
Genetics of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) and Role of Sacsin in Neurodegeneration




Abstract
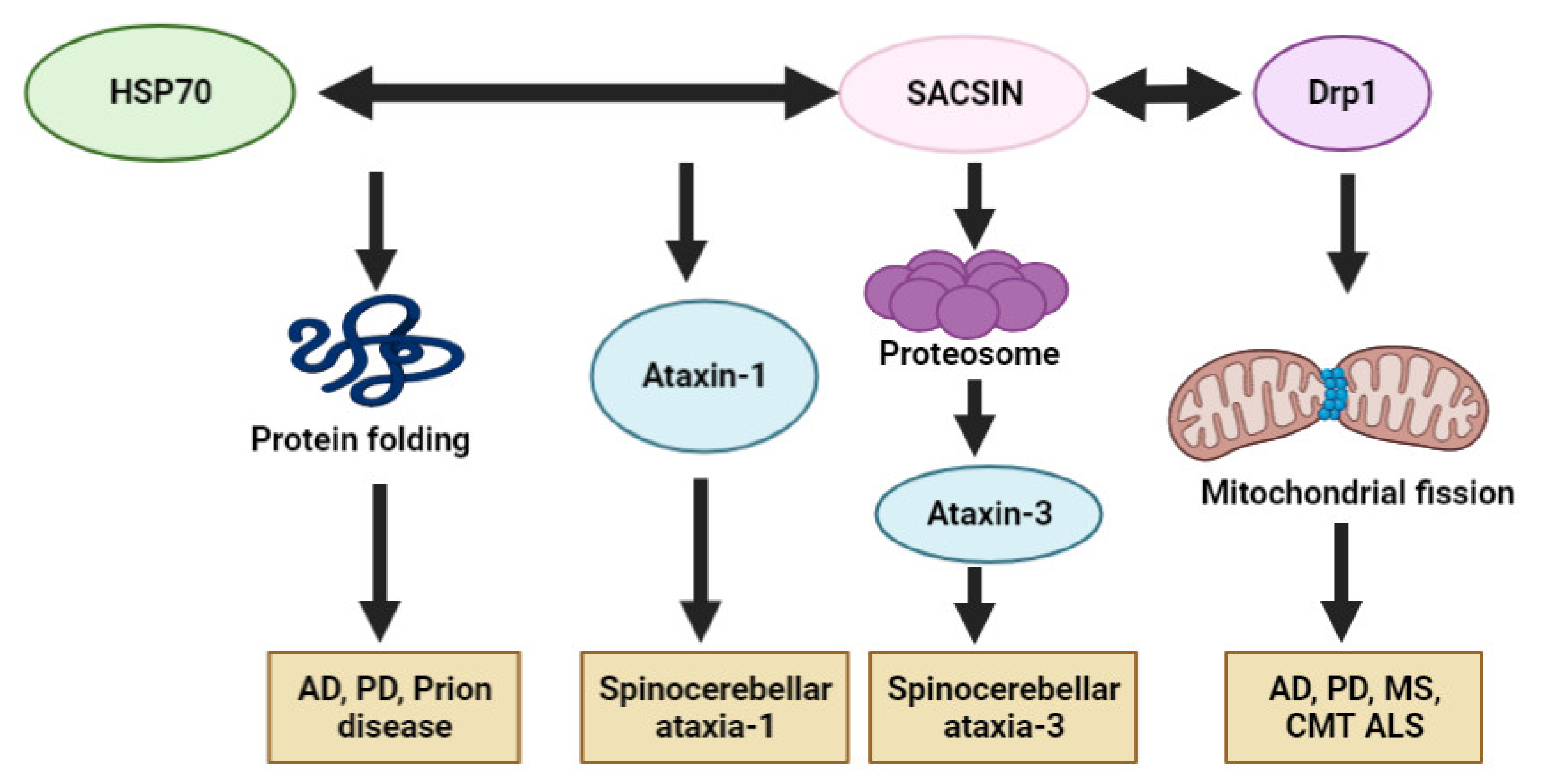
1. Introduction: Sacsin (SACS) Gene and ARSACS
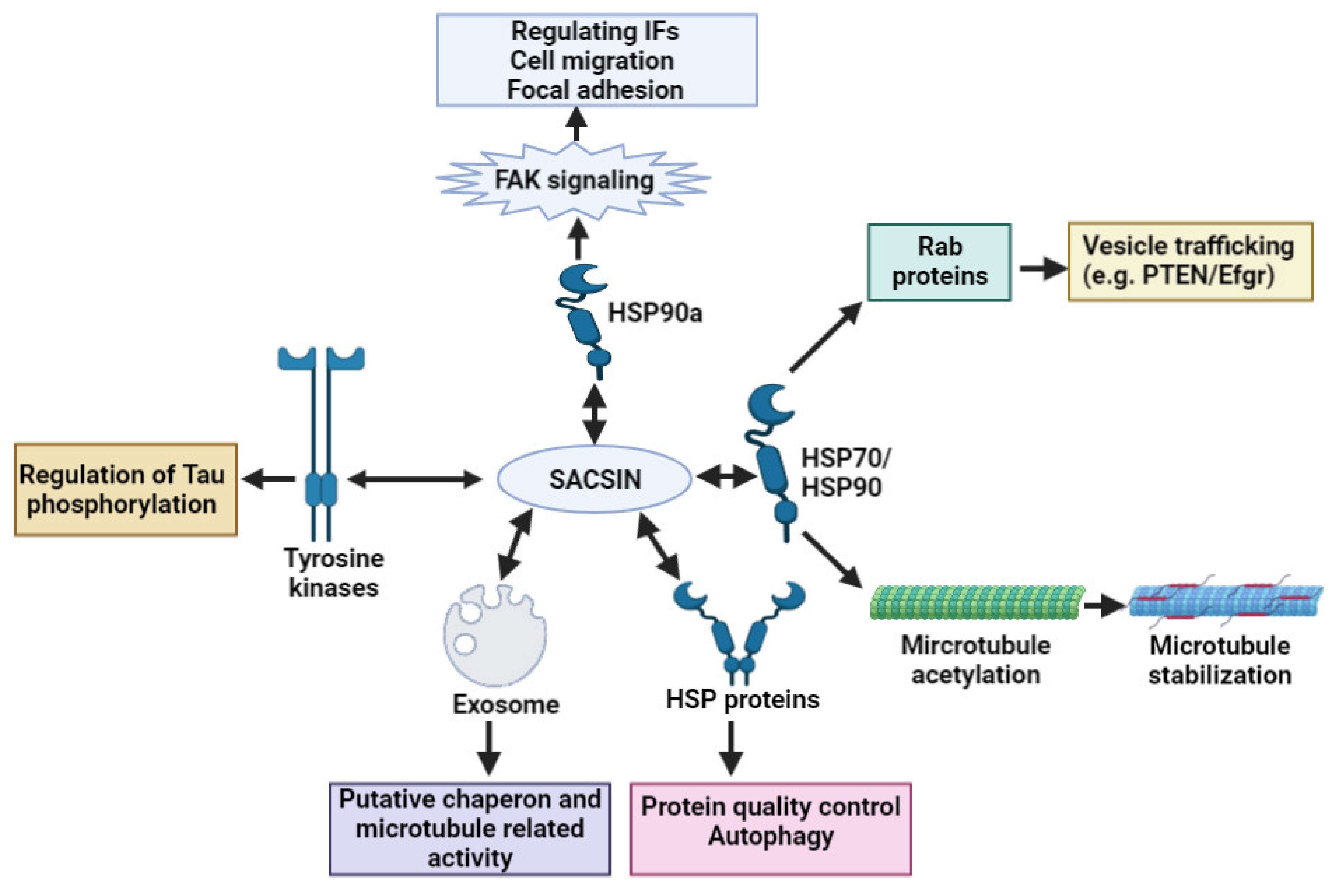
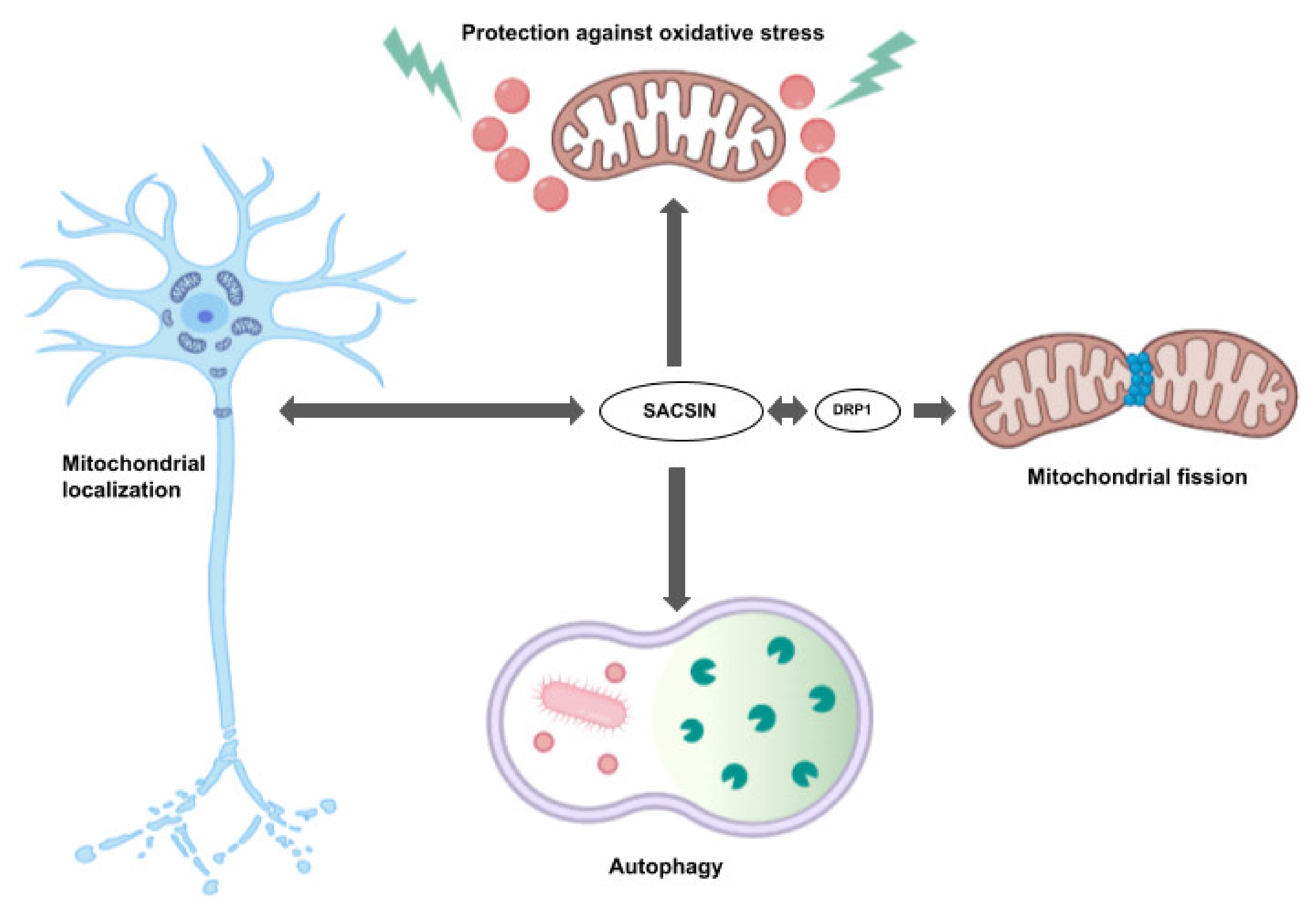
2. Sacsin Functions and Cell/Animal Models of ARSACS
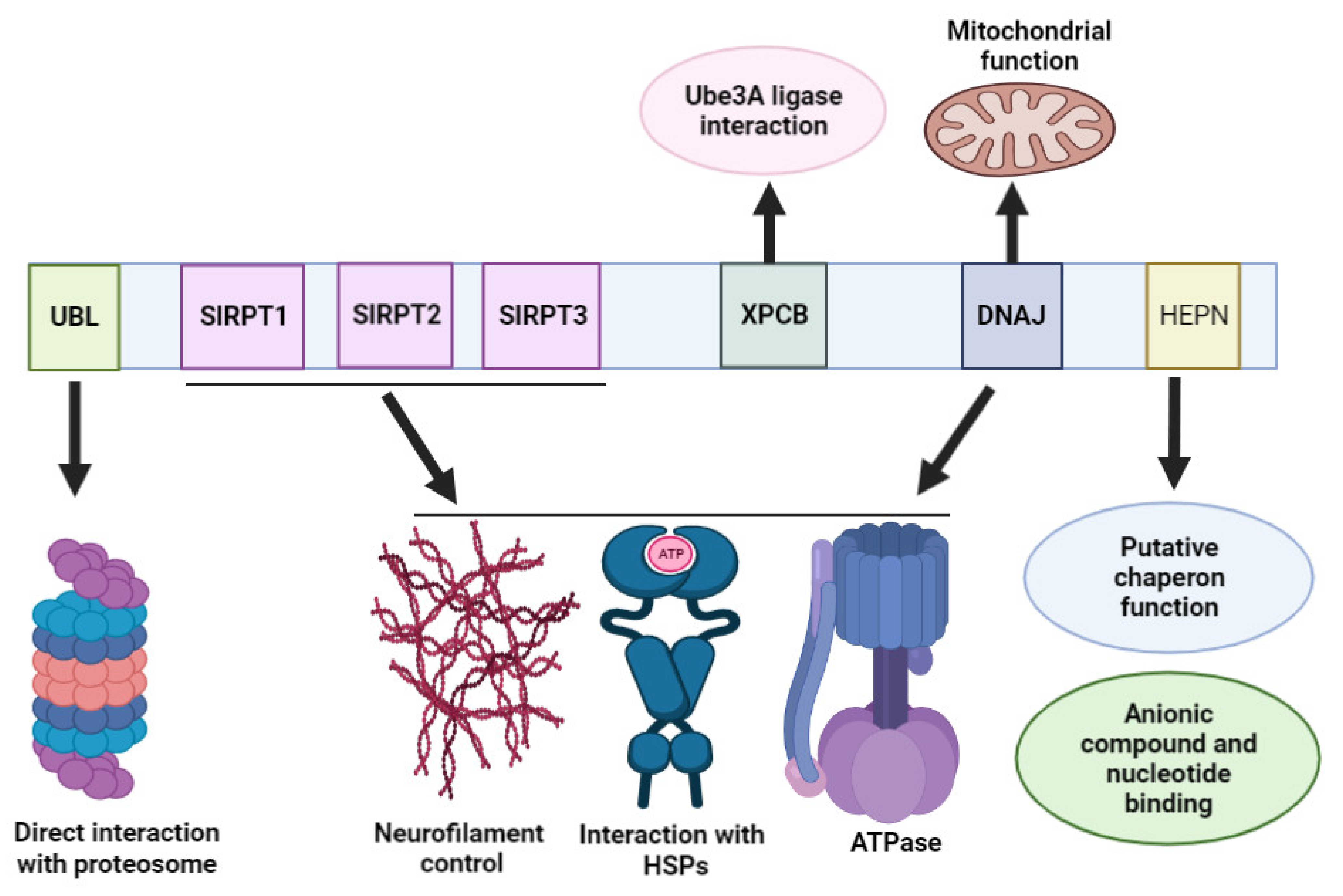
3. Sacsin Protein Domains
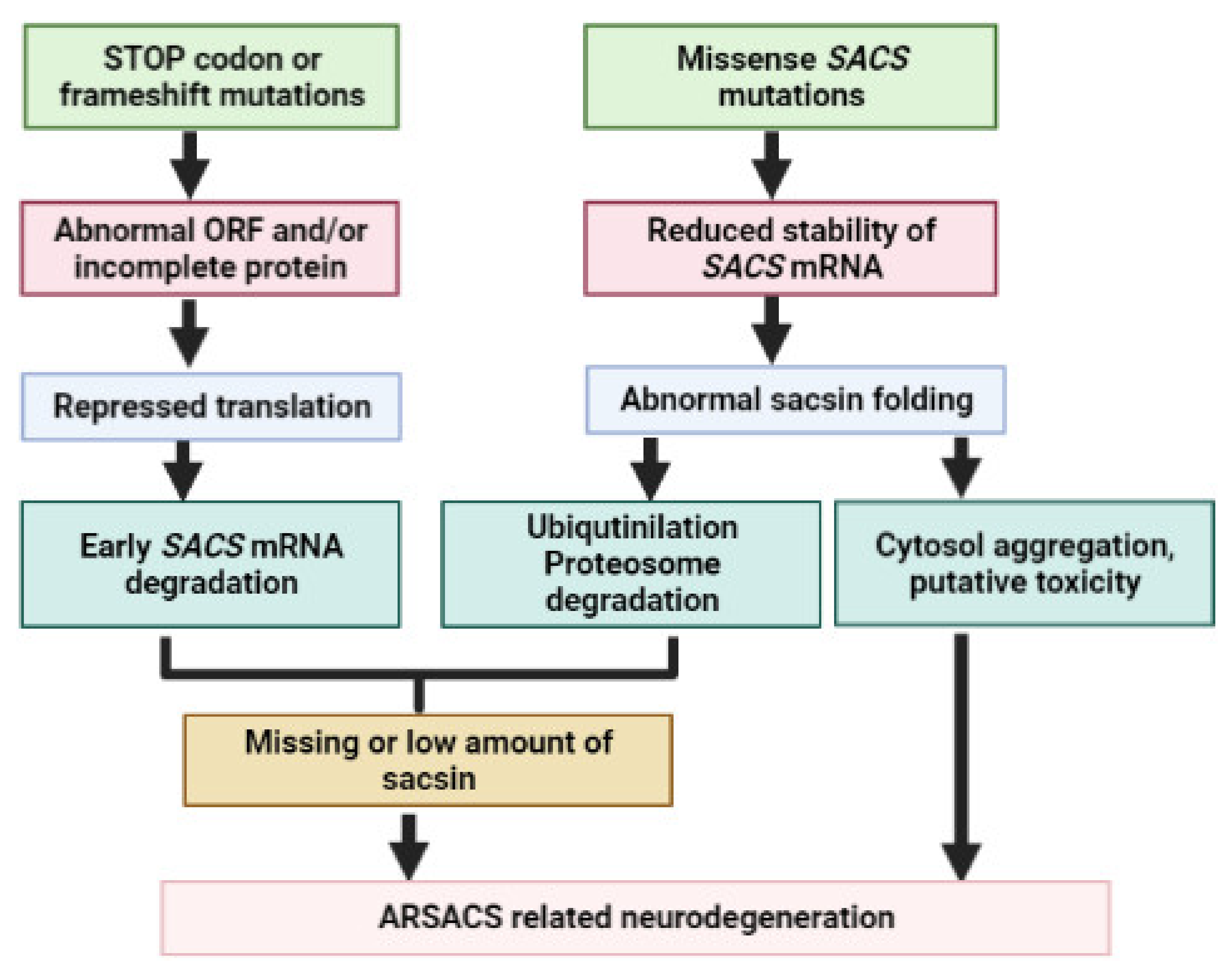
4. SACS Genetics and Mutations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Exon | Domain | AOO | Clinical Symptoms | Neurological Changes | Population (Ethnicity) | Refs. |
|---|---|---|---|---|---|---|---|
| c.6594delT (I2949Ffs*4,) or c.5254C > T (p.Q1752X), hm or c-het | 10 | SIRPT1 + SIRPT3 rs | 12–18 mths |
|
| Canada (French Canadian) | [5,38] |
| several variants, c.8844delT (p.I2949FfsX2952): most common, hm or c-het | 8 or 10 | SIRPT1, 2 or 3, XPBC or DNAJ | NA |
| NA | [42] | |
| c.9284dupC 9p.Ala3096Cysfs*2), hm | 10 | SIRPT3 | early child- hood |
