
Nuclear Cytoskeleton in Virus Infection


Abstract
1. Nuclear Cytoskeleton
2. Lamins and Nuclear Lamina
3. Lamins in Virus Infection
3.1. Herpesviruses
3.1.1. Betaherpesviruses
3.1.2. Alpha- and Gammaherpesviruses
3.2. Baculoviruses
3.3. Polyomaviruses
3.4. Parvoviruses
3.5. Circoviruses
4. Nuclear Actin
5. Nuclear Actin and Virus Infection
5.1. Role of Nuclear Actin in Movement of Capsids in the Nucleus of Infected Cells
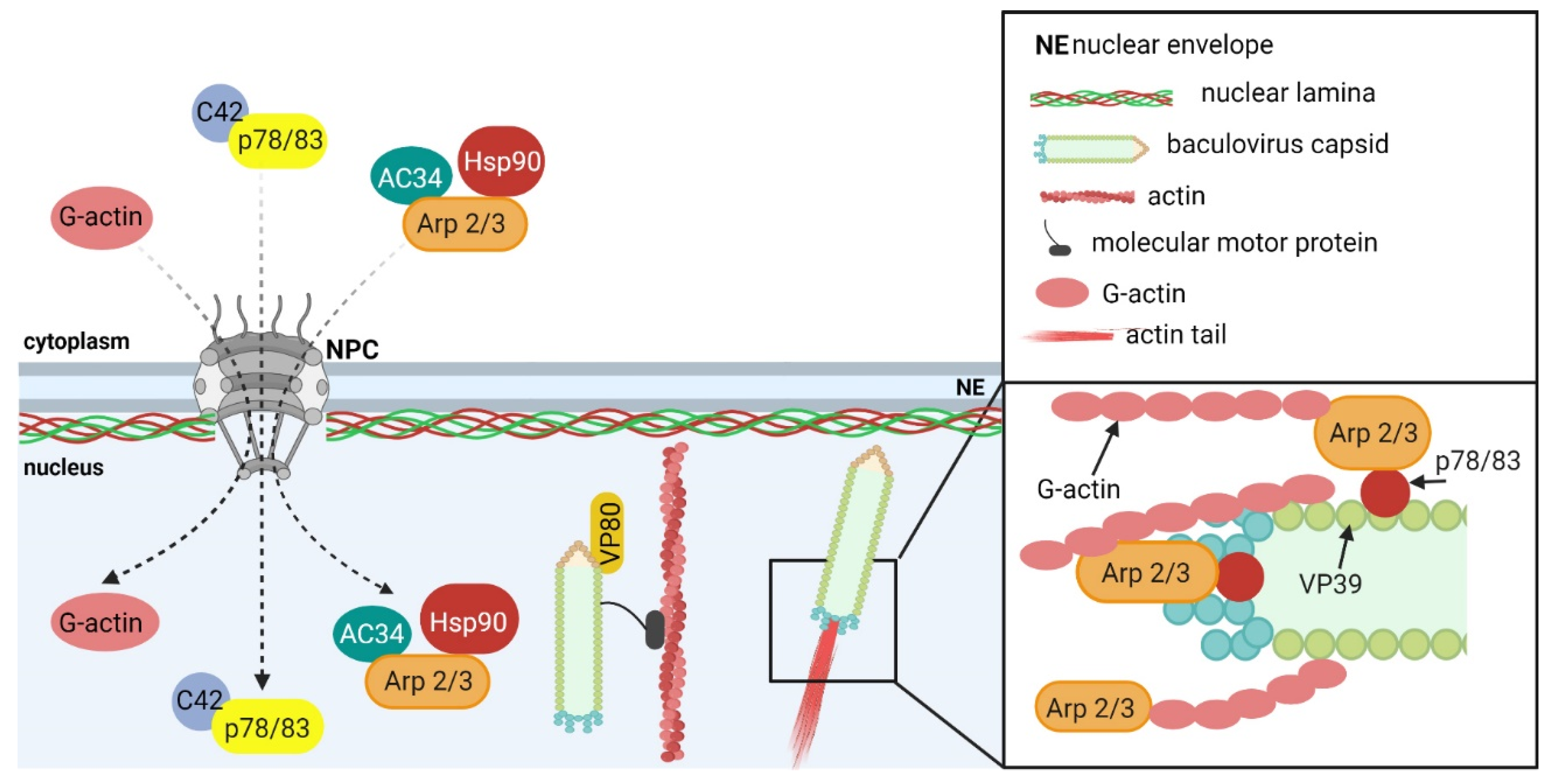
5.1.1. Baculoviruses
5.1.2. Herpesviruses
5.2. Role of Nuclear Actin in Other Aspects of Virus Replication Cycle
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nowak, G.; Pestic-Dragovich, L.; Hozák, P.; Philimonenko, A.; Simerly, C.; Schatten, G.; de Lanerolle, P. Evidence for the presence of myosin I in the nucleus. J. Biol. Chem. 1997, 272, 17176–17181. [Google Scholar] [CrossRef]
- Pestic-Dragovich, L.; Stojiljkovic, L.; Philimonenko, A.A.; Nowak, G.; Ke, Y.; Settlage, R.E.; Shabanowitz, J.; Hunt, D.F.; Hozak, P.; de Lanerolle, P. A myosin I isoform in the nucleus. Science 2000, 290, 337–341. [Google Scholar] [CrossRef]
- Akoumianaki, T.; Kardassis, D.; Polioudaki, H.; Georgatos, S.D.; Theodoropoulos, P.A. Nucleocytoplasmic shuttling of soluble tubulin in mammalian cells. J. Cell Sci. 2009, 122, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Ruksha, K.; Mezheyeuski, A.; Nerovnya, A.; Bich, T.; Tur, G.; Gorgun, J.; Luduena, R.; Portyanko, A. Over-expression of ΒII-tubulin and especially its localization in cell nuclei correlates with poorer outcomes in colorectal cancer. Cells 2019, 8, E25. [Google Scholar] [CrossRef] [PubMed]
- Kırlı, K.; Karaca, S.; Dehne, H.J.; Samwer, M.; Pan, K.T.; Lenz, C.; Urlaub, H.; Görlich, D. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. Elife 2015, 4, e11466. [Google Scholar] [CrossRef] [PubMed]
- Aebi, U.; Cohn, J.; Buhle, L.; Gerace, L. The Nuclear lamina is a meshwork of intermediate-type filaments. Nature 1986, 323, 560–564. [Google Scholar] [CrossRef]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef]
- Gerace, L.; Burke, B. Functional organization of the nuclear envelope. Annu. Rev. Cell Biol. 1988, 4, 335–374. [Google Scholar] [CrossRef]
- Nmezi, B.; Xu, J.; Fu, R.; Armiger, T.J.; Rodriguez-Bey, G.; Powell, J.S.; Ma, H.; Sullivan, M.; Tu, Y.; Chen, N.Y.; et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315. [Google Scholar] [CrossRef]
- Wilson, K.L.; Foisner, R. Lamin-binding proteins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000554. [Google Scholar] [CrossRef]
- Simon, D.N.; Wilson, K.L. Partners and post-translational modifications of nuclear lamins. Chromosoma 2013, 122, 13–31. [Google Scholar] [CrossRef]
- Casey, P.J. Biochemistry of protein prenylation. J. Lipid Res. 1992, 33, 1731–1740. [Google Scholar] [CrossRef]
- Casey, P.J.; Seabra, M.C. Protein prenyltransferases. J. Biol. Chem. 1996, 271, 5289–5292. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S. Protein isoprenylation and methylation at carboxyl-terminal cysteine residues. Annu. Rev. Biochem. 1992, 61, 355–386. [Google Scholar] [CrossRef] [PubMed]
- Sinensky, M.; Fantle, K.; Trujillo, M.; McLain, T.; Kupfer, A.; Dalton, M. The processing pathway of prelamin A. J. Cell Sci. 1994, 107, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Fong, L.G.; Michaelis, S. Prelamin A, zmpste24, misshapen cell nuclei, and progeria—New evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res. 2005, 46, 2531–2558. [Google Scholar] [CrossRef]
- Liu, S.Y.; Ikegami, K. Nuclear lamin phosphorylation: An emerging role in gene regulation and pathogenesis of laminopathies. Nucleus 2020, 11, 299–314. [Google Scholar] [CrossRef]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior—Life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef]
- Prokocimer, M.; Davidovich, M.; Nissim-Rafinia, M.; Wiesel-Motiuk, N.; Bar, D.Z.; Barkan, R.; Meshorer, E.; Gruenbaum, Y. Nuclear lamins: Key regulators of nuclear structure and activities. J. Cell Mol. Med. 2009, 13, 1059–1085. [Google Scholar] [CrossRef]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex Virus 1 capsids to the nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Döhner, K.; Cornelius, A.; Serrero, M.C.; Sodeik, B. The journey of herpesvirus capsids and genomes to the host cell nucleus. Curr. Opin. Virol. 2021, 50, 147–158. [Google Scholar] [CrossRef]
- Copeland, A.M.; Newcomb, W.W.; Brown, J.C. Herpes simplex virus replication: Roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 2009, 83, 1660–1668. [Google Scholar] [CrossRef]
- Abaitua, F.; O’Hare, P. Identification of a highly conserved, functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1-2 tegument protein. J. Virol. 2008, 82, 5234–5244. [Google Scholar] [CrossRef] [PubMed]
