
Regulation of Heterogenous LexA Expression in Staphylococcus aureus by an Antisense RNA Originating from Transcriptional Read-Through upon Natural Mispairings in the sbrB Intrinsic Terminator

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The lexA Antisense RNA Originates from a Transcriptional Read-Through Event of an Upstream Terminator
2.2. Alkaline Stress Increases lexA-asRNA Expression through SigB Activation
2.3. Translation of SbpB Does Not Affect the Transcriptional Read-Through of TTsbrB
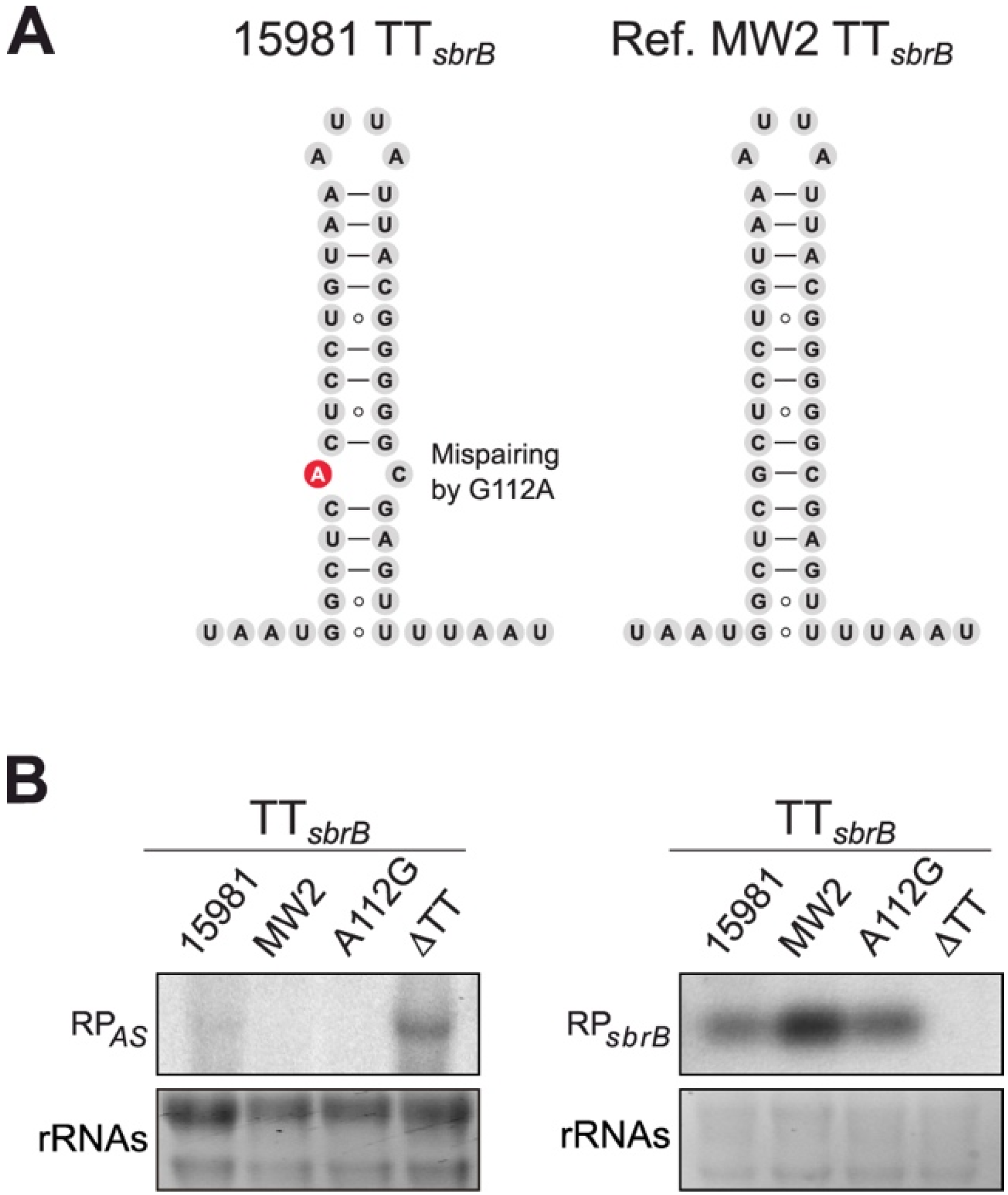
2.4. A Single Nucleotide Change (G112A) in TTsbrB of S. aureus Is Responsible for Its Transcriptional Read-Through
2.5. Variations in the TTsbrB Sequence in Other Staphylococcus Strains Produce Different TTsbrB Read-Through Levels
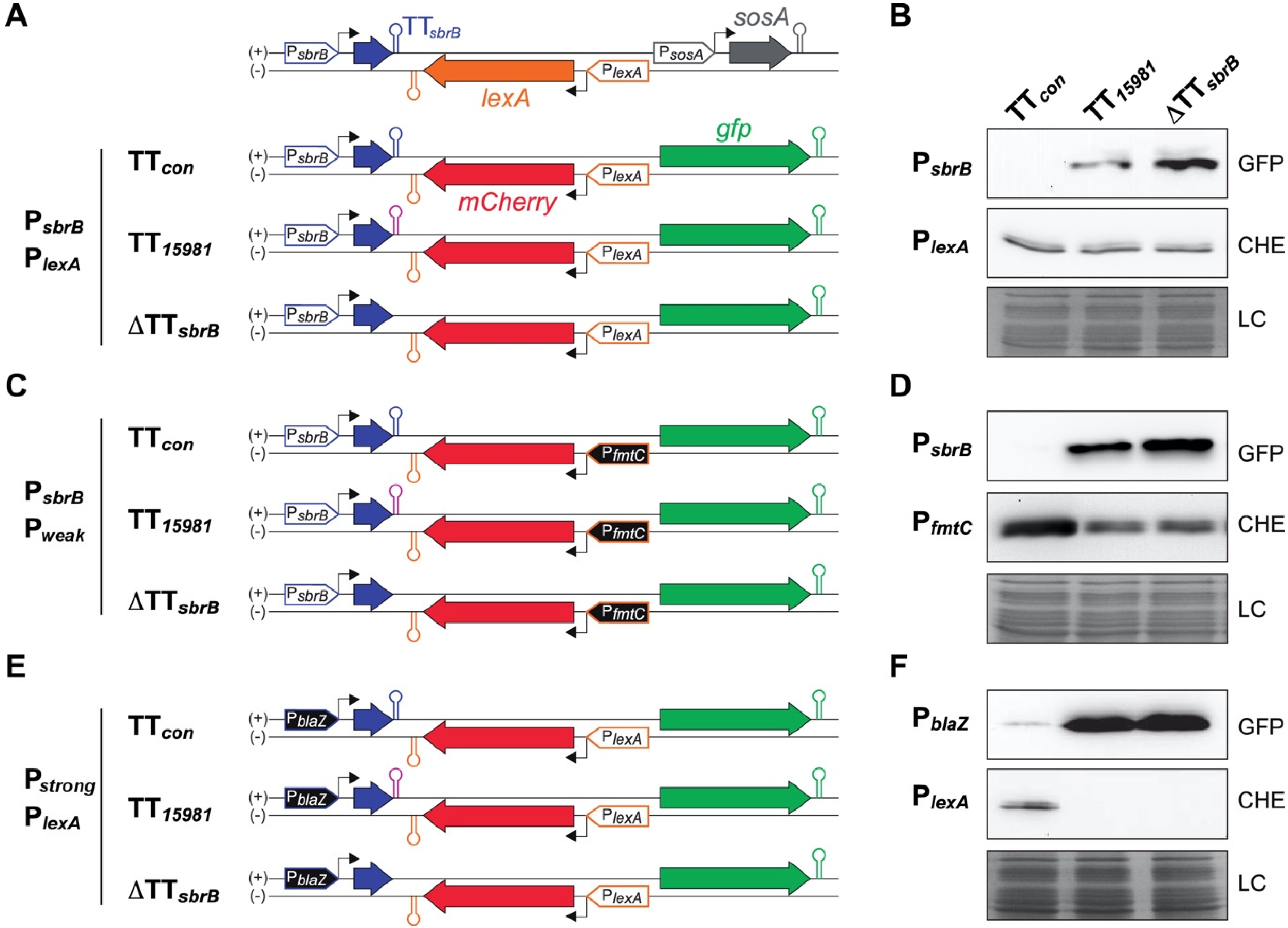
2.6. The asRNA/mRNA Ratio Drives the LexA Reporter Expression
2.7. Heterogeneity on the LexA Reporter Expression Is Reduced by lexA-asRNA
2.8. Mispairing Nucleotides in Intrinsic Terminators Transcriptionally Connect Contiguous Genes
3. Discussion
4. Materials and Methods
4.1. Strains, Plasmids, Oligonucleotides and Growth Conditions
4.2. Generation of Chromosomal Mutants by Homologous Recombination
4.3. RNA Extraction and Northern Blotting
4.4. Simultaneous Mapping of the 5′ and 3′ Ends (mRACE)
4.5. Plasmid Construction
4.6. Bacterial Cultures for Total Protein Extraction and Western Blotting
4.7. Time-Lapse Fluorescence Microscopy
4.8. Determination of Transcriptional Read-Through on Predicted TTs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution-Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef]
- Georg, J.; Hess, W.R. Widespread antisense transcription in prokaryotes. Microbiol. Spectr. 2018, 6, 1–20. [Google Scholar] [CrossRef]
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiss, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermüller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255. [Google Scholar] [CrossRef]
- Lasa, I.; Toledo-Arana, A.; Dobin, A.; Villanueva, M.; de los Mozos, I.R.; Vergara-Irigaray, M.; Segura, V.; Fagegaltier, D.; Penadés, J.R.; Valle, J.; et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 20172–20177. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Förstner, K.U.; Cong, J.P.; Sharma, C.M.; Bassler, B.L. Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc. Natl. Acad. Sci. USA 2015, 112, E766–E775. [Google Scholar] [CrossRef] [PubMed]
