Rehabilitation Strategies. PMR 2016, 8, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Sathornsumetee, S.; Rich, J.N. Molecularly Targeted Therapy in Neuro-Oncology. In Handbook of Clinical Neurology, 1st ed.; Aminoff, M.J., Boller, F., Swaab, D.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 104, pp. 255–278. ISBN 1216636079. [Google Scholar]
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 22, 1831. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.A.; Groot, J.F. De Cell Signaling Pathways in Brain Tumors. Top. Magn. Reson. Imaging 2017, 26, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Krol, S.K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin. Epigenet. 2019, 11, 11. [Google Scholar] [CrossRef]
- Chessum, N.; Jones, K.; Pasqua, E.; Tucker, M. Recent Advances in Cancer Therapeutics. In Progress in Medicinal Chemistry, 1st ed.; Lawton, G., Witty, D.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 54, pp. 1–63. [Google Scholar]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Ellert-miklaszewska, A.; Poleszak, K.; Pasierbinska, M. Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 888. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Voutsadakis, I.A.; Papandreou, C.N. The ubiquitin-proteasome system in glioma cell cycle control. Cell Div. 2012, 7, 18. [Google Scholar] [CrossRef]
- Young Woo, H.; Na, K.; Yoo, J.; Chang, J.H.; Park, Y.N.; Hyo, S.; Kim, S.H. Glioblastomas harboring gene fusions detected by next-generation sequencing. Brain Tumor Pathol. 2020, 37, 136–144. [Google Scholar]
- Ferguson, S.D.; Zhou, S.; Huse, J.T.; de Groot, J.F.; Xiu, J.; Subramaniam, D.S.; Mehta, S.; Gatalica, Z.; Swensen, J.; Sanai, N.; et al. Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J. Neuropathol. Exp. Neurol. 2018, 77, 437–442. [Google Scholar] [CrossRef] [PubMed]
- You, G.; Fan, X.; Hu, H.; Jiang, T.; Chen, C.C. Fusion Genes Altered in Adult Malignant Gliomas. Front. Neurol. 2021, 12, 715206. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y. Management of Glioblastoma: State of the Art and Future Directions. CA A Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Karpel-massler, G.; Nguyen, T.; Shang, E.; Siegelin, M.D.; Biology, C. Novel IDH1 Targeted Glioma Therapies. CNS Drugs. 2020, 33, 1155–1166. [Google Scholar] [CrossRef]
- Chen, R.; Cohen, A.L.; Colman, H. Targeted Therapeutics in Patients With High-Grade Gliomas: Past, Present, and Future. Curr. Treat. Options Oncol. 2016, 17, 42. [Google Scholar] [CrossRef]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Miranda-Goncalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro-Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef]
- Pérez-escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Smith, C.P.; Wilbur, R.R.; Cain, C.P.; Kallu, S.R.; Valasapalli, S.; Sahoo, A.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. An overview of MCT1 and MCT4 in GBM: Small molecule transporters with large implications. Am. J. Cancer Res. 2018, 8, 1967–1976. [Google Scholar] [PubMed]
- Khalyfa, A.; Qiao, Z.; Raju, M.; Shyu, C.R.; Coghill, L.; Ericsson, A.; Gozal, D. Monocarboxylate transporter-2 expression restricts tumor growth in a murine model of lung cancer: A multi-omic analysis. Int. J. Mol. Sci. 2021, 22, 10616. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.M.; Deitmer, J.W. Proton Transport in Cancer Cells: The Role of Carbonic Anhydrases. Int. J. Mol. Sci. 2021, 22, 3171. [Google Scholar] [CrossRef]
- National Cancer Institute Cancer Drug Resistance: Unraveling Its Complexity. Available online: https://www.cancer.gov/research/annual-plan/scientific-topics/drug-resistance (accessed on 1 June 2021).
- El Demerdash, N.; Kedda, J.; Ram, N.; Brem, H.; Tyler, B. Novel therapeutics for brain tumors: Current practice and future prospects. Expert Opin. Drug Deliv. 2020, 17, 9–21. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Haider, T.; Pandey, V.; Banjare, N.; Gupta, P.N.; Soni, V. Drug resistance in cancer: Mechanisms and tackling strategies. Pharmacol. Rep. 2020, 72, 1125–1151. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Sá-pereira, I.; Brites, D.; Brito, M.A. Neurovascular Unit: A Focus on Pericytes. Mol. Neurobiol. 2012, 45, 327–347. [Google Scholar] [CrossRef]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood–brain barrier: Molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef] [PubMed]
- Etame, A.B.; Diaz, R.J.D.; Smith, C.A.; Mainprize, T.; Kullervo, H.; Rutka, J.T. Focused ultrasound disruption of the blood brain barrier: A new frontier for therapeutic delivery in molecular neuro-oncology. Neurosurg. Focus 2014, 32, E3. [Google Scholar] [CrossRef] [PubMed]
- Karmur, B.S.; Philteos, J.; Abbasian, A.; Zacharia, B.E.; Lipsman, N. Blood-Brain Barrier Disruption in Neuro-Oncology: Strategies, Failures, and Challenges to Overcome. Front. Oncol. 2020, 10, 563840. [Google Scholar] [CrossRef] [PubMed]
- Jouyban, A.; Soltani, S. Blood Brain Barrier Permeation. In Toxicity and Drug Testing; Acree, W., Ed.; IntechOpen: Rijeka, Croatia, 2012; pp. 1–24. ISBN 978-953-51-0004-1. [Google Scholar] [CrossRef]
- Weller, M.; Bent, M.V.D.; Preusser, M.; Dirven, L.; Smits, M.; Westphal, M.; Taphoorn, M.J.B.; Soffietti, R. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Karisa, C.; Schreck, S.A.G. Role of Temozolomide in the Treatment of Cancers Involving the Central Nervous System. Available online: https://www.cancernetwork.com/view/role-temozolomide-cns (accessed on 14 March 2021).
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Vargo, M.M. Brain Tumors and Metastases. Phys. Med. Rehabil. Clin. 2017, 28, 115–141. [Google Scholar] [CrossRef]
- Barciszewska, A.M.; Gurda, D.; Głodowicz, P.; Nowak, S.; Naskręt-Barciszewska, M.Z. A new epigenetic mechanism of temozolomide action in glioma cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef]
- RCM-Temodal. Available online: https://www.ema.europa.eu/en/documents/product-information/temodal-epar-product-information_pt.pdf (accessed on 4 June 2021).
- Lopes, I.C.; De Oliveira, S.C.B.; Oliveira-brett, A.M. Temozolomide chemical degradation to 5-aminoimidazole-4-carboxamide–Electrochemical study. J. Electroanal. Chem. 2013, 704, 183–189. [Google Scholar] [CrossRef]
- 3D Conformer Image of CID 5394 (Temozolomide). Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Temozolomide (accessed on 14 March 2021).
- 3D Conformer Image of CID 9679 [5-Aminoimidazole-4-carboxamide]. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/9679 (accessed on 14 March 2021).
- 3D Conformer Image of CID 76953 [5-(3-Methyl-1-triazeno)imidazole-4-carboxamide]. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5-_3-Methyl-1-triazeno_imidazole-4-carboxamide (accessed on 14 March 2021).
- 3D Conformer Image of CID 115287 (Methanediazonium). Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Methanediazonium (accessed on 4 June 2021).
- 3D Conformer Image of CID 280 [Carbon dioxide]. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/280 (accessed on 10 February 2022).