- Pasdeloup, D.; Blondel, D.; Isidro, A.L.; Rixon, F.J. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein PUL25. J. Virol. 2009, 83, 6610–6623. [Google Scholar] [CrossRef] [PubMed]
- Preston, V.G.; Murray, J.; Preston, C.M.; McDougall, I.M.; Stow, N.D. The UL25 gene product of herpes simplex virus type 1 Is Involved in Uncoating of the Viral Genome. J. Virol. 2008, 82, 6654–6666. [Google Scholar] [CrossRef] [PubMed]
- Jovasevic, V.; Liang, L.; Roizman, B. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 2008, 82, 3311–3319. [Google Scholar] [CrossRef]
- Radsak, K.D.; Brücher, K.H.; Georgatos, S.D. Focal nuclear envelope lesions and specific nuclear lamin A/C Dephosphorylation during infection with human cytomegalovirus. Eur. J. Cell Biol. 1991, 54, 299–304. [Google Scholar]
- Scott, E.S.; O’Hare, P. Fate of the inner nuclear membrane protein lamin B receptor and nuclear lamins in herpes simplex virus type 1 infection. J. Virol. 2001, 75, 8818–8830. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Haas, J.; Wagner, M.; Krohne, G.; Koszinowski, U.H. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 2002, 297, 854–857. [Google Scholar] [CrossRef]
- Dal Monte, P.; Pignatelli, S.; Zini, N.; Maraldi, N.M.; Perret, E.; Prevost, M.C.; Landini, M.P. Analysis of Intracellular and Intraviral Localization of the Human Cytomegalovirus UL53 Protein. J. Gen. Virol. 2002, 83, 1005–1012. [Google Scholar] [CrossRef]
- Marschall, M.; Marzi, A.; aus dem Siepen, P.; Jochmann, R.; Kalmer, M.; Auerochs, S.; Lischka, P.; Leis, M.; Stamminger, T. Cellular P32 recruits cytomegalovirus kinase PUL97 to redistribute the nuclear lamina. J. Biol. Chem. 2005, 280, 33357–33367. [Google Scholar] [CrossRef]
- Sharma, M.; Kamil, J.P.; Coughlin, M.; Reim, N.I.; Coen, D.M. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J. Virol. 2014, 88, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Camozzi, D.; Pignatelli, S.; Valvo, C.; Lattanzi, G.; Capanni, C.; Dal Monte, P.; Landini, M.P. Remodelling of the nuclear lamina during human cytomegalovirus infection: Role of the viral proteins PUL50 and PUL53. J. Gen. Virol. 2008, 89, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Kuan, M.I.; O’Dowd, J.M.; Fortunato, E.A. The absence of P53 during human cytomegalovirus infection leads to decreased UL53 expression, disrupting UL50 localization to the inner nuclear membrane, and thereby inhibiting capsid nuclear egress. Virology 2016, 497, 262–278. [Google Scholar] [CrossRef]
- Hamirally, S.; Kamil, J.P.; Ndassa-Colday, Y.M.; Lin, A.J.; Jahng, W.J.; Baek, M.-C.; Noton, S.; Silva, L.A.; Simpson-Holley, M.; Knipe, D.M.; et al. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009, 5, e1000275. [Google Scholar] [CrossRef]
- Milbradt, J.; Webel, R.; Auerochs, S.; Sticht, H.; Marschall, M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 2010, 285, 13979–13989. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Auerochs, S.; Marschall, M. Cytomegaloviral proteins PUL50 and PUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 2007, 88, 2642–2650. [Google Scholar] [CrossRef]
- Sonntag, E.; Hamilton, S.T.; Bahsi, H.; Wagner, S.; Jonjic, S.; Rawlinson, W.D.; Marschall, M.; Milbradt, J. Cytomegalovirus PUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J. Gen. Virol. 2016, 97, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Kuny, C.V.; Chinchilla, K.; Culbertson, M.R.; Kalejta, R.F. Cyclin-dependent kinase-like function is shared by the beta- and gamma-subset of the conserved herpesvirus protein kinases. PLoS Pathog. 2010, 6, e1001092. [Google Scholar] [CrossRef]
- Milbradt, J.; Hutterer, C.; Bahsi, H.; Wagner, S.; Sonntag, E.; Horn, A.H.C.; Kaufer, B.B.; Mori, Y.; Sticht, H.; Fossen, T.; et al. The prolyl isomerase pin1 promotes the herpesvirus-induced phosphorylation-dependent disassembly of the nuclear lamina required for nucleocytoplasmic egress. PLoS Pathog. 2016, 12, e1005825. [Google Scholar] [CrossRef]
- Milbradt, J.; Sonntag, E.; Wagner, S.; Strojan, H.; Wangen, C.; Lenac Rovis, T.; Lisnic, B.; Jonjic, S.; Sticht, H.; Britt, W.J.; et al. Human cytomegalovirus nuclear capsids associate with the core nuclear egress complex and the viral protein kinase PUL97. Viruses 2018, 10, E35. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Furlong, W.E.; Pennell, L.; Geadah, M.; Hertel, L. RASCAL is a new human cytomegalovirus-encoded protein that localizes to the nuclear lamina and in cytoplasmic vesicles at late times postinfection. J. Virol. 2010, 84, 6483–6496. [Google Scholar] [CrossRef]