- Thomason, M.K.; Bischler, T.; Eisenbart, S.K.; Förstner, K.U.; Zhang, A.; Herbig, A.; Nieselt, K.; Sharma, C.M.; Storz, G. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J. Bacteriol. 2015, 197, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Sáenz-Lahoya, S.; Bitarte, N.; Garcia, B.; Burgui, S.; Vergara-Irigaray, M.; Valle, J.; Solano, C.; Toledo-Arana, A.; Lasa, I. Noncontiguous operon is a genetic organization for coordinating bacterial gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Arana, A.; Lasa, I. Advances in bacterial transcriptome understanding: From overlapping transcription to the excludon concept. Mol. Microbiol. 2020, 113, 593–602. [Google Scholar] [CrossRef]
- de Los Mozos, I.R.; Vergara-Irigaray, M.; Segura, V.; Villanueva, M.; Bitarte, N.; Saramago, M.; Domingues, S.; Arraiano, C.M.; Fechter, P.; Romby, P.; et al. Base pairing interaction between 5′- and 3′-UTRs controls icaR mRNA translation in Staphylococcus aureus. PLoS Genet. 2013, 9, e1004001. [Google Scholar] [CrossRef]
- Wade, J.T.; Grainger, D.C. Pervasive transcription: Illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014, 12, 647–653. [Google Scholar] [CrossRef]
- Schultze, T.; Izar, B.; Qing, X.; Mannala, G.K.; Hain, T. Current status of antisense RNA-mediated gene regulation in Listeria monocytogenes. Front. Cell. Infect. Microbiol. 2014, 4, 135. [Google Scholar] [CrossRef] [PubMed]
- Mars, R.A.T.; Nicolas, P.; Denham, E.L.; van Dijl, J.M. Regulatory RNAs in Bacillus subtilis: A Gram-Positive Perspective on Bacterial RNA-Mediated Regulation of Gene Expression. Microbiol. Mol. Biol. Rev. 2016, 80, 1029–1057. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, P.; Mäder, U.; Dervyn, E.; Rochat, T.; Leduc, A.; Pigeonneau, N.; Bidnenko, E.; Marchadier, E.; Hoebeke, M.; Aymerich, S.; et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 2012, 335, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Lybecker, M.; Zimmermann, B.; Bilusic, I.; Tukhtubaeva, N.; Schroeder, R. The double-stranded transcriptome of Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, 3134–3139. [Google Scholar] [CrossRef] [PubMed]
- Bidnenko, V.; Nicolas, P.; Grylak-Mielnicka, A.; Delumeau, O.; Auger, S.; Aucouturier, A.; Guérin, C.; Francis, R.; Bardowski, J.; Aymerich, S.; et al. Termination factor Rho: From the control of pervasive transcription to cell fate determination in Bacillus subtilis. PLoS Genet. 2017, 13, e1006909. [Google Scholar] [CrossRef] [PubMed]
- Bidnenko, E.; Bidnenko, V. Transcription termination factor Rho and microbial phenotypic heterogeneity. Curr. Genet. 2018, 64, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Vangeloff, A.D.; Landick, R. Bacterial transcription terminators: The RNA 3′-end chronicles. J. Mol. Biol. 2011, 412, 793–813. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Yakhnin, A.V.; Sebastian, A.; Albert, I.; Babitzke, P. NusA-dependent transcription termination prevents misregulation of global gene expression. Nat. Microbiol. 2016, 1, 15007. [Google Scholar] [CrossRef]