- McFaline-Figueroa, J.R.; Lee, E.Q. Brain Tumors. Am. J. Med. 2018, 131, 874–882. [Google Scholar] [CrossRef]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M.; Tumor, B.; Program, I.; Metastasis, S. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2021, 20, 12–25. [Google Scholar] [CrossRef]
- Avendaño, C.; Menendez, J.C. Medicinal Chemistry of Anticancer Drugs; Elsevier: Oxford, UK, 2015. [Google Scholar]
- Antonini, I.; Lin, T.S.; Cosby, L.A.; Dai, Y.R.; Sartorelli, A.C. Journal of Medicinal Chemistry: DNA-Interactive Agents; ACS Publications: Columbus, OH, USA, 2014. [Google Scholar]
- Nikolova, T.; Hennekes, F.; Bhatti, A.; Kaina, B. Chloroethylnitrosourea-induced cell death and genotoxicity. Cell Cycle 2012, 11, 2606–2619. [Google Scholar] [CrossRef] [PubMed]
- 3D Conformer Image of CID 75394 [1-(2-Chloroethyl)-1-nitrosourea]. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/75394 (accessed on 10 June 2021).
- 3D Conformer Image of CID 23662428 [cis-(2-Chloroethyl)hydroxydiazene potassium salt]. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/23662428 (accessed on 10 June 2021).
- 3D Conformer Image of CID 637659 [Isocyanatocyclopropane]. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/637659 (accessed on 10 June 2021).
- Brandes, A.A.; Bartolotti, M.; Tosoni, A.; Franceschi, E. Nitrosoureas in the Management of Malignant Gliomas. Curr. Neurol. Neurosci. Rep. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, T.; Roos, W.P.; Kr, O.H.; Strik, H.M. Chloroethylating nitrosoureas in cancer therapy: DNA damage, repair and cell death signaling. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2017, 1868, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Bethesda, M.; PDQ® Adult Treatment Editorial Board. PDQ Adult Central Nervous System Tumors Treatment. Available online: https://www.cancer.gov/types/brain/hp/adult-brain-treatment-pdq (accessed on 10 June 2021).
- Vasan, N.; Hyman, D.M.; Kettering, S. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Solimando, D.; Waddel, A. Procarbazine, Lomustine, and Vincristine (PCV) Regimen for Central Nervous System Tumors. Hosp. Pharm. 2017, 52, 98–104. [Google Scholar] [CrossRef]
- Lassman, A.B. Procarbazine, lomustine and vincristine or temozolomide: Which is the better regimen? CNS Oncol. 2015, 4, 341–346. [Google Scholar] [CrossRef]
- Procarbazine. CCO Formul. 2019, pp. 1–11. Available online: https://www.cancercareontario.ca/en/drugformulary/drugs/monograph/44016 (accessed on 26 February 2022).
- Vardanyan, R.S.; Hruby, V.J. Synthesis of Essential Drugs; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Avendaño, C.; Menéndez, C. Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Chua, J.; Nafziger, E.; Leung, D. Evidence-Based Practice: Temozolomide Beyond Glioblastoma. Curr. Oncol. Rep. 2019, 21, 30. [Google Scholar] [CrossRef]
- Schiff, D.; Van Den Bent, M.; Vogelbaum, M.A.; Wick, W.; Miller, C.R.; Taphoorn, M.; Pope, W.; Brown, P.D.; Platten, M.; Jalali, R.; et al. Recent developments and future directions in adult lower-grade gliomas: Society for Neuro-Oncology (SNO) and European Association of Neuro-Oncology (EANO) consensus. Neuro-Oncol. 2019, 21, 838–854. [Google Scholar] [CrossRef]
- Wick, W.; Clinic, N.; Cooperation, C.; Neurooncology, U.; Consortium, C.; Winkler, F.; Clinic, N. Regimen of Procarbazine, Lomustine, and Vincristine Versus Temozolomide for Gliomas. Cancer 2018, 124, 2674–2677. [Google Scholar] [CrossRef]
- Hospital, T.; Hospital, P.W. Radiotherapy Plus Procarbazine, Lomustine, and Vincristine Versus Radiotherapy Plus Temozolomide for IDH-Mutant Anaplastic Astrocytoma: A Retrospective Multicenter Analysis of the French POLA Cohort. Oncologist 2021, 26, 838–846. [Google Scholar]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Vogelbaum, M.A. Targeted Therapies for Brain Tumors: Will They Ever Deliver ? Clin. Cancer Res. 2018, 24, 3790–3792. [Google Scholar] [CrossRef] [PubMed]
- Bolcaen, J.; Nair, S.; Driver, C.H.S.; Boshomane, T.M.G.; Ebenhan, T.; Vandevoorde, C. Novel Receptor Tyrosine Kinase Pathway Inhibitors for Targeted Radionuclide Therapy of Glioblastoma. Pharmaceuticals 2021, 14, 626. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Lamborn, K.R.; Chang, S.; Burton, E.; Malec, M.; Kapadia, A.; Rabbitt, J.; Page, M.S.; Fedoroff, A.; Xie, D.; et al. Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma 1. Neuro-Oncol. 2006, 8, 67–78. [Google Scholar] [CrossRef]
- Van Den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.M.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.; et al. Randomized Phase II Trial of Erlotinib Versus Temozolomide or Carmustine in Recurrent Glioblastoma: EORTC Brain Tumor Group Study 26034. J. Neurooncol. 2021, 27, 1268–1274. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Shepard, D.R.; Ahluwalia, M.S.; Barnett, G.H. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J. Neurooncol. 2010, 98, 93–99. [Google Scholar] [CrossRef]
- Uhm, J.; Ballman, K.; Wu, W.; Giannini, C.; Krauss, J.; Buckner, J.; James, C.D.; Scheithauer, B.; Behrens, R.; Flynn, P.; et al. Phase II evaluation of gefitinib in patients with newly diagnosed grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group study N0074. Int. J. Radiat. Oncol. Biol. Phys. 2018, 80, 347–353. [Google Scholar] [CrossRef]
- Reardon, D.A.; Vredenburgh, J.J.; Ii, J.E.H.; Friedman, H.S. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neurooncol. 2010, 96, 219–230. [Google Scholar] [CrossRef]
- Vivanco, I.; Robins, H.I.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Tao, H.; et al. Differential Sensitivity of Glioma-versus Lung Cancer-specific EGFR mutations to EGFR Kinase Inhibitors. Cancer Discov. 2013, 2, 458–471. [Google Scholar] [CrossRef]
- Vengoji, R.; Macha, M.A.; Nimmakayala, R.K.; Rachagani, S.; Siddiqui, J.A.; Mallya, K.; Gorantla, S.; Jain, M.; Ponnusamy, M.P.; Batra, S.K.; et al. Afatinib and Temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 266. [Google Scholar] [CrossRef]
- Wagle, N.; Juarez, T.; Carrillo, J.; Heng, A.; Gill, J.; Nguyen, M.; Truong, J.; Boucher, N.; Archer, A.; Nguyen, H.; et al. CTNI-46. Phase I Open-Label, Single-Center, Dose Escalation Study (Nct02423525) Of Pulsatile Afatinib In Patients With Brain Cancer. Neuro-Oncol. 2021, 23, vi70. [Google Scholar] [CrossRef]
- Chi, A.S.; Cahill, D.P.; Reardon, D.A.; Wen, P.Y.; Mikkelsen, T. Exploring Predictors of Response to Dacomitinib in EGFR-Amplified Recurrent Glioblastoma. JCO Precis. Oncol. 2020, 4, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Laramy, J.K.; Mohammad, A.S.; Talele, S.; Fisher, J.; Sarkaria, J.N.; Elmquist, W.F. Brain distribution of a panel of epidermal growth factor receptor inhibitors using cassette dosing in wild-type and ABCB1/ABCG2-deficient mice. Drug Metab. Dispos. 2019, 47, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Colclough, N.; Chen, K.; Johnstrom, P.; Strittmatter, N.; Yan, Y.; Wrigley, G.L.; Schou, M.; Goodwin, R.; Varnas, K.; Adua, S.J.; et al. Preclinical comparison of the blood-brain barrier permeability of osimertinib with other EGFR TKIs. Clin. Cancer Res. 2021, 27, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 219. [Google Scholar] [CrossRef]
- Chagoya, G.; Kwatra, S.G.; Nanni, C.W.; Roberts, C.M.; Phillips, S.M.; Nullmeyergh, S.; Gilmore, S.P.; Spasojevic, I.; Corcoran, D.L.; Young, C.C.; et al. Efficacy of osimertinib against EGFRvIII+glioblastoma. Oncotarget 2020, 11, 2074–2082. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Q.; Zhang, G.; Dong, X.; Yang, C.-T.; Song, Y.; Chang, G.-C.; Lu, Y.; Pan, H.; Chiu, C.-H.; et al. Abstract CT190: A Multicenter, Open-Label, Single-Arm, Phase II Study: The Third Generation Egfr Tyrosine Kinase Inhibitor Almonertinib for Pretreated Egfr t790m-Positive Locally Advanced or Metastatic Non-Small Cell Lung Cancer (APOLLO). Available online: https://aacrjournals.org/cancerres/article/80/16_Supplement/CT190/645131/Abstract-CT190-A-multicenter-open-label-single-arm (accessed on 17 April 2022).