- Murray, L.A.; Sheng, X.; Cristea, I.M. Orchestration of protein acetylation as a toggle for cellular defense and virus replication. Nat. Commun. 2018, 9, 4967. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.E.; Wills, E.G.; Roller, R.J.; Ryckman, B.J.; Baines, J.D. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 2002, 76, 8939–8952. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Holley, M.; Baines, J.; Roller, R.; Knipe, D.M. Herpes simplex virus 1 U(L)31 and U(L)34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 2004, 78, 5591–5600. [Google Scholar] [CrossRef]
- Simpson-Holley, M.; Colgrove, R.C.; Nalepa, G.; Harper, J.W.; Knipe, D.M. Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J. Virol. 2005, 79, 12840–12851. [Google Scholar] [CrossRef]
- Roller, R.J.; Zhou, Y.; Schnetzer, R.; Ferguson, J.; DeSalvo, D. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J. Virol. 2000, 74, 117–129. [Google Scholar] [CrossRef]
- Farina, A.; Santarelli, R.; Bloise, R.; Gonnella, R.; Granato, M.; Bei, R.; Modesti, A.; Cirone, M.; Bengtsson, L.; Angeloni, A.; et al. KSHV ORF67 encoded lytic protein localizes on the nuclear membrane and alters emerin distribution. Virus Res. 2013, 175, 143–150. [Google Scholar] [CrossRef]
- Lee, C.-P.; Huang, Y.-H.; Lin, S.-F.; Chang, Y.; Chang, Y.-H.; Takada, K.; Chen, M.-R. Epstein-barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 2008, 82, 11913–11926. [Google Scholar] [CrossRef]
- Gonnella, R.; Farina, A.; Santarelli, R.; Raffa, S.; Feederle, R.; Bei, R.; Granato, M.; Modesti, A.; Frati, L.; Delecluse, H.-J.; et al. Characterization and intracellular localization of the epstein-barr virus protein BFLF2: Interactions with BFRF1 and with the nuclear lamina. J. Virol. 2005, 79, 3713–3727. [Google Scholar] [CrossRef]
- Mou, F.; Forest, T.; Baines, J.D. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 2007, 81, 6459–6470. [Google Scholar] [CrossRef]
- Bjerke, S.L.; Roller, R.J. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 2006, 347, 261–276. [Google Scholar] [CrossRef]
- Kato, A.; Yamamoto, M.; Ohno, T.; Tanaka, M.; Sata, T.; Nishiyama, Y.; Kawaguchi, Y. Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J. Virol. 2006, 80, 1476–1486. [Google Scholar] [CrossRef]
- Cano-Monreal, G.L.; Wylie, K.M.; Cao, F.; Tavis, J.E.; Morrison, L.A. Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology 2009, 392, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; Baines, J.D. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the Nuclear membrane and increased phosphorylation of lamin B. J. Virol. 2006, 80, 494–504. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Wu, S.; Pan, S.; Zhou, C.; Ma, Y.; Ru, Y.; Dong, S.; He, B.; Zhang, C.; et al. P32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J. Biol. Chem. 2014, 289, 35795–35805. [Google Scholar] [CrossRef] [PubMed]
- Leach, N.R.; Roller, R.J. Significance of host cell kinases in herpes simplex virus type 1 egress and lamin-associated protein disassembly from the nuclear lamina. Virology 2010, 406, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes simplex virus 1 induces phosphorylation and reorganization of lamin A/C through the Γ134.5 protein that facilitates nuclear egress. J. Virol. 2016, 90, 10414–10422. [Google Scholar] [CrossRef]
- Leach, N.; Bjerke, S.L.; Christensen, D.K.; Bouchard, J.M.; Mou, F.; Park, R.; Baines, J.; Haraguchi, T.; Roller, R.J. Emerin Is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J. Virol. 2007, 81, 10792–10803. [Google Scholar] [CrossRef]
- Morris, J.B.; Hofemeister, H.; O’Hare, P. Herpes simplex virus infection induces phosphorylation and delocalization of emerin, a key inner nuclear membrane protein. J. Virol. 2007, 81, 4429–4437. [Google Scholar] [CrossRef]
- Mou, F.; Wills, E.G.; Park, R.; Baines, J.D. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J. Virol. 2008, 82, 8094–8104. [Google Scholar] [CrossRef] [PubMed]
- Turan, A.; Grosche, L.; Krawczyk, A.; Mühl-Zürbes, P.; Drassner, C.; Düthorn, A.; Kummer, M.; Hasenberg, M.; Voortmann, S.; Jastrow, H.; et al. Autophagic degradation of lamins facilitates the nuclear egress of herpes simplex virus type 1. J. Cell Biol. 2019, 218, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.; Cliffe, A.; Chang, L.; Knipe, D.M. Role for A-type lamins in herpesviral DNA targeting and heterochromatin modulation. PLoS Pathog. 2008, 4, e1000071. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.; Oh, H.S.; Chang, L.; Yan, Z.; Triezenberg, S.J.; Knipe, D.M. Roles of the nuclear lamina in stable nuclear association and assembly of a herpesviral transactivator complex on viral immediate-early genes. mBio 2012, 3, e00300-11. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Vanarsdall, A.L.; Mikhailov, V.S.; Rohrmann, G.F. Conserved molecular systems of the baculoviridae. Virology 2006, 344, 77–87. [Google Scholar] [CrossRef]
- Wilson, M.E.; Price, K.H. Association of autographa californica nuclear polyhedrosis virus (AcMNPV) with the nuclear matrix. Virology 1988, 167, 233–241. [Google Scholar] [CrossRef]