|
| [41] | |
| c.10046G > C (p.A3324P), hm | 10 | Between SIRPT3 and XPBC | 2–4 yrs |
|
| Tunisian (Tunisian) | [43] |
| c.1411delT, hm | 8 | SIRPT1 | 5 yrs |
| |||
| c.1155insA, hm | 8 | SIRPT1 | 1–9 yrs |
| |||
| c.3662T > C (p.W1196R), hm | 10 | SIRPT1 | 10–14 yrs |
| |||
| c.12846_12850delAGAG, hm | 10 | Between XPBC and DNAJ | 1–3 yrs |
|
| [44] | |
| c.2439-2440delAT(V815Gfs*4), hm | 10 | SIRPT1 | 13–19 yrs |
|
| [45] |
| Mutation | Exon | Domain | AOO | Clinical Symptoms | Neurological Changes | Country (Ethnicity) | Refs. |
|---|---|---|---|---|---|---|---|
| c.1859insC, hm | 8 | SIRPT1 | 2 yrs |
|
| Italy (Italian) | [48] |
| c.4999del CAGAA5000-(p.C1679X), hm | 10 | SIRPT2 | before 10 yrs |
|
| [47] | |
| c.1858C>T (p.Q620X), c.4585insA:V1528→p.fsX1540), c.het | 8 & 10, rs | SIRPT1 & SIRPT2, rs | before 2 yrs |
|
| ||
| c.563G>A (p.G188E) + c.7394C>T (S2465L), c.het | 7 & 10, rs | SIRPT1 & SIRPT3, rs | 15 yrs |
|
| [49] | |
| c.962G>A; p.R321Q + c.8330G>A, (R2777L), c.het | 8 & 10, rs | SIRPT1 & SIRPT3, rs | 15 mths |