- Papini, F.; Sundaresan, J.; Leonetti, A.; Tiseo, M.; Rolfo, C.; Peters, G.J.; Giovannetti, E. Hype or hope–Can combination therapies with third-generation EGFR-TKIs help overcome acquired resistance and improve outcomes in EGFR-mutant advanced/metastatic NSCLC ? Crit. Rev. Oncol. Hematol. 2021, 166, 103454. [Google Scholar] [CrossRef]
- Black Diamond Therapeutics IND for BDTX-1535 Cleared by FDA. Available online: https://www.precisiononcologynews.com/cancer/black-diamond-therapeutics-ind-bdtx-1535-cleared-fda#.YlxHYovMJPY (accessed on 15 April 2022).
- Zhong, W.; Zhang, J.; Mu, A.; Sun, C. Abstract 1326: WSD0922: A BBB penetrable EGFR/EGFRVIII inhibitor for the treatment of GBM and metastatic CNS tumor. Cancer Res. 2019, 79, 1326. [Google Scholar] [CrossRef]
- Combs, S.E.; Heeger, S.; Haselmann, R.; Edler, L.; Debus, J.; Schulz-Ertner, D. Treatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (GERT)-Phase I/II trial: Study protocol. BMC Cancer 2006, 6, 133. [Google Scholar] [CrossRef]
- Neyns, B.; Sadones, J.; Joosens, E.; Bouttens, F.; Verbeke, L.; Baurain, J.F.; D’Hondt, L.; Strauven, T.; Chaskis, C.; In’t Veld, P.; et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 2009, 20, 1596–1603. [Google Scholar] [CrossRef]
- Hasselbalch, B.; Lassen, U.; Hansen, S.; Holmberg, M.; Soørensen, M.; Kosteljanetz, M.; Broholm, H.; Stockhausen, M.T.; Poulsen, H.S. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: A phase II trial. Neuro-Oncol. 2010, 12, 508–516. [Google Scholar] [PubMed]
- Solomon, M.T.; Miranda, N.; Jorrín, E.; Chon, I.; Marinello, J.J.; Alert, J.; Lorenzo-Luaces, P.; Crombet, T. Nimotuzumab in combination with radiotherapy in high grade glioma patients: A single institution experience. Cancer Biol. Ther. 2014, 15, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Heese, O.; Steinbach, J.P.; Schnell, O.; Schackert, G.; Mehdorn, M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur. J. Cancer 2015, 51, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, L.; Sheng, X.F.; Chen, S.; Dai, J.Z. Nimotuzumab, a humanized monoclonal antibody specific for the EGFR, in combination with temozolomide and radiation therapy for newly diagnosed glioblastoma multiforme: First results in Chinese patients. Asia. Pac. J. Clin. Oncol. 2016, 12, e23–e29. [Google Scholar] [CrossRef] [PubMed]
- Du, X.J.; Li, X.M.; Cai, L.B.; Sun, J.C.; Wang, S.Y.; Wang, X.C.; Pang, X.L.; Deng, M.L.; Chen, F.F.; Wang, Z.Q.; et al. Efficacy and safety of nimotuzumab in addition to radiotherapy and temozolomide for cerebral glioblastoma: A phase II multicenter clinical trial. J. Cancer 2019, 10, 3214–3223. [Google Scholar] [CrossRef]
- Yeini, E.; Ofek, P.; Albeck, N.; Rodriguez Ajamil, D.; Neufeld, L.; Eldar-Boock, A.; Kleiner, R.; Vaskovich, D.; Koshrovski-Michael, S.; Dangoor, S.I.; et al. Targeting Glioblastoma: Advances in Drug Delivery and Novel Therapeutic Approaches. Adv. Ther. 2021, 4, 2000124. [Google Scholar] [CrossRef]
- Van Den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.S.; Franceschi, E.; Clement, P.M.; Chinot, O.; De Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncol. 2020, 22, 684–693. [Google Scholar] [CrossRef]
- Li, L.; Quang, T.S.; Gracely, E.J.; Kim, J.H.; Emrich, J.G.; Yaeger, T.E.; Jenrette, J.M.; Cohen, S.C.; Black, P.; Brady, L.W. A phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J. Neurosurg. 2010, 113, 192–198. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Kozlosky, C.J.; Siu, S.; Chang, W.S.; Liu, H.; Foltz, I.N.; Trueblood, E.S.; Meininger, D.; Arora, T.; Twomey, B.; et al. AMG 595, an anti-EGFRvIII antibody-drug conjugate, induces potent antitumor activity against EGFRvIII-expressing glioblastoma. Mol. Cancer Ther. 2015, 14, 1614–1624. [Google Scholar] [CrossRef]
- Rosenthal, M.; Curry, R.; Reardon, D.A.; Rasmussen, E.; Upreti, V.V.; Damore, M.A.; Henary, H.A.; Hill, J.S.; Cloughesy, T. Safety, tolerability, and pharmacokinetics of anti-EGFRvIII antibody–drug conjugate AMG 595 in patients with recurrent malignant glioma expressing EGFRvIII. Cancer Chemother. Pharmacol. 2019, 84, 327–336. [Google Scholar] [CrossRef]
- Oprita, A.; Baloi, S.; Staicu, G.; Alexandru, O. Updated Insights on EGFR Signaling Pathways in Glioma. Int. J. Mol. Sci. 2021, 22, 587. [Google Scholar] [CrossRef] [PubMed]
- Hu-lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Cancer Therapy: Preclinical Nonclinical Antiangiogenesis and Antitumor Activities of Axitinib (AG-013736), an Oral, Potent, and Selective Inhibitor of Vascular Endothelial Growth Factor Receptor Tyrosine Kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7284. [Google Scholar] [CrossRef] [PubMed]
- Du, J.D.S.; Vandervorst, F.F.; Le, N.D.H.M. Randomized phase II study of axitinib versus physicians best alternative choice of therapy in patients with recurrent glioblastoma. J. Neurooncol. 2016, 128, 147–155. [Google Scholar]
- Du, J.D.S.; Bouttens, F.F.; Verschaeve, C.A.V. Randomized phase II trial comparing axitinib with the combination of axitinib and lomustine in patients with recurrent glioblastoma. J. Neurooncol. 2017, 136, 115–125. [Google Scholar] [CrossRef]
- Awada, G.; Salama, L.B.; Cremer, J.D.; Schwarze, J.K.; Vanbinst, M.; Fischbuch, L.; Seynaeve, L.; Four, S.D.; Michotte, A.; Everaert, H.; et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: A stratified. J. Immunother. Cancer 2020, 8, e001146. [Google Scholar] [CrossRef]