- Wei, W.; Wang, H.; Li, X.; Fang, N.; Yang, S.; Liu, H.; Kang, X.; Sun, X.; Ji, S. Cloning and characterization of Sf9 cell lamin and the lamin conformational changes during autographa californica multiple nucleopolyhedrovirus infection. Viruses 2016, 8, E126. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, K.; Wei, D.; Wu, W.; Yang, K.; Yuan, M. Baculovirus Infection induces disruption of the nuclear lamina. Sci. Rep. 2017, 7, 7823. [Google Scholar] [CrossRef]
- Wei, W.; Hu, Z.; Jia, Y.; Gu, T.; Zhao, W.; Ji, S. Characterization of lamin b receptor of Sf9 cells and its fate during autographa californica nucleopolyhedrovirus infection. Cytotechnology 2020, 72, 315–325. [Google Scholar] [CrossRef]
- Cook, L. Polyomaviruses. Microbiol. Spectr. 2016, 4, 10. [Google Scholar] [CrossRef]
- Ehlers, B.; Moens, U. Genome analysis of non-human primate polyomaviruses. Infect. Genet. Evol. 2014, 26, 283–294. [Google Scholar] [CrossRef]
- Prado, J.C.M.; Monezi, T.A.; Amorim, A.T.; Lino, V.; Paladino, A.; Boccardo, E. Human polyomaviruses and cancer: An overview. Clinics 2018, 73, e558s. [Google Scholar] [CrossRef]
- Gerits, N.; Moens, U. Agnoprotein of mammalian polyomaviruses. Virology 2012, 432, 316–326. [Google Scholar] [CrossRef]
- Okada, Y.; Suzuki, T.; Sunden, Y.; Orba, Y.; Kose, S.; Imamoto, N.; Takahashi, H.; Tanaka, S.; Hall, W.W.; Nagashima, K.; et al. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: Role in nuclear egress of viral particles. EMBO Rep. 2005, 6, 452–457. [Google Scholar] [CrossRef]
- Panou, M.-M.; Prescott, E.L.; Hurdiss, D.L.; Swinscoe, G.; Hollinshead, M.; Caller, L.G.; Morgan, E.L.; Carlisle, L.; Müller, M.; Antoni, M.; et al. Agnoprotein is an essential egress factor during BK polyomavirus infection. Int. J. Mol. Sci. 2018, 19, 902. [Google Scholar] [CrossRef]
- Horníková, L.; Bruštíková, K.; Forstová, J. Microtubules in polyomavirus infection. Viruses 2020, 12, E121. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Barger, E.K.; Magnuson, B.; Tsai, B. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J. Virol. 2007, 81, 12996–13004. [Google Scholar] [CrossRef] [PubMed]
- Huérfano, S.; Ryabchenko, B.; Španielová, H.; Forstová, J. Hydrophobic domains of mouse polyomavirus minor capsid proteins promote membrane association and virus exit from the ER. FEBS J. 2017, 284, 883–902. [Google Scholar] [CrossRef]
- Soldatova, I.; Prilepskaja, T.; Abrahamyan, L.; Forstová, J.; Huérfano, S. Interaction of the mouse polyomavirus capsid proteins with importins is required for efficient import of viral DNA into the cell nucleus. Viruses 2018, 10, E165. [Google Scholar] [CrossRef]
- Butin-Israeli, V.; Ben-nun-Shaul, O.; Kopatz, I.; Adam, S.A.; Shimi, T.; Goldman, R.D.; Oppenheim, A. Simian virus 40 induces lamin A/C fluctuations and nuclear envelope deformation during cell entry. Nucleus 2011, 2, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Black, P.H.; Crawford, E.M.; Crawford, L.V. The purification of simian virus 40. Virology 1964, 24, 381–387. [Google Scholar] [CrossRef]
- Ye, Q.; Worman, H.J. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to drosophila HP1. J. Biol. Chem. 1996, 271, 14653–14656. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Callebaut, I.; Pezhman, A.; Courvalin, J.C.; Worman, H.J. Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J. Biol. Chem. 1997, 272, 14983–14989. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Chipman, P.R.; Agbandje-McKenna, M.; Kajigaya, S.; Brown, K.E.; Young, N.S.; Baker, T.S.; Rossmann, M.G. Cryo-electron microscopy studies of empty capsids of human parvovirus B19 complexed with its cellular receptor. Proc. Natl. Acad. Sci. USA 1996, 93, 7502–7506. [Google Scholar] [CrossRef]
- Summerford, C.; Samulski, R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 Virions. J. Virol. 1998, 72, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small does not mean simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Qing, K.; Srivastava, A. Infection of purified nuclei by adeno-associated virus 2. Mol. Ther. 2001, 4, 289–296. [Google Scholar] [CrossRef]
- Cohen, S.; Panté, N. Pushing the envelope: Microinjection of minute virus of mice into xenopus oocytes causes damage to the nuclear envelope. J. Gen. Virol. 2005, 86, 3243–3252. [Google Scholar] [CrossRef]
- Cohen, S.; Behzad, A.R.; Carroll, J.B.; Panté, N. Parvoviral nuclear import: Bypassing the host nuclear-transport machinery. J. Gen. Virol. 2006, 87, 3209–3213. [Google Scholar] [CrossRef]
- Cohen, S.; Marr, A.K.; Garcin, P.; Panté, N. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J. Virol. 2011, 85, 4863–4874. [Google Scholar] [CrossRef] [PubMed]