|
| ||
| 16 novel mutations, including a large deletion, hm or c-het | 8 & 10 | SIRPT 1-2-3 or XPCB or DNAJ | 1–32 yrs |
|
| [50] | |
| c.4198T>A/c.5719C>T, c.het | 10 | SIRPT2 | 15–16 yrs |
|
| [51] | |
| c.13132C >T (p.R4378X), hm | 10 | DNAJ | 2 yrs |
|
| [52] | |
| c.5629C>T- (p.R1877X), hm | 10 | SIRPT2 | 26 mths |
|
| [13] | |
| c.600_604+1delAACAGG (p.I202fsX6), hm; 1.5 MB large deletion | 8 | SIRPT1 | 42 yrs |
|
| [53] | |
| c.6680T>C (p.L2374S), hm | 10 | SIRPT2 | early childhood |
|
| [13] | |
| c.10743C>T (p.Q3582X), hm | 10 | XPCPB | early childhood |
|
| [54] | |
| c.11471A>G, (p.N3799D), hm | 10 | SIRPT3 | 2.5–3.5 yrs |
|
| Turkey (Turkish) | [55,56] |
| c.9655_9658 delAGTT), truncation of I3199, hm | 10 | SIRPT3 | 1.5 yrs |
|
| ||
| c. 2018T>C (p.C648R), hm | 8 | SIRPT1 | 2.5 yrs |
|
| ||
| c. 8124delC-truncation of p.A2708, hm | 10 | SIRPT3 | 2 yrs |
|
| ||
| c.1160A>G (p.F4011S), hm | 10 | between SIRPT3 & DNAJ | 1 yr |
|
| [57] | |