- Batchelor, T.; Duda, D.G.; di Tomaso, E.; Ancukiewicz, M.; Plotkin, S.R.; Gerstner, E.; Eichler, A.F.; Drappatz, J.; Hochberg, F.H.; Benner, T.; et al. Phase II Study of Cediranib, an Oral Pan–Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor, in Patients With Recurrent Glioblastoma. Am. Soc. Clin. Oncol. 2010, 28, 2817–2823. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III Randomized Trial Comparing the Efficacy of Cediranib As Monotherapy, and in Combination With Lomustine, Versus Lomustine Alone in Patients With Recurrent Glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef]
- Brown, N.; McBain, C.; Nash, S.; Hopkins, K.; Sanhera, P.; Saran, F.; Phillips, M.; Dungey, F.; Clifton-hadley, L.; Wanek, K.; et al. Multi-Center Randomized Phase II Study Comparing Cediranib plus Gefitinib with Cediranib plus Placebo in Subjects with Recurrent/Progressive Glioblastoma. PLoS ONE 2016, 11, e0156369. [Google Scholar] [CrossRef]
- Gerstner, E.R.; Eichler, A.F. Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide. J. Neurooncol. 2011, 103, 325–332. [Google Scholar] [CrossRef]
- Brandes, A.A.; Stupp, R.; Hau, P.; Lacombe, D.; Gorlia, T.; Mirimanoff, O.; Kros, J.M.; Van Den Bent, M.J.; Tosoni, A. EORTC study 26041-22041: Phase I/II study on concomitant and adjuvant temozolomide (TMZ) and radiotherapy (RT) with PTK787/ZK222584 (PTK/ZK) in newly diagnosed glioblastoma. Eur. J. Cancer 2009, 6, 348–354. [Google Scholar] [CrossRef]
- Huang, Y.; Lieu, A. Treatment response of bevacizumab combination chemotherapy in recurrent glioblastoma. Medicine 2020, 99, e19226. [Google Scholar] [CrossRef] [PubMed]
- Refusal of a Change to the Marketing Authorisation for Avastin (Bevacizumab). Available online: https://www.ema.europa.eu/en/documents/smop/questions-answers-refusal-change-marketing-authorisation-avastin-bevacizumab_en.pdf (accessed on 5 June 2021).
- Carneiro, A.V.; Costa, J. A prescrição fora das indicações aprovadas (off-label): Prática e problemas. Rev. Port. Cardiol. 2013, 32, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Nishikawa, R.; Narita, Y.; Kobayashi, H.; Takano, S.; Shinoura, N.; Aoki, T.; Sugiyama, K.; Kuratsu, J.; Muragaki, Y. Phase II Study of Single-agent Bevacizumab in Japanese Patients with Recurrent Malignant Glioma. Jpn. J. Clin. Oncol. 2012, 42, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.L.; Rouge, T.d.l.M.; Moore, N.; Zeaiter, A.; Phillips, H.; Modrusan, Z.; Cloughesy, T. AVAglio: Phase 3 Trial of Bevacizumab Plus Temozolomide and Radiotherapy AVAglio: Phase 3 Trial of Bevacizumab Plus Temozolomide and Radiotherapy in Newly Diagnosed Glioblastoma Multiforme. Adv. Ther. 2011, 28, 334–340. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II Trial of Single-Agent Bevacizumab Followed by Bevacizumab Plus Irinotecan at Tumor Progression in Recurrent Glioblastoma. J. Clin. Oncol. 2021, 27, 740–745. [Google Scholar] [CrossRef]
- Reyes-Botero, G.; Cartalat-Carel, S.; Chinot, O.L.; Barrie, M.; Taillandier, L.; Beauchesne, P.; Catry-Thomas, I.; Barriere, J.; Guillamo, J.-S.; Fabbro, M.; et al. Temozolomide Plus Bevacizumab in Elderly Patients with Newly Diagnosed Glioblastoma and Poor Performance Status: An ANOCEF Phase II Trial (ATAG). Oncologist 2018, 23, 524–544. [Google Scholar] [CrossRef]
- Cruz, E.; Silva, D.; Mercier, M.; Etienne-selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Dresemann, G.; Weller, M.; Rosenthal, M.A.; Wedding, U.; Wagner, W.; Engel, E.; Heinrich, B.; Øystein, A.K.; Anna, F.; Maximilian, N.; et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J. Neurooncol. 2010, 96, 393–402. [Google Scholar] [CrossRef]
- Raymond, E.; Brandes, A.A.; Dittrich, C.; Fumoleau, P.; Coudert, B.; Clement, P.M.J.; Frenay, M.; Rampling, R.; Stupp, R.; Kros, J.M.; et al. Phase II Study of Imatinib in Patients With Recurrent Gliomas of Various Histologies: A European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J. Clin. Oncol. 2008, 26, 4659–4665. [Google Scholar] [CrossRef]
- Alexandru, O.; Sevastre, A.; Castro, J.; Artene, S.; Tache, D.E.; Purcaru, O.S.; Sfredel, V.; Tataranu, L.G.; Dricu, A. Platelet-Derived Growth Factor Receptor and Ionizing Radiation in High Grade Glioma Cell Lines. Int. J. Mol. Sci. 2019, 20, 4663. [Google Scholar] [CrossRef]
- Popescu, A.M.; Alexandru, O.; Brindusa, C.; Purcaru, S.O.; Elise, D.; Tataranu, L.G.; Taisescu, C.; Dricu, A. Targeting the VEGF and PDGF signaling pathway in glioblastoma treatment. Int. J. Clin. Exp. Pathol. 2015, 8, 7825–7837. [Google Scholar] [PubMed]
- Lassman, A.B.; Pugh, S.L.; Gilbert, M.R.; Aldape, K.D.; Geinoz, S.; Beumer, J.H.; Christner, S.M.; Komaki, R.; Deangelis, L.M.; Gaur, R.; et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627) Andrew. Neuro-Oncol. 2015, 17, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Twohy, E.L.; Carrero, X.; Dixon, J.; Tran, D.D.; Jeyapalan, S.A.; Anderson, D.M.; Kaufmann, T.J.; Feathers, R.W.; et al. A Phase 1 and Randomized, Placebo-Controlled Phase 2 Trial of Bevacizumab Plus Dasatinib in Patients With Recurrent Glioblastoma: Alliance/North Central Cancer Treatment Group N0872. Cancer 2019, 125, 3790–9800. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Sampetrean, O.; Shimizu, T.; Saga, I.; Onishi, N.; Sugihara, E.; Okubo, J.; Fujita, S.; Takano, S.; Matsumura, A.; et al. IGF1 Receptor Signaling Regulates Adaptive Radioprotection in Glioma Stem Cells. Stem Cells 2013, 31, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Andersson, S.; Yin, S.; Girnita, A.; Stro, T.; Larsson, O.; Zheng, H.; Ericsson, C.; Axelson, M.; Niste, M.; et al. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro-Oncol. 2010, 12, 19–27. [Google Scholar]
- Aiken, R.; Axelson, M.; Harmenberg, J.; Klockare, M.; Wassberg, C. Phase I clinical trial of AXL1717 for treatment of relapsed malignant astrocytomas: Analysis of dose and response. Oncotarget 2017, 8, 81501–81510. [Google Scholar] [CrossRef]