- Hao, Z.; Epshtein, V.; Kim, K.H.; Proshkin, S.; Svetlov, V.; Kamarthapu, V.; Bharati, B.; Mironov, A.; Walz, T.; Nudler, E. Pre-termination Transcription Complex: Structure and Function. Mol. Cell 2021, 81, 281–292.e8. [Google Scholar] [CrossRef]
- Said, N.; Hilal, T.; Sunday, N.D.; Khatri, A.; Bürger, J.; Mielke, T.; Belogurov, G.A.; Loll, B.; Sen, R.; Artsimovitch, I.; et al. Steps toward translocation-independent RNA polymerase inactivation by terminator ATPase p. Science 2021, 371, eabd1673. [Google Scholar] [CrossRef]
- Hao, Z.; Svetlov, V.; Nudler, E. Rho-dependent transcription termination: A revisionist view. Transcription 2021, 12, 171–181. [Google Scholar] [CrossRef]
- Mäder, U.; Nicolas, P.; Depke, M.; Pane-Farre, J.; Débarbouillé, M.; van der Kooi-Pol, M.M.; Guérin, C.; Dérozier, S.; Hiron, A.; Jarmer, H.; et al. Staphylococcus aureus Transcriptome Architecture: From Laboratory to Infection-Mimicking Conditions. PLoS Genet. 2016, 12, e1005962-32. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Mooney, R.A.; Grass, J.A.; Jessen, E.D.; Tran, F.; Landick, R. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev. 2012, 26, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Botella, L.; Vaubourgeix, J.; Livny, J.; Schnappinger, D. Depleting Mycobacterium tuberculosis of the transcription termination factor Rho causes pervasive transcription and rapid death. Nat. Commun. 2017, 8, 14731. [Google Scholar] [CrossRef] [PubMed]
- Lasa, I.; Toledo-Arana, A.; Gingeras, T.R. An effort to make sense of antisense transcription in bacteria. RNA Biol. 2012, 9, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Sesto, N.; Wurtzel, O.; Archambaud, C.; Sorek, R.; Cossart, P. The excludon: A new concept in bacterial antisense RNA-mediated gene regulation. Nat. Rev. Microbiol. 2012, 11, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Butala, M.; Zgur-Bertok, D.; Busby, S.J.W. The bacterial LexA transcriptional repressor. Cell. Mol. Life Sci. 2009, 66, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.S.; Christiansen, M.H.G.; Bonde, M.; Gottschalk, S.; Frees, D.; Thomsen, L.E.; Kallipolitis, B.H. Searching for small σB-regulated genes in Staphylococcus aureus. Arch. Microbiol. 2010, 193, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherr, L.; Bischoff, M.; Lattar, S.M.; Noto Llana, M.; Pförtner, H.; Niemann, S.; Geraci, J.; Van de Vyver, H.; Fraunholz, M.J.; Cheung, A.L.; et al. Sigma factor SigB is crucial to mediate Staphylococcus aureus adaptation during chronic infections. PLoS Pathog. 2015, 11, e1004870. [Google Scholar] [CrossRef]
- Koch, G.; Yepes, A.; Förstner, K.U.; Wermser, C.; Stengel, S.T.; Modamio, J.; Ohlsen, K.; Foster, K.R.; Lopez, D. Evolution of resistance to a last-resort antibiotic in Staphylococcus aureus via bacterial competition. Cell 2014, 158, 1060–1071. [Google Scholar] [CrossRef]