| c. 6945A>G (p.V3369A), hm | 10 | SIRPT3 | 3–7 yrs |
|
| ||
| c.12841T>A (p.K1404), c.6945A>G (p.V3369A), c.het | 10 | SIRPT2 & SIRPT3,rs | 2 yrs |
|
| ||
| c.6945A>G (p.V3369A), c.12020C (p.S4007F), c.het | 10 | SIRPT3 & loop between XPCB-DNAJ | 4 yrs |
|
| ||
| c.8346–8347insT (p.G2902V), hm | 10 | SIRPT2 | 2 yrs |
|
| ||
| c.5677G>A (p.R3801X), hm | 10 | SIRPT3 | 3 yrs |
|
| ||
| c. c.13485delC (p.K4495N), hm | 10 | DNAJ | 3 yrs |
|
| ||
| p.G2772A, hm | 10 | SIRPT3 | early childhood |
|
| [58] | |
| c.2182C>T (p.R728), hm | 8 | SIRPT1 | 4 yrs |
|
| [59] | |
| c.12461delC (p.P4154QfsX20), hm | 10 | between XPCB & DNAJ | After 1 yr |
|
| [60] | |
| Several mutations, hm or c het | 8 or 10 | SIRPT2 or 3 or between SIRPT3 & DNAJ | 1–13 yrs | patients had similar symptoms:
|
| Netherlands (Dutch, British, Turkish) | [61] |
| c.3491T>A, p.M1164K, hm | 10 | SIRPT1 | 12–13 yrs |
|
| Belgium (Belgian) | [62] |
| Several mutations, hm or c -het | 8 or 10, deletion of exon 3–5 | SIRPT1 or 2 or 3 or HEPN | 1–24 yrs |
|