- Higano, C.S.; Berlin, J.; Gordon, M.; Lorusso, P. Safety, tolerability, and pharmacokinetics of single and multiple doses of intravenous cixutumumab (IMC-A12), an inhibitor of the insulin-like growth factor-I receptor, administered weekly or every 2 weeks in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 450–462. [Google Scholar] [CrossRef]
- Zamykal, M.; Martens, T.; Matschke, J.; Gu, H.S.; Kathagen, A.; Schulte, A.; Peters, R.; Westphal, M.; Lamszus, K. Inhibition of intracerebral glioblastoma growth by targeting the insulin-like growth factor 1 receptor involves different context-dependent mechanisms. Neuro-Oncol. 2015, 17, 1076–1085. [Google Scholar] [CrossRef]
- Chaponis, D.; Barnes, J.W.; Dellagatta, J.L.; Kesari, S.; Kieran, M.W. Lonafarnib (SCH66336) improves the activity of temozolomide and radiation for orthotopic malignant gliomas. J. Neurooncol. 2011, 104, 179–189. [Google Scholar] [CrossRef]
- Glass, T.L.; Liu, T.; Yung, W.K.A. Inhibition of cell growth in human glioblastoma cell lines by farnesyltransferase inhibitor SCH66336. Neuro-Oncol. 2000, 2, 151–158. [Google Scholar] [CrossRef]
- Jakubowicz-gil, J.; Badziul, D.; Langner, E.; Wertel, I.; Zajac, A.; Rzeski, W. Temozolomide and sorafenib as programmed cell death inducers of human glioma cells. Pharmacol. Rep. 2017, 69, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II Study of Bevacizumab in Combination with Sorafenib in Recurrent Glioblastoma (N0776): A North Central Cancer Treatment Group Trial. Clin. Cancer Res. 2013, 19, 4816–4824. [Google Scholar] [CrossRef] [PubMed]
- Schiff, D.; Jaeckle, K.A.; Anderson, S.K.; Galanis, E.; Giannini, C. Phase 1/2 Trial of Temsirolimus and Sorafenib in the Treatment of Patients With Recurrent Glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer 2018, 124, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Kuhn, J.; Lamborn, K.R.; Abrey, L.; Deangelis, L.M.; Lieberman, F.; Robins, H.I.; Chang, S.M.; Yung, W.K.A.; Drappatz, J.; et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro-Oncol. 2012, 14, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Zustovich, F.; Landi, L.; Lombardi, G.; Galli, L.; Porta, C.; Amoroso, A.; Fontana, D.; Andreuccetti, M.; Galli, C.; Falcone, A.; et al. Sorafenib Plus Daily Low Dose Temozolomide for Relapsed Glioblastoma: A Phase II Study. Anticancer. Res. 2013, 33, 3487–3494. Available online: https://ascopubs.org/doi/abs/10.1200/jco.2011.29.15_suppl.2080 (accessed on 15 August 2021). [CrossRef]
- Hainsworth, J.D.; Ervin, T.; Friedman, E.; Priego, V.; Murphy, P.B. Concurrent Radiotherapy and Temozolomide Followed by Temozolomide and Sorafenib in the First-Line Treatment of Patients With Glioblastoma Multiforme. Cancer 2010, 116, 3663–3669. [Google Scholar] [CrossRef]
- Reardon, D.A.; Vredenburgh, J.J.; Friedman, H.S. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: Results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J. Neuro-Oncol. 2011, 101, 57–66. [Google Scholar] [CrossRef]
- Fondevila, F.; Méndez-Blanco, C.; Fernández-Palanca, P.; González-Gallego, J.; Mauriz, J.L. Anti-tumoral activity of sin-gle and combined regorafenib treatments in preclinical models of liver and gastrointestinal cancers. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Detti, B.; Scoccianti, S.; Lucidi, S.; Maragna, V.; Teriaca, M.A.; Ganovelli, M.; Desideri, I.; Lorenzetti, V.; Scoccimarro, E.; Greto, D.; et al. Regorafenib in glioblastoma recurrence: A case report. Cancer Treat. Res. Commun. 2021, 26, 100263. [Google Scholar] [CrossRef]
- Lombardi, G.; Caccese, M.; Padovan, M.; Cerretti, G.; Pintacuda, G.; Manara, R.; Di Sarra, F.; Zagonel, V. Regorafenib in recurrent glioblastoma patients: A large and monocentric real-life study. Cancers 2021, 13, 4731. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Wei, W.; Long, C.; Li, J. Atorvastatin augments temozolomide’s efficacy in glioblastoma via prenylation-dependent inhibition of Ras signaling. Biochem. Biophys. Res. Commun. 2017, 489, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Altwairgi, A.K.; Alghareeb, W.; Alnajjar, F.; Alsaeed, E.; Balbaid, A.; Alhussain, H.; Aldanan, S.; Orz, Y.; Lari, A.; Alsharm, A. Phase II study of atorvastatin in combination with radiotherapy and temozolomide In patients with glioblastoma (ART): Interim analysis report. Ann. Oncol. 2016, 27, 103–113. [Google Scholar] [CrossRef]
- Altwairgi, A.K.; Alnajjar, F.; Alhussain, H.; Alsaeed, E.; Balbaid, A.; Aldandan, S.; Orz, Y.; Lary, A.; Alghareeb, W.; Alsharm, A. Phase II study of atorvastatin in combination with radiotherapy and temozolomide in patients with glioblastoma (ART): Final analysis report. Ann. Oncol. 2019, 30, ix20–ix21. [Google Scholar] [CrossRef]
- Azaro, A.; Plummer, E.R.; Urruticoechea, A.; Rodon, J.; Md Haris, N.R.; Veal, G.; Perier, A.; Tur, V.; Escriba, P.V.; Busquets, X.; et al. Final Report of a Phase I Study of 2-Hydroxyoleic Acid (2OHOA) a Novel Sphingomyelin Synthase Activator in Patients (pt) with Advanced Solid Tumors (AST) Including Recurrent High Grade Gliomas (rHGG). J. Clin. Oncol. 2017, 35, 2554. Available online: https://ascopubs.org/doi/10.1200/JCO.2017.35.15_suppl.2554 (accessed on 15 August 2021). [CrossRef]
- EMA EU/3/11/916. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu311916 (accessed on 15 August 2021).
- A Clinical Phase IIB Trial with 2OHOA in Patients with Newly-Diagnosed Malignant Glioma. Available online: https://cordis.europa.eu/project/id/755179 (accessed on 15 August 2021).