- Britton, R.A.; Wen, T.; Schaefer, L.; Pellegrini, O.; Uicker, W.C.; Mathy, N.; Tobin, C.; Daou, R.; Szyk, J.; Condon, C. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol. Microbiol. 2007, 63, 127–138. [Google Scholar] [CrossRef]
- Naville, M.; Gautheret, D. Transcription attenuation in bacteria: Theme and variations. Brief. Funct. Genom. Proteom. 2010, 9, 178–189. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaiser, J.C.; King, A.N.; Grigg, J.C.; Sheldon, J.R.; Edgell, D.R.; Murphy, M.E.P.; Brinsmade, S.R.; Heinrichs, D.E. Repression of branched-chain amino acid synthesis in Staphylococcus aureus is mediated by isoleucine via CodY, and by a leucine-rich attenuator peptide. PLoS Genet. 2018, 14, e1007159. [Google Scholar] [CrossRef]
- Orr, M.W.; Mao, Y.; Storz, G.; Qian, S.-B. Alternative ORFs and small ORFs: Shedding light on the dark proteome. Nucleic Acids Res. 2021, 48, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Andre, G.; Even, S.; Putzer, H.; Burguiere, P.; Croux, C.; Danchin, A.; Martin-Verstraete, I.; Soutourina, O. S-box and T-box riboswitches and antisense RNA control a sulfur metabolic operon of Clostridium acetobutylicum. Nucleic Acids Res. 2008, 36, 5955–5969. [Google Scholar] [CrossRef] [PubMed]
- Mellin, J.R.; Tiensuu, T.; Bécavin, C.; Gouin, E.; Johansson, J.; Cossart, P. A riboswitch-regulated antisense RNA in Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 2013, 110, 13132–13137. [Google Scholar] [CrossRef]
- Cheng, S.W.C.; Lynch, E.C.; Leason, K.R.; Court, D.L.; Shapiro, B.A.; Friedman, D.I. Functional importance of sequence in the stem-loop of a transcription terminator. Science 1991, 254, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Cambray, G.; Guimaraes, J.C.; Mutalik, V.K.; Lam, C.; Mai, Q.-A.; Thimmaiah, T.; Carothers, J.M.; Arkin, A.P.; Endy, D. Measurement and modeling of intrinsic transcription terminators. Nucleic Acids Res. 2013, 41, 5139–5148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Liu, P.; Nielsen, A.A.K.; Brophy, J.A.N.; Clancy, K.; Peterson, T.; Voigt, C.A. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 2013, 10, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Safina, K.R.; Mironov, A.A.; Bazykin, G.A. Compensatory Evolution of Intrinsic Transcription Terminators in Bacillus cereus. Genome Biol. Evol. 2017, 9, 340–349. [Google Scholar] [CrossRef][Green Version]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Gil, P.; Caballero, C.J.; Catalan-Moreno, A.; Irurzun, N.; Barrio-Hernandez, I.; Caldelari, I.; Toledo-Arana, A. Differential evolution in 3′UTRs leads to specific gene expression in Staphylococcus. Nucleic Acids Res. 2020, 48, 2544–2563. [Google Scholar] [CrossRef] [PubMed]
- Kamenšek, S.; Podlesek, Z.; Gillor, O.; Žgur-Bertok, D. Genes regulated by the Escherichia coli SOS repressor LexA exhibit heterogenous expression. BMC Microbiol. 2010, 10, 283. [Google Scholar] [CrossRef]