- Porwal, M.; Cohen, S.; Snoussi, K.; Popa-Wagner, R.; Anderson, F.; Dugot-Senant, N.; Wodrich, H.; Dinsart, C.; Kleinschmidt, J.A.; Panté, N.; et al. Parvoviruses cause nuclear envelope breakdown by activating key enzymes of mitosis. PLoS Pathog. 2013, 9, e1003671. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Cao, C.X.; Shao, R.G.; Pommier, Y. Lamin B phosphorylation by protein kinase calpha and proteolysis during apoptosis in human leukemia HL60 cells. J. Biol. Chem. 1998, 273, 8669–8674. [Google Scholar] [CrossRef]
- Jin, Y.H.; Yoo, K.J.; Lee, Y.H.; Lee, S.K. Caspase 3-mediated cleavage of P21WAF1/CIP1 associated with the cyclin A-cyclin-dependent kinase 2 complex is a prerequisite for apoptosis in SK-HEP-1 cells. J. Biol. Chem. 2000, 275, 30256–30263. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Mitra, J.; van den Heuvel, S.; Enders, G.H. S and G2 phase roles for Cdk2 revealed by inducible expression of a dominant-negative mutant in human cells. Mol. Cell Biol. 2001, 21, 2755–2766. [Google Scholar] [CrossRef]
- Mäntylä, E.; Niskanen, E.A.; Ihalainen, T.O.; Vihinen-Ranta, M. Reorganization of nuclear pore complexes and the lamina in late-stage parvovirus infection. J. Virol. 2015, 89, 11706–11710. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tischer, I.; Gelderblom, H.; Vettermann, W.; Koch, M.A. A very small porcine virus with circular single-stranded DNA. Nature 1982, 295, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Delwart, E.; Rosario, K.; Segalés, J.; Varsani, A. ICTV report consortium, null ICTV Virus Taxonomy Profile: Circoviridae. J. Gen. Virol. 2017, 98, 1997–1998. [Google Scholar] [CrossRef]
- Biagini, P. Human circoviruses. Vet. Microbiol. 2004, 98, 95–101. [Google Scholar] [CrossRef]
- Finsterbusch, T.; Mankertz, A. Porcine circoviruses—Small but powerful. Virus Res. 2009, 143, 177–183. [Google Scholar] [CrossRef]
- Finsterbusch, T.; Steinfeldt, T.; Doberstein, K.; Rödner, C.; Mankertz, A. Interaction of the replication proteins and the capsid protein of porcine circovirus type 1 and 2 with host proteins. Virology 2009, 386, 122–131. [Google Scholar] [CrossRef]
- Soltys, B.J.; Kang, D.; Gupta, R.S. Localization of P32 protein (GC1q-R) in mitochondria and at specific extramitochondrial locations in normal tissues. Histochem. Cell Biol. 2000, 114, 245–255. [Google Scholar] [CrossRef]
- Wang, T.; Du, Q.; Niu, Y.; Zhang, X.; Wang, Z.; Wu, X.; Yang, X.; Zhao, X.; Liu, S.-L.; Tong, D.; et al. Cellular P32 is a critical regulator of porcine circovirus type 2 nuclear egress. J. Virol. 2019, 93, e00979-19. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.J. Intranuclear fibrillar bodies in actinomycin D-treated oocytes. J. Cell Biol. 1969, 40, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Douvas, A.S.; Harrington, C.A.; Bonner, J. Major nonhistone proteins of rat liver chromatin: Preliminary identification of myosin, actin, tubulin, and tropomyosin. Proc. Natl. Acad. Sci. USA 1975, 72, 3902–3906. [Google Scholar] [CrossRef] [PubMed]
- Fukui, Y.; Katsumaru, H. Nuclear actin bundles in amoeba, dictyostelium and human HeLa cells induced by dimethyl sulfoxide. Exp. Cell Res. 1979, 120, 451–455. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, W.; Rando, O.J.; Xue, Y.; Swiderek, K.; Kuo, A.; Crabtree, G.R. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell 1998, 95, 625–636. [Google Scholar] [CrossRef]
- Xie, X.; Almuzzaini, B.; Drou, N.; Kremb, S.; Yousif, A.; Farrants, A.-K.Ö.; Gunsalus, K.; Percipalle, P. β-actin-dependent global chromatin organization and gene expression programs control cellular identity. FASEB J. 2018, 32, 1296–1314. [Google Scholar] [CrossRef]
- Almuzzaini, B.; Sarshad, A.A.; Farrants, A.-K.Ö.; Percipalle, P. Nuclear myosin 1 contributes to a chromatin landscape compatible with RNA polymerase II transcription activation. BMC Biol. 2015, 13, 35. [Google Scholar] [CrossRef]
- Plessner, M.; Melak, M.; Chinchilla, P.; Baarlink, C.; Grosse, R. Nuclear F-actin formation and reorganization upon cell spreading. J. Biol. Chem. 2015, 290, 11209–11216. [Google Scholar] [CrossRef]
- Gonsior, S.M.; Platz, S.; Buchmeier, S.; Scheer, U.; Jockusch, B.M.; Hinssen, H. Conformational difference between nuclear and cytoplasmic actin as detected by a monoclonal antibody. J. Cell Sci. 1999, 112, 797–809. [Google Scholar] [CrossRef]
- Schoenenberger, C.-A.; Buchmeier, S.; Boerries, M.; Sütterlin, R.; Aebi, U.; Jockusch, B.M. Conformation-specific antibodies reveal distinct actin structures in the nucleus and the cytoplasm. J. Struct Biol. 2005, 152, 157–168. [Google Scholar] [CrossRef]
- Dopie, J.; Skarp, K.-P.; Rajakylä, E.K.; Tanhuanpää, K.; Vartiainen, M.K. Active maintenance of nuclear actin by importin 9 supports transcription. Proc. Natl. Acad. Sci. USA 2012, 109, E544–E552. [Google Scholar] [CrossRef] [PubMed]