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Nethland, I.A.; Førde, H.E.; Sleire, L.; Leiss, L.; Rahman, M.A.; Skeie, B.S. Treatment with the PI3K inhibitor buparlisib (NVP-BKM120) suppresses the growth of established patient-derived GBM xenografts and prolongs survival in nude rats. J. Neurooncol. 2016, 129, 57–66. [Google Scholar] [CrossRef]
- Speranza, M.; Nowicki, M.O.; Behera, P.; Cho, C.; Chiocca, E.A.; Sean, E.L. BKM-120 (Buparlisib): A Phosphatidyl-Inositol-3 Kinase Inhibitor with Anti-Invasive Properties in Glioblastoma. Sci. Rep. 2016, 6, 20189. [Google Scholar] [CrossRef]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2021, 37, 741–761. [Google Scholar] [CrossRef]
- Koul, D.; Shen, R.; Kim, Y.; Kondo, Y.; Lu, Y.; Bankson, J.; Ronen, S.M.; Kirkpatrick, D.L.; Powis, G.; Yung, W.K.A. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro-Oncol. 2010, 12, 559–569. [Google Scholar] [CrossRef]
- Pitz, M.W.; Eisenhauer, E.A.; Macneil, M.V.; Thiessen, B.; Easaw, J.C.; Macdonald, D.R.; Eisenstat, D.D.; Kakumanu, A.S.; Salim, M.; Chalchal, H.; et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro-Oncol. 2015, 17, 1270–1274. [Google Scholar] [CrossRef] [PubMed]
- Lassen, U.; Sorensen, M.; Gaziel, T.B.; Hasselbalch, B.; Poulsen, H.S. Phase II Study of Bevacizumab and Temsirolimus Combination Therapy for Recurrent Glioblastoma Multiforme. Anticancer Res. 2013, 33, 1657–1660. [Google Scholar] [PubMed]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; Van Den Bent, M.J.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 2016, 22, 4797–4807. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.J.; Galanis, E.; Anderson, S.K.; Schiff, D.; Kaufmann, T.J.; Peller, P.J.; Giannini, C.; Brown, P.D.; Uhm, J.H.; Mcgraw, S.; et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-Oncol. 2015, 17, 1261–1269. [Google Scholar] [CrossRef]
- Wick, W.; Puduvalli, V.K.; Chamberlain, M.C.; Van Den Bent, M.J.; Carpentier, A.F.; Cher, L.M.; Mason, W.; Weller, M.; Hong, S.; Musib, L.; et al. Phase III Study of Enzastaurin Compared With Lomustine in the Treatment of Recurrent Intracranial Glioblastoma. Am. Soc. Clin. Oncol. 2021, 28, 1168–1174. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Kotliarova, S.; Butman, J.A.; Albert, P.S.; Kim, L.; Musib, L.; Thornton, D.; Fine, H.A.; Branch, N.; Maryland, T.N.K. A phase I/II trial of enzastaurin in patients with recurrent high-grade gliomas. Neuro-Oncol. 2010, 12, 181–189. [Google Scholar] [CrossRef]
- Bota, D.A.; Alexandru, D.; Keir, S.T.; Bigner, D.; Vredenburgh, J.; Friedman, H.S.; Robert, P.; Brain, T.; Carolina, N. Proteasome inhibition with bortezomib induces cell death in GBM stem-like cells and temozolomide-resistant glioma cell lines, but stimulates GBM stem-like cells’ VEGF production and angiogenesis. J. Neurooncol. 2015, 119, 1415–1423. [Google Scholar] [CrossRef]
- Tang, J.H.; Yang, L.; Chen, J.X.; Li, Q.R.; Zhu, L.R.; Xu, Q.F.; Huang, G.H. Bortezomib inhibits growth and sensitizes glioma to temozolomide (TMZ) via down-regulating the FOXM1–Survivin axis. Cancer Commun. 2019, 39, 81. [Google Scholar] [CrossRef]
- Su, Z.; Han, S.; Jin, Q.; Zhou, N.; Lu, J.; Shangguan, F.; Yu, S.; Liu, Y.; Wang, L.; Lu, J.; et al. Ciclopirox and bortezomib synergistically inhibits glioblastoma multiforme growth via simultaneously enhancing JNK/p38 MAPK and NF-κ B signaling. Cell Death Dis. 2021, 12, 251. [Google Scholar] [CrossRef]
- Di, K.; Lloyd, G.K.; Abraham, V.; Maclaren, A.; Burrows, F.J.; Desjardins, A.; Trikha, M.; Bota, D.A.; California, K.D.; Corporation, T.A.; et al. Marizomib activity as a single agent in malignant gliomas: Ability to cross the blood-brain barrier. Neuro-Oncol. 2016, 18, 840–848. [Google Scholar] [CrossRef]
- Boccellato, C.; Kolbe, E.; Peters, N.; Juric, V.; Fullstone, G. Marizomib sensitizes primary glioma cells to apoptosis induced by a latest-generation TRAIL receptor agonist. Cell Death Dis. 2021, 12, 647. [Google Scholar] [CrossRef] [PubMed]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.; Herndon, J.; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncologist 2018, 23, 157-e21. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: A north central cancer treatment group study. Neuro-Oncol. 2012, 14, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, F.M.; Lamborn, K.R.; Kuhn, J.G.; Wen, P.Y.; Yung, W.K.A.; Gilbert, M.R.; Chang, S.M.; Lieberman, F.S.; Prados, M.D.; Fine, H.A. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neuro-Oncol. 2011, 13, 509–516. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.; Neyns, B.; Goldbrunner, R.; Schlegel, U.; Clement, P.M.J.; Grabenbauer, G.G.; Ochsenbein, A.F.; Simon, M.; Dietrich, P.; et al. Phase I/IIa Study of Cilengitide and Temozolomide With Concomitant Radiotherapy Followed by Cilengitide and Temozolomide Maintenance Therapy in Patients With Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2010, 28, 2712–2718. [Google Scholar] [CrossRef]
- Reardon, D.A.; Fink, K.L.; Mikkelsen, T.; Cloughesy, T.F.; Neill, A.O.; Plotkin, S.; Glantz, M.; Ravin, P.; Raizer, J.J.; Rich, K.M.; et al. Randomized Phase II Study of Cilengitide, an Integrin-Targeting Arginine-Glycine-Aspartic Acid Peptide, in Recurrent Glioblastoma Multiforme. J. Clin. Oncol. 2021, 26, 5610–5617. [Google Scholar] [CrossRef]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: Results of the open-label, controlled, randomized phase II CORE study. Neuro-Oncol. 2015, 17, 708–717. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Molecular characterization of cancers with NTRK gene fusions. Mod. Pathol. 2018, 32, 147–153. [Google Scholar] [CrossRef]
- Drilon, A.; Ou, S.I.; Cho, B.C.; Kim, D.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent-Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion–Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.; Sepúlveda-Sánchez, J.; Cloughesy, T.; Gil-Gil, J.; Puduvalli, V.; Raizer, J.; De Vos, F.; Wen, P.; Butowski, N.; Clement, P.; et al. ACTR-33. Infigratinib (bgj398) in Patients with Recurrent Gliomas with Fibroblast Growth Factor Receptor (FGFR) Alterations. Available online: https://academic.oup.com/neuro-oncology/article-abstract/21/Supplement_6/vi20/5620107?redirectedFrom=fulltext (accessed on 15 August 2021).