- Helfrich, S.; Pfeifer, E.; Krämer, C.; Sachs, C.C.; Wiechert, W.; Kohlheyer, D.; Nöh, K.; Frunzke, J. Live cell imaging of SOS and prophage dynamics in isogenic bacterial populations. Mol. Microbiol. 2015, 98, 636–650. [Google Scholar] [CrossRef]
- Cirz, R.T.; Jones, M.B.; Gingles, N.A.; Minogue, T.D.; Jarrahi, B.; Peterson, S.N.; Romesberg, F.E. Complete and SOS-Mediated Response of Staphylococcus aureus to the Antibiotic Ciprofloxacin. J. Bacteriol. 2006, 189, 531–539. [Google Scholar] [CrossRef]
- de Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef]
- Kingsford, C.L.; Ayanbule, K.; Salzberg, S.L. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007, 8, R22. [Google Scholar] [CrossRef]
- Price, I.R.; Gaballa, A.; Ding, F.; Helmann, J.D.; Ke, A. Mn2+-sensing mechanisms of yybP-ykoY orphan riboswitches. Mol. Cell 2015, 57, 1110–1123. [Google Scholar] [CrossRef] [PubMed]
- Cohn, M.T.; Kjelgaard, P.; Frees, D.; Penadés, J.R.; Ingmer, H. Clp-dependent proteolysis of the LexA N-terminal domain in Staphylococcus aureus. Microbiology 2011, 157, 677–684. [Google Scholar] [CrossRef]
- Bojer, M.S.; Wacnik, K.; Kjelgaard, P.; Gallay, C.; Bottomley, A.L.; Cohn, M.T.; Lindahl, G.; Frees, D.; Veening, J.-W.; Foster, S.J.; et al. SosA inhibits cell division in Staphylococcus aureus in response to DNA damage. Mol. Microbiol. 2019, 112, 1116–1130. [Google Scholar] [CrossRef]
- Maiques, E.; Ubeda, C.; Campoy, S.; Salvador, N.; Lasa, I.; Novick, R.P.; Barbé, J.; Penadés, J.R. Beta-lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J. Bacteriol. 2006, 188, 2726–2729. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Toledo-Arana, A.; Berasain, C.; Ghigo, J.-M.; Amorena, B.; Penadés, J.R.; Lasa, I. SarA and not sigma B is essential for biofilm development by Staphylococcus aureus. Mol. Microbiol. 2003, 48, 1075–1087. [Google Scholar] [CrossRef]
- Cuirolo, A.; Plata, K.; Rosato, A.E. Development of homogeneous expression of resistance in methicillin-resistant Staphylococcus aureus clinical strains is functionally associated with a β-lactam-mediated SOS response. J. Antimicrob. Chemother. 2009, 64, 37–45. [Google Scholar] [CrossRef]
- Plata, K.B.; Rosato, R.R.; Rosato, A.E. Fate of mutation rate depends on agr locus expression during oxacillin-mediated heterogeneous-homogeneous selection in methicillin-resistant Staphylococcus aureus clinical strains. Antimicrob. Agents Chemother. 2011, 55, 3176–3186. [Google Scholar] [CrossRef]
- Traber, K.E.; Lee, E.; Benon, S.; Corrigan, R.; Cantera, M.; Shopsin, B.; Novick, R.P. agr function in clinical Staphylococcus aureus isolates. Microbiology 2008, 154, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, R.; Sloan, D.B.; Ochman, H. Antisense transcription is pervasive but rarely conserved in enteric bacteria. mBio 2012, 3, e00156-12. [Google Scholar] [CrossRef]