- Stüven, T.; Hartmann, E.; Görlich, D. Exportin 6: A novel nuclear export receptor that is specific for profilin·actin complexes. EMBO J. 2003, 22, 5928–5940. [Google Scholar] [CrossRef]
- Kapoor, P.; Shen, X. Mechanisms of nuclear actin in chromatin-remodeling complexes. Trends Cell Biol. 2014, 24, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Serebryannyy, L.; de Lanerolle, P. Nuclear actin: The new normal. Mutat. Res. 2020, 821, 111714. [Google Scholar] [CrossRef] [PubMed]
- Egly, J.M.; Miyamoto, N.G.; Moncollin, V.; Chambon, P. Is actin a transcription initiation factor for RNA polymerase B? EMBO J. 1984, 3, 2363–2371. [Google Scholar] [CrossRef]
- Scheer, U.; Hinssen, H.; Franke, W.W.; Jockusch, B.M. Microinjection of actin-binding proteins and actin antibodies demonstrates involvement of nuclear actin in transcription of lampbrush chromosomes. Cell 1984, 39, 111–122. [Google Scholar] [CrossRef]
- Percipalle, P.; Zhao, J.; Pope, B.; Weeds, A.; Lindberg, U.; Daneholt, B. Actin bound to the heterogeneous nuclear ribonucleoprotein Hrp36 is associated with balbiani ring MRNA from the gene to polysomes. J. Cell Biol. 2001, 153, 229–236. [Google Scholar] [CrossRef]
- Percipalle, P.; Jonsson, A.; Nashchekin, D.; Karlsson, C.; Bergman, T.; Guialis, A.; Daneholt, B. Nuclear actin is associated with a specific subset of HnRNP A/B-type proteins. Nucleic Acids Res. 2002, 30, 1725–1734. [Google Scholar] [CrossRef]
- Hofmann, W.A.; Stojiljkovic, L.; Fuchsova, B.; Vargas, G.M.; Mavrommatis, E.; Philimonenko, V.; Kysela, K.; Goodrich, J.A.; Lessard, J.L.; Hope, T.J.; et al. Actin is part of pre-initiation complexes and is necessary for transcription by RNA polymerase II. Nat. Cell Biol. 2004, 6, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Wu, S.; Hernandez, N. A role for beta-actin in RNA polymerase III transcription. Genes Dev. 2004, 18, 3010–3015. [Google Scholar] [CrossRef] [PubMed]
- Philimonenko, V.V.; Zhao, J.; Iben, S.; Dingová, H.; Kyselá, K.; Kahle, M.; Zentgraf, H.; Hofmann, W.A.; de Lanerolle, P.; Hozák, P.; et al. Nuclear actin and myosin I are required for RNA polymerase I transcription. Nat. Cell Biol. 2004, 6, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yoo, Y.; Okuhama, N.N.; Tucker, P.W.; Liu, G.; Guan, J.-L. Regulation of RNA-polymerase-II-dependent transcription by N-WASP and its nuclear-binding partners. Nat. Cell Biol. 2006, 8, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, L.; Nourani, A.; Boudreault, A.A.; Zhang, Y.; Héliot, L.; Allard, S.; Savard, J.; Lane, W.S.; Stillman, D.J.; Côté, J. Multiple links between the NuA4 histone acetyltransferase complex and epigenetic control of transcription. Mol. Cell 2000, 5, 927–937. [Google Scholar] [CrossRef]
- Shen, X.; Mizuguchi, G.; Hamiche, A.; Wu, C. A chromatin remodelling complex involved in transcription and DNA processing. Nature 2000, 406, 541–544. [Google Scholar] [CrossRef]
- Fenn, S.; Breitsprecher, D.; Gerhold, C.B.; Witte, G.; Faix, J.; Hopfner, K.-P. Structural biochemistry of nuclear actin-related proteins 4 and 8 reveals their interaction with actin. EMBO J. 2011, 30, 2153–2166. [Google Scholar] [CrossRef]
- Kapoor, P.; Chen, M.; Winkler, D.D.; Luger, K.; Shen, X. Evidence for monomeric actin function in INO80 chromatin remodeling. Nat. Struct. Mol. Biol. 2013, 20, 426–432. [Google Scholar] [CrossRef]
- Serebryannyy, L.A.; Cruz, C.M.; de Lanerolle, P. A role for nuclear actin in HDAC 1 and 2 regulation. Sci. Rep. 2016, 6, 28460. [Google Scholar] [CrossRef]
- Oza, P.; Jaspersen, S.L.; Miele, A.; Dekker, J.; Peterson, C.L. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 2009, 23, 912–927. [Google Scholar] [CrossRef]
- Andrin, C.; McDonald, D.; Attwood, K.M.; Rodrigue, A.; Ghosh, S.; Mirzayans, R.; Masson, J.-Y.; Dellaire, G.; Hendzel, M.J. A requirement for polymerized actin in DNA double-strand break repair. Nucleus 2012, 3, 384–395. [Google Scholar] [CrossRef]
- Schrank, B.R.; Aparicio, T.; Li, Y.; Chang, W.; Chait, B.T.; Gundersen, G.G.; Gottesman, M.E.; Gautier, J. Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 2018, 559, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Caridi, C.P.; D’Agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and spatial uncoupling of DNA double strand break repair pathways within mammalian heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Zastrow, M.S.; Flaherty, D.B.; Benian, G.M.; Wilson, K.L. Nuclear titin interacts with A- and B-type lamins in vitro and in vivo. J. Cell Sci. 2006, 119, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar] [CrossRef]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef]