| Belgium (Belgian, Moroccan, Serbian, Hungarian) | [63] |
| c.10517T>C (p.F3506S) + chromosomal deletion, c-het | 10 | SIRPT3 | 16 yrs |
|
| [76] | |
| NA | NA | NA | 1 yr |
|
| Spain (Spanish) | [77] |
| c.7848C>T (p.R2556C), hm | 10 | SIRPT3 | before 1 yr |
|
| [64] | |
| c.826C>T (p.R276C) + c.3904C>T (p.P1302S), c-het | 8 & 10, rs | SIRPT1+ between SIRPT3 & DNAJ, rs | early childhood |
|
| [65] | |
| c.9695delC (p.T3232KfsX24),hm | 10 | SIRPT3 | 8–10 yrs |
|
| Greece (Greek) | [66] |
| c.9804_9805insC, (p.S3268 _Ifs), hm | 10 | SIRPT3 | 1.5–5 yrs |
|
| Poland (Polish) | [67] |
| c.72276C>T (p.R2426X) | 10 | SIRPT3 | 32 yrs |
|
| Russia (Russian) | [68] |
| c.13352T>C (p.L4451P) + c.6890T>G (p.L2297W), c.het | 10 | HEPN+ SIRPT2, rs | before 10 yrs |
|
| Norway (Norwegian) | [69] |
| 17 novel SACS mutations, hm or c-het | 8 + 9 + 10 | SIRT1 or 2 or 3 or DNAJ or HEPN | 1–30 yrs |
|
| Germany (German, Turkish, Greek, Macedonian) | [4] |
| 9 different mutations in 6 families, hm or c-het | 8 or 10 | SIRT1 or 2 or 3 or DNAJ or HEPN | 2–15 yrs |