- Das, A.; Cheng, R.R.; Hilbert, M.L.T.; Dixon-moh, Y.N.; Decandio, M.; Alex, W.; Iii, V.; Banik, N.L.; Lindhorst, S.M.; Cachia, D.; et al. Synergistic Effects of Crizotinib and Temozolomide in Experimental FIG-ROS1 Fusion-Positive Glioblastoma. Cancer Growth Metastasis 2015, 8, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Cloughesy, T.F.; Wen, P.Y.; Taylor, J.W.; Maher, E.A.; Arrillaga-romany, I.; Peters, K.B.; Choi, C.; Ellingson, B.M.; Lin, A.P.; et al. A Phase 1, Open-Label, Perioperative Study of Ivosidenib (AG-120) and Vorasidenib (AG-881) in Recurrent, IDH1-Mutant, Low-Grade Glioma: Results from Cohort 1. 2019. Available online: https://www.servier.us/sites/default/files/2021-04/SNO19_phase1.pdf (accessed on 25 August 2021).
- Machida, Y.; Nakagawa, M.; Matsunaga, H.; Yamaguchi, M.; Ogawara, Y.; Shima, Y.; Yamagata, K.; Katsumoto, T.; Hattori, A.; Itoh, M.; et al. A Potent Blood–Brain Barrier-Permeable Mutant IDH1 Inhibitor Suppresses the Growth of Glioblastoma with IDH1 Mutation in a Patient-Derived Orthotopic Xenograft Model. Mol. Cancer Ther. 2020, 19, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Ovens, M.J.; Davies, A.J.; Wilson, M.C.; Murray, C.M.; Halestrap, A.P. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10. Biochem. J. 2010, 530, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Voss, D.M.; Spina, R.; Carter, D.L.; Suan Lim, K.; Jeffery, C.J.; Bar, E.E. Disruption of the monocarboxylate transporter-4-basigin interaction inhibits the hypoxic response, proliferation, and tumor progression. Sci. Rep. 2017, 7, 4292. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2021, 11, 603911. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Kettenmann, H. Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron 2020, 104, 442–449. [Google Scholar] [CrossRef]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2021, 10, 18. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2020, 79, 4557–4566. [Google Scholar] [CrossRef]
- Sokratous, G.; Polyzoidis, S.; Ashkan, K. Immune infiltration of tumor microenvironment following immunotherapy for glioblastoma multiforme. Hum. Vaccin. Immunother. 2017, 13, 2575–2582. [Google Scholar] [CrossRef]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Baranska, J. Advances in Experimental Medicine and Biology: Glioma Signaling, 2nd ed.; Springer Nature: Cham, Switzerland, 2020; ISBN 9783030306502. [Google Scholar]
- Desland, F.A.; Hormigo, A. The CNS and the Brain Tumor Microenvironment: Implications for Glioblastoma Immunotherapy. Int. J. Mol. Sci 2020, 21, 7358. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Prabhu, V.C.; Lukas, R.V.; Peters, K.B. What is New in Neuro-oncology? Neurol. Clin. NA 2021, 39, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Hajjar, J. Advances in Experimental Medicine and Biology: Immunotherapy; Springer Nature: Cham, Switzerland, 2018; ISBN 9783030410070. [Google Scholar]
- Altshuler, D.B.; Kadiyala, P.; Núñez, F.J.; Núñez, F.M.; Alghamri, M.S.; Garcia-fabiani, M.B.; Asad, A.S.; Candia, A.J.N.; Candolfi, M.; Lahann, J.; et al. Prospects of Biological and Synthetic Pharmacotherapies for Glioblastoma. Expert. Opin. Biol. Ther. 2021, 20, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Raizer, J.J.; Lim, M.; Sughrue, M.; Komotar, R.; AbrahamsDonald O’Rourke, J.; D’Ambrosio, A.; Bruce, J.N.; Parsa, A. Newly Diagnosed Glioblastoma Patients Treated with an Autologous Heat Shock Protein Peptide Vaccine: PD-L1 Expression and Response to Therapy. Available online: https://ascopubs.org/doi/abs/10.1200/jco.2015.33.15_suppl.2011 (accessed on 25 July 2021).
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; Mcdermott, M.W.; et al. Heat-shock protein peptide complex–96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-Oncol. 2014, 16, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Campian, J.; Ghiaseddin, A.; Rahman, M.; Ansstas, G.; Kim, A.; Leuthardt, E.; Tran, D. ATIM-17. Early results of a multicenter phase I and open-label, randomized phase II study testing the toxicities and efficacy of MK-3475 (pembrolizumab) in combination with MRI-guided laser interstitial thermal therapy (LITT) in recurrent malignant glioma. Neuro-Oncol. 2017, 29, vi29. [Google Scholar] [CrossRef]
- Lukas, R.V.; Wainwright, D.A.; Horbinski, C.M.; Sonabend, A.M. Immunotherapy against gliomas-is the breakthrough near? Drugs 2020, 79, 1839–1848. [Google Scholar] [CrossRef]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef]
- Sakai, K.; Shimodaira, S.; Maejima, S.; Udagawa, N.; Sano, K.; Higuchi, Y.; Koya, T.; Ochiai, T.; Koide, M.; Uehara, S.; et al. Dendritic cell–based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J. Neurosurg. 2015, 123, 989–997. [Google Scholar] [CrossRef]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-perez, L.; Smita, K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Ii, J.E.H.; Mclendon, R.E.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; Andre, S.D.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Núñez, F.J.; Haase, S.; Mcclellan, B.L.; Faisal, S.M.; Carney, S.V.; Yu, J.; Alghamri, M.S.; Candolfi, M.; Lowenstein, P.R.; et al. Current Approaches for Glioma Gene Therapy and Virotherapy. Front. Mol. Neurosci. 2021, 14, 621831. [Google Scholar] [CrossRef] [PubMed]
- Tietze, S.; Michen, S.; Schackert, G.; Temme, A. Prospects of immune checkpoint blockade and vaccine-based immunotherapy for glioblastoma. Innov. Surg. Sci. 2021, 6, 35–48. [Google Scholar] [CrossRef]
- Wang, X.; Lu, J.; Guo, G.; Yu, J. Immunotherapy for recurrent glioblastoma: Practical insights and challenging prospects. Cell Death Dis. 2021, 12, 299. [Google Scholar] [CrossRef]
- Yang, T.; Kong, Z.; Ma, W. PD-1/PD-L1 immune checkpoint inhibitors in glioblastoma: Clinical studies, challenges and potential. Hum. Vaccin. Immunother. 2020, 17, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Nayak, L.; Peters, K.B.; Leigh Clarke, J.; Jordan, J.T.; Frederick De Groot, J.; Leia Nghiemphu, P.; Joseph Kaley, T.; Colman, H.; Gaffey, S.C.; et al. Phase II Study of Pembrolizumab or Pembrolizumab Plus Bevacizumab for Recurrent Glioblastoma (rGBM) Patients. Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2018.36.15_suppl.2006 (accessed on 25 July 2021).