- Menendez-Gil, P.; Toledo-Arana, A. Bacterial 3′UTRs: A Useful Resource in Post-transcriptional Regulation. Front. Mol. Biosci. 2021, 7, 617633. [Google Scholar] [CrossRef] [PubMed]
- Phadtare, S.; Inouye, M.; Severinov, K. The nucleic acid melting activity of Escherichia coli CspE is critical for transcription antitermination and cold acclimation of cells. J. Biol. Chem. 2002, 277, 7239–7245. [Google Scholar] [CrossRef]
- Goodson, J.R.; Klupt, S.; Zhang, C.; Straight, P.; Winkler, W.C. LoaP is a broadly conserved antiterminator protein that regulates antibiotic gene clusters in Bacillus amyloliquefaciens. Nat. Microbiol. 2017, 2, 17003–17010. [Google Scholar] [CrossRef] [PubMed]
- Goodson, J.R.; Winkler, W.C. Processive antitermination. Microbiol. Spectr. 2018, 6, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Bossi, L.; Schwartz, A.; Guillemardet, B.; Boudvillain, M.; Figueroa-Bossi, N. A role for Rho-dependent polarity in gene regulation by a noncoding small RNA. Genes Dev. 2012, 26, 1864–1873. [Google Scholar] [CrossRef]
- Wang, X.; Ji, S.C.; Jeon, H.J.; Lee, Y.; Lim, H.M. Two-level inhibition of galK expression by Spot 42: Degradation of mRNA mK2 and enhanced transcription termination before the galK gene. Proc. Natl. Acad. Sci. USA 2015, 112, 7581–7586. [Google Scholar] [CrossRef]
- Chen, J.; Morita, T.; Gottesman, S. Regulation of transcription termination of small RNAs and by small RNAs: Molecular mechanisms and biological functions. Front. Cell. Infect. Microbiol. 2019, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, M.; Chastanet, A.; Debarbouille, M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, Gram-positive bacteria. Appl. Environ. Microbiol. 2004, 70, 6887–6891. [Google Scholar] [CrossRef] [PubMed]
- Catalan-Moreno, A.; Caballero, C.J.; Irurzun, N.; Cuesta, S.; López Sagaseta, J.; Toledo-Arana, A. One evolutionarily selected amino acid variation is sufficient to provide functional specificity in the cold shock protein paralogs of Staphylococcus aureus. Mol. Microbiol. 2020, 113, 826–840. [Google Scholar] [CrossRef]
- Toledo-Arana, A.; Dussurget, O.; Nikitas, G.; Sesto, N.; Guet-Revillet, H.; Balestrino, D.; Loh, E.; Gripenland, J.; Tiensuu, T.; Vaitkevicius, K.; et al. The Listeria transcriptional landscape from saprophytism to virulence. Nature 2009, 459, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Caballero, C.J.; Menendez-Gil, P.; Catalan-Moreno, A.; Vergara-Irigaray, M.; García, B.; Segura, V.; Irurzun, N.; Villanueva, M.; de Los Mozos, I.R.; Solano, C.; et al. The regulon of the RNA chaperone CspA and its auto-regulation in Staphylococcus aureus. Nucleic Acids Res. 2018, 46, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C. Electrotransformation of Staphylococci. Methods Mol. Biol. 1995, 47, 209–216. [Google Scholar] [PubMed]