- Mislow, J.M.K.; Kim, M.S.; Davis, D.B.; McNally, E.M. Myne-1, a spectrin repeat transmembrane protein of the myocyte inner nuclear membrane, interacts with lamin A/C. J. Cell Sci. 2002, 115, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Holaska, J.M.; Wilson, K.L. An emerin “proteome”: Purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, MRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 2007, 46, 8897–8908. [Google Scholar] [CrossRef]
- Lammerding, J.; Hsiao, J.; Schulze, P.C.; Kozlov, S.; Stewart, C.L.; Lee, R.T. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J. Cell Biol. 2005, 170, 781–791. [Google Scholar] [CrossRef]
- Charlton, C.A.; Volkman, L.E. Sequential rearrangement and nuclear polymerization of actin in baculovirus-infected spodoptera frugiperda cells. J. Virol. 1991, 65, 1219–1227. [Google Scholar] [CrossRef]
- Newsome, T.P.; Marzook, N.B. Viruses that ride on the coat-tails of actin nucleation. Semin. Cell Dev. Biol. 2015, 46, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Dowd, G.C.; Mortuza, R.; Ireton, K. Molecular mechanisms of intercellular dissemination of bacterial pathogens. Trends Microbiol. 2021, 29, 127–141. [Google Scholar] [CrossRef]
- Ohkawa, T.; Volkman, L.E.; Welch, M.D. Actin-based motility drives baculovirus transit to the nucleus and cell surface. J. Cell Biol. 2010, 190, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Au, S.; Wu, W.; Zhou, L.; Theilmann, D.A.; Panté, N. A new mechanism for nuclear import by actin-based propulsion used by a baculovirus nucleocapsid. J. Cell Sci. 2016, 129, 2905–2911. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, T.; Rowe, A.R.; Volkman, L.E. Identification of six autographa californica multicapsid nucleopolyhedrovirus early genes that mediate nuclear localization of G-actin. J. Virol. 2002, 76, 12281–12289. [Google Scholar] [CrossRef]
- Ohkawa, T.; Volkman, L.E. Nuclear F-actin is required for AcMNPV nucleocapsid morphogenesis. Virology 1999, 264, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Goley, E.D.; Ohkawa, T.; Mancuso, J.; Woodruff, J.B.; D’Alessio, J.A.; Cande, W.Z.; Volkman, L.E.; Welch, M.D. Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-like protein. Science 2006, 314, 464–467. [Google Scholar] [CrossRef]
- Volkman, L.E.; Goldsmith, P.A.; Hess, R.T. Evidence for microfilament involvement in budded autographa californica nuclear polyhedrosis virus production. Virology 1987, 156, 32–39. [Google Scholar] [CrossRef]
- Ohkawa, T.; Welch, M.D. Baculovirus actin-based motility drives nuclear envelope disruption and nuclear egress. Curr. Biol. 2018, 28, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Zhang, Y.; Hu, Y.; Hu, X.; Zhou, Y.; Chen, X.; Wang, Y. The role of viral protein Ac34 in nuclear relocation of subunits of the actin-related protein 2/3 complex. Virol. Sin. 2016, 31, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Y.; Hou, D.; Guan, Z.; Shen, S.; Peng, K.; Deng, F.; Chen, X.; Hu, Z.; Wang, H.; et al. Host factor heat-shock protein 90 contributes to baculovirus budded virus morphogenesis via facilitating nuclear actin polymerization. Virology 2019, 535, 200–209. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Zhao, S.; Wu, X. Identification of A functional region in bombyx mori nucleopolyhedrovirus VP39 that is essential for nuclear actin polymerization. Virology 2020, 550, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Q.; Liang, C.; Song, J.; Li, N.; Shi, H.; Chen, X. Autographa californica multiple nucleopolyhedrovirus nucleocapsid protein BV/ODV-C42 mediates the nuclear entry of P78/83. J. Virol. 2008, 82, 4554–4561. [Google Scholar] [CrossRef]
- Marek, M.; Merten, O.-W.; Galibert, L.; Vlak, J.M.; van Oers, M.M. Baculovirus VP80 protein and the F-actin cytoskeleton interact and connect the viral replication factory with the nuclear periphery. J. Virol. 2011, 85, 5350–5362. [Google Scholar] [CrossRef]
- Guan, Z.; Zhong, L.; Li, C.; Wu, W.; Yuan, M.; Yang, K. The autographa californica multiple nucleopolyhedrovirus Ac54 gene is crucial for localization of the major capsid protein VP39 at the site of nucleocapsid assembly. J. Virol. 2016, 90, 4115–4126. [Google Scholar] [CrossRef]
- Mu, J.; Zhang, Y.; Hu, Y.; Hu, X.; Zhou, Y.; Zhao, H.; Pei, R.; Wu, C.; Chen, J.; Zhao, H.; et al. Autographa californica multiple nucleopolyhedrovirus Ac34 protein retains cellular actin-related protein 2/3 complex in the nucleus by subversion of CRM1-dependent nuclear export. PLoS Pathog. 2016, 12, e1005994. [Google Scholar] [CrossRef]
- Gandhi, K.M.; Ohkawa, T.; Welch, M.D.; Volkman, L.E. Nuclear localization of actin requires AC102 in autographa californica multiple nucleopolyhedrovirus-infected cells. J. Gen. Virol. 2012, 93, 1795–1803. [Google Scholar] [CrossRef]