|
| Germany (German, Italian, Romanian, Turkish, Arabic) | [70] |
| c.2076delG (p.T692) + c.3965_3966del (p.G1322Vfs*1343), c-het | 8 & 10 | SIRT1+ SIRPT2, rs | 19–26 yrs |
|
| UK (British) | [71] |
| 20 mutations, 11 novel mutations, hm or c-het | 10 | SIRT1 or 2 or 3 or DNAJ or HEPN | 1–46 yrs |
|
| [72] | |
| c.5744_5745delAT (p.H1915Rfs*19),hm | 10 | SIRPT2 | 9–19 yrs |
|
| France (French) | [73] |
| p.E1100K + p.N1489S + p.M1359T, c-het | 10 | SIRT1+ SIRPT2, rs | 6–15 yrs |
|
| Finland (Finnish) | [74] |
| c.13721T/G (p.F4574C), hm | 10 | HEPN | 37 yrs |
|
| Macedonia (Macedonian) | [75] |
| Mutation | Exon | Domain | AOO | Clinical Symptoms | Neurological Changes | Country (Ethnicity) | References |
|---|---|---|---|---|---|---|---|
| p.3774C>T, p.Q1198X, c.het | 10 | XPCB & SIRPT1, rs | 9 yrs |
|
| Japan (Japanese) | [78] |
| c.2951_2952delAG(p.Q984GfsX986)+3922delT(p.1308LfsX1326), c. het | 10 | SIRPT1 + between SIRPT3 & XPBC, rs | 15–20 yrs |
|