- Reardon, D.; Kaley, T.; Dietrich, J.; Clarke, J.; Dunn, G.; Lim, M.; Cloughesy, T.; Gan, H.; Park, A.; Schwarzenberger, P.; et al. ATIM-38. Phase 2 Study to Evaluate the Clinical Efficacy and Safety of medi4736 (Durvalumab, Durva)+Bevacizumab (bev) in Bev-Naïve Patients with Recurrent Glioblastoma (GBM). Neuro-Oncol. 2018, 20, vi10. Available online: https://academic.oup.com/neuro-oncology/article/20/suppl_6/vi10/5153790 (accessed on 25 July 2021). [CrossRef][Green Version]
- Reardon, D.A.; Kaley, T.J.; Dietrich, J.; Clarke, J.L.; Dunn, G.; Lim, M.; Cloughesy, T.F.; Gan, H.K.; Park, A.J.; Schwarzenberger, P.; et al. Phase II Study to Evaluate Safety and Efficacy of MEDI4736 (Durvalumab)+Radiotherapy in Patients with Newly Diagnosed Unmethylated MGMT Glioblastoma (New Unmeth GBM). Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2019.37.15_suppl.2032 (accessed on 25 July 2021).
- Qi, Y.; Liu, B.; Sun, Q.; Xiong, X.; Chen, Q. Immune Checkpoint Targeted Therapy in Glioma: Status and Hopes. Front. Immunol. 2020, 11, 578877. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, L.; Ye, M.; Kang, C.; You, H. Treatment Progress of Immune Checkpoint Blockade Therapy for Glioblastoma. Front. Immunol. 2020, 11, 592612. [Google Scholar] [CrossRef]
- Kelly, E.; Russell, S.J. History of Oncolytic Viruses: Genesis to Genetic Engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef]
- Varela-guruceaga, M.; Tejada-sol, S.; Garc, M.; Fueyo, J.; Gomez-manzano, C.; Patiño-garc, A.; Alonso, M.M. Oncolytic Viruses as Therapeutic Tools for Pediatric Brain Tumors. Cancers 2018, 10, 226. [Google Scholar] [CrossRef]
- Rius-rocabert, S.; Garc, A. Oncolytic Virotherapy in Glioma Tumors. Int. J. Mol. Sci. 2020, 21, 7604. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.M.; Shah, A.H.; Vallejo, F.A.; Eichberg, D.G.; Luther, E.M.; Shah, S.S.; Komotar, R.J.; Ivan, M.E. Clinical trials using oncolytic viral therapy to treat adult glioblastoma: A progress report. Neurosurg. Focus 2021, 50, E3. [Google Scholar] [CrossRef] [PubMed]
- Cheema, T.A.; Wakimoto, H.; Fecci, P.E.; Ning, J.; Kuroda, T.; Jeyaretna, D.S.; Martuza, R.L.; Rabkin, S.D. Multifaceted Oncolytic Virus Therapy for Glioblastoma in an Immunocompetent Cancer Stem Cell Model. Proc. Natl. Acad. Sci. USA 2013, 110, 12006–12011. Available online: https://doi.org/10.1073/pnas.1307935110/-/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1307935110 (accessed on 15 March 2022). [CrossRef] [PubMed]
- Todo, T. ATIM-14. Results of phase ii clinical trial of oncolytic herpes virus g47δ in patients with glioblastoma. Neuro-Oncol. 2019, 11, vi4. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, G.; Lang, F.; Daras, M.; Cloughesy, T.; Colman, H.; Ong, S.; Ramakrishna, R.; Vogelbaum, M.; Groves, M.; Nassiri, F.; et al. ATIM-24. interim results of a phase ii multicenter study of the conditionally replicative oncolytic adenovirus dnx-2401 with pembrolizumab (keytruda) for recurrent glioblastoma; captive study (keynote-192). Neuro-Oncol. 2018, 20, vi6. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus Annick. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Landolfi, J.; Hogan, D.J.; Bloomfield, S.; Chen, C.C.; Elder, J.B.; Kalkanis, S.N.; Kesari, S.; Lee, I.Y.; Liau, L.M.; et al. Phase 1 Trial of Vocimagene Amiretrorepvec and 5-Fluorocytosine for Recurrent High Grade Glioma. Sci. Transl. Med. 2016, 8, 341ra75. [Google Scholar] [CrossRef]
- Perez, O.D.; Logg, C.R.; Hiraoka, K.; Diago, O.; Burnett, R.; Inagaki, A.; Jolson, D.; Amundson, K.; Buckley, T.; Lohse, D.; et al. Design and Selection of Toca 511 for Clinical Use: Modified Retroviral Replicating Vector With Improved Stability and Gene Expression. Mol. Ther. 2012, 20, 1689–1698. [Google Scholar] [CrossRef]
- Tocagen Reports Results of Toca 5 Phase 3 Trial in Recurrent Brain Cancer. Available online: https://www.prnewswire.com/news-releases/tocagen-reports-results-of-toca-5-phase-3-trial-in-recurrent-brain-cancer-300916705.html (accessed on 24 July 2021).
- Feldman, L.; Badie, B. Chimeric Antigen Receptor T-Cell Therapy: Updates in Glioblastoma Treatment. Neurosurgery 2021, 88, 1056–1064. [Google Scholar] [CrossRef]
- Gatto, L.; Di Nunno, V.; Franceschi, E.; Tosoni, A.; Bartolini, S.; Brandes, A.A. Pharmacotherapeutic Treatment of Glioblastoma: Where Are We to Date? Drugs 2022, 82, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Gwak, H.S.; Han, N.; Hong, E.K.; Choi, B.K.; Lee, S.; Choi, S.; Park, J.H.; Seok, J.H.; Jeon, Y.; et al. Chimeric Antigen Receptor T Cells With Modified Interleukin-13 Preferentially Recognize IL13Rα2 and Suppress Malignant Glioma: A Preclinical Study. Front. Immunol. 2021, 12, 715000. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.Y.; Fang, D.; Latha, K.; et al. Opening of the blood–brain barrier using low-intensity pulsed ultrasound enhances responses to immunotherapy in preclinical glioma models. Clin. Cancer Res. 2021, 27, 4325–4337. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ErbB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Sakao, S.; Tatsumi, K. Molecular mechanisms of lung-specific toxicity induced by epidermal growth factor receptor tyrosine kinase inhibitors (Review). Oncol. Lett. 2012, 4, 865–867. [Google Scholar] [CrossRef]
- Lin, M.; Xiong, W.; Wang, S.; Li, Y.; Hou, C.; Li, C.; Li, G. The Research Progress of Trastuzumab-Induced Cardiotoxicity in HER-2-Positive Breast Cancer Treatment. Front. Cardiovasc. Med. 2022, 8, 821663. [Google Scholar] [CrossRef]
- Hahn, V.S.; Zhang, K.W.; Sun, L.; Narayan, V.; Lenihan, D.J.; Ky, B. Heart Failure with Targeted Cancer Therapies: Mechanisms and Cardioprotection. Circ. Res. 2021, 128, 1576–1593. [Google Scholar] [CrossRef]
- Hedge, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.; Orange, J.S.; Sumazin, P.; Man, T.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonso, M.; Brito, M.A. Therapeutic Options in Neuro-Oncology. Int. J. Mol. Sci. 2022, 23, 5351. https://doi.org/10.3390/ijms23105351
Afonso M, Brito MA. Therapeutic Options in Neuro-Oncology. International Journal of Molecular Sciences. 2022; 23(10):5351. https://doi.org/10.3390/ijms23105351
Chicago/Turabian StyleAfonso, Mariana, and Maria Alexandra Brito. 2022. "Therapeutic Options in Neuro-Oncology" International Journal of Molecular Sciences 23, no. 10: 5351. https://doi.org/10.3390/ijms23105351
APA StyleAfonso, M., & Brito, M. A. (2022). Therapeutic Options in Neuro-Oncology. International Journal of Molecular Sciences, 23(10), 5351. https://doi.org/10.3390/ijms23105351