- Hepp, S.E.; Borgo, G.M.; Ticau, S.; Ohkawa, T.; Welch, M.D. Baculovirus AC102 is a nucleocapsid protein that is crucial for nuclear actin polymerization and nucleocapsid morphogenesis. J. Virol. 2018, 92, e00111-18. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, X.; Mu, J.; Hu, Y.; Zhou, Y.; Zhao, H.; Wu, C.; Pei, R.; Chen, J.; Chen, X.; et al. Ac102 participates in nuclear actin polymerization by modulating BV/ODV-C42 ubiquitination during autographa californica multiple nucleopolyhedrovirus infection. J. Virol. 2018, 92, e00005-18. [Google Scholar] [CrossRef] [PubMed]
- Feierbach, B.; Piccinotti, S.; Bisher, M.; Denk, W.; Enquist, L.W. Alpha-herpesvirus infection induces the formation of nuclear actin filaments. PLoS Pathog. 2006, 2, e85. [Google Scholar] [CrossRef]
- Forest, T.; Barnard, S.; Baines, J.D. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 2005, 7, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, A.R.; Lawler, J.L.; Coen, D.M. A role for nuclear F-actin induction in human cytomegalovirus nuclear egress. mBio 2016, 7, e01254-16. [Google Scholar] [CrossRef]
- Wilkie, A.R.; Sharma, M.; Pesola, J.M.; Ericsson, M.; Fernandez, R.; Coen, D.M. A role for myosin Va in human cytomegalovirus nuclear egress. J. Virol. 2018, 92, e01849-17. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Godinez, W.J.; Kim, I.-H.; Tektonidis, M.; de Lanerolle, P.; Eils, R.; Rohr, K.; Knipe, D.M. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late MRNA. Proc. Natl. Acad. Sci. USA 2011, 108, E136–E144. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.B.; Virding, S.; Thiberge, S.Y.; Scherer, J.; Wodrich, H.; Ruzsics, Z.; Koszinowski, U.H.; Enquist, L.W. Nuclear herpesvirus capsid motility is not dependent on F-actin. mBio 2014, 5, e01909–e01914. [Google Scholar] [CrossRef]
- Bosse, J.B.; Hogue, I.B.; Feric, M.; Thiberge, S.Y.; Sodeik, B.; Brangwynne, C.P.; Enquist, L.W. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc. Natl. Acad. Sci. USA 2015, 112, E5725–E5733. [Google Scholar] [CrossRef]
- Fuchsova, B.; Serebryannyy, L.A.; de Lanerolle, P. Nuclear actin and myosins in adenovirus infection. Exp. Cell Res. 2015, 338, 170–182. [Google Scholar] [CrossRef]
- Sankovski, E.; Abroi, A.; Ustav, M.; Ustav, M. Nuclear myosin 1 associates with papillomavirus E2 regulatory protein and influences viral replication. Virology 2018, 514, 142–155. [Google Scholar] [CrossRef]
- Kimura, T.; Hashimoto, I.; Yamamoto, A.; Nishikawa, M.; Fujisawa, J.I. Rev-dependent association of the intron-containing HIV-1 gag MRNA with the nuclear actin bundles and the inhibition of its nucleocytoplasmic transport by latrunculin-B. Genes Cells 2000, 5, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.; Reichart, B.; Ewald, A.; Müller, E.; Schmitt, I.; Stauber, R.H.; Lottspeich, F.; Jockusch, B.M.; Scheer, U.; Hauber, J.; et al. Cofactor requirements for nuclear export of rev response element (RRE)- and constitutive transport element (CTE)-containing retroviral RNAs. An unexpected role for actin. J. Cell Biol. 2001, 152, 895–910. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Modification | Protein(s) Responsible for Modification | Reference |
|---|---|---|
| lamins phosphorylation | UL97 (HCMV) | [31,35,39] |
| US3 (HSV-1) | [51,52] | |
| UL13 (HSV-2) | [54] | |
| BGLF4 (EBV) | [49] | |
| PKC | [29,36,55,56,93,103] | |
| cdk-1 | [95] | |
| lamins A/C cleavage | caspase-6 | [80] |
| caspase-3 | [91] | |
| conformation change of lamin A/C leading to its disassembly | Pin-1 | [40] |
| lamin B1 acetylation | unknown (HCMV) | [43] |
| emerin phosphorylation | PKC | [59,60] |
| unknown (KSV) | [48] | |
| p32 phosphorylation | UL97 (HCMV) | [29] |
| UL34 phosphorylation (HSV-1) | US3 (HSV-1) | [44] |
| US3 phosphorylation (HSV-1) | UL13 (HSV-1) | [53] |
| UL50 (HCMV) | PKC | [37] |
| Cdk2 phosphorylation | PKC, caspase-3 | [94] |
| Cdk1 phosphorylation | cdk2 | [95] |
| PKC phosphorylation | phospholipase C, ERK and JNK kinases | [103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horníková, L.; Bruštíková, K.; Huérfano, S.; Forstová, J. Nuclear Cytoskeleton in Virus Infection. Int. J. Mol. Sci. 2022, 23, 578. https://doi.org/10.3390/ijms23010578
Horníková L, Bruštíková K, Huérfano S, Forstová J. Nuclear Cytoskeleton in Virus Infection. International Journal of Molecular Sciences. 2022; 23(1):578. https://doi.org/10.3390/ijms23010578
Chicago/Turabian StyleHorníková, Lenka, Kateřina Bruštíková, Sandra Huérfano, and Jitka Forstová. 2022. "Nuclear Cytoskeleton in Virus Infection" International Journal of Molecular Sciences 23, no. 1: 578. https://doi.org/10.3390/ijms23010578
APA StyleHorníková, L., Bruštíková, K., Huérfano, S., & Forstová, J. (2022). Nuclear Cytoskeleton in Virus Infection. International Journal of Molecular Sciences, 23(1), 578. https://doi.org/10.3390/ijms23010578