- Charpentier, E.; Anton, A.I.; Barry, P.; Alfonso, B.; Fang, Y.; Novick, R.P. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 2004, 70, 6076–6085. [Google Scholar] [CrossRef] [PubMed]
- Catalan-Moreno, A.; Cela, M.; Menendez-Gil, P.; Irurzun, N.; Caballero, C.J.; Caldelari, I.; Toledo-Arana, A. RNA thermoswitches modulate Staphylococcus aureus adaptation to ambient temperatures. Nucleic Acids Res. 2021, 49, 3409–3426. [Google Scholar] [CrossRef]
- Balestrino, D.; Hamon, M.A.; Dortet, L.; Nahori, M.-A.; Pizarro-Cerda, J.; Alignani, D.; Dussurget, O.; Cossart, P.; Toledo-Arana, A. Single-cell techniques using chromosomally tagged fluorescent bacteria to study Listeria monocytogenes infection processes. Appl. Environ. Microbiol. 2010, 76, 3625–3636. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastet, L.; Bustos-Sanmamed, P.; Catalan-Moreno, A.; Caballero, C.J.; Cuesta, S.; Matilla-Cuenca, L.; Villanueva, M.; Valle, J.; Lasa, I.; Toledo-Arana, A. Regulation of Heterogenous LexA Expression in Staphylococcus aureus by an Antisense RNA Originating from Transcriptional Read-Through upon Natural Mispairings in the sbrB Intrinsic Terminator. Int. J. Mol. Sci. 2022, 23, 576. https://doi.org/10.3390/ijms23010576
Bastet L, Bustos-Sanmamed P, Catalan-Moreno A, Caballero CJ, Cuesta S, Matilla-Cuenca L, Villanueva M, Valle J, Lasa I, Toledo-Arana A. Regulation of Heterogenous LexA Expression in Staphylococcus aureus by an Antisense RNA Originating from Transcriptional Read-Through upon Natural Mispairings in the sbrB Intrinsic Terminator. International Journal of Molecular Sciences. 2022; 23(1):576. https://doi.org/10.3390/ijms23010576
Chicago/Turabian StyleBastet, Laurène, Pilar Bustos-Sanmamed, Arancha Catalan-Moreno, Carlos J. Caballero, Sergio Cuesta, Leticia Matilla-Cuenca, Maite Villanueva, Jaione Valle, Iñigo Lasa, and Alejandro Toledo-Arana. 2022. "Regulation of Heterogenous LexA Expression in Staphylococcus aureus by an Antisense RNA Originating from Transcriptional Read-Through upon Natural Mispairings in the sbrB Intrinsic Terminator" International Journal of Molecular Sciences 23, no. 1: 576. https://doi.org/10.3390/ijms23010576
APA StyleBastet, L., Bustos-Sanmamed, P., Catalan-Moreno, A., Caballero, C. J., Cuesta, S., Matilla-Cuenca, L., Villanueva, M., Valle, J., Lasa, I., & Toledo-Arana, A. (2022). Regulation of Heterogenous LexA Expression in Staphylococcus aureus by an Antisense RNA Originating from Transcriptional Read-Through upon Natural Mispairings in the sbrB Intrinsic Terminator. International Journal of Molecular Sciences, 23(1), 576. https://doi.org/10.3390/ijms23010576