| [81] | |
| c.32627-32636delACACTGTTAC(p.W395-fsX407), c.31760delT (p.V687-fsX713), c.het | 8 | SIRPT1 | under 10 yrs |
|
| [79] | |
| c.6543delA, (p.R2002X), hm | 10 | SIRPT2 | early childhood |
|
| [80] | |
| c.5988-9del CT, hm | 10 | SIRPT2 | early childhood |
|
| [81] | |
| c. 987C>T (p.F304S), hm | 8 | SIRPT1 | before 10 yrs |
|
| [82] | |
| c. 6355C>T (p.R2119X), hm | 10 | SIRPT2 | 20s |
|
| [83] | |
| c.482delA (p.L802P), c.2405T>C (p.N161fsX175), c-het | 10 & 7 rs | SIRPT1 | late 10s- early 20s |
|
| [85] | |
| c.12976A/G (p.K4326Q), c.4233-4236 delACTT (p.L1412Kfs*16), c-het | 10 | DNAJ + SIRPT2, rs | ~22 yrs |
|
| [85] | |
| c.3769 G>T (p.G1257X)+11361–2insT(p.R3788SfsX3820), c-het | 10 | SIRPT2 & SIRPT3, rs | 12 yrs |
|
| [86] | |
| c.414 C>G (p.Y138X) +5263–4delAA (p.K1755VfsX1775), c-het | 7 & 10, rs | SIRPT1 & SIRPT2, rs | 12–19 yrs |
|
| [87,88] | |
| c.4756_4760del (p.N1586Yfs*3)+ putative noncoding mutation, c-het | 10 | SIRPT2 | early childhood |
|
| Korea (Korean) | [89] |
| c.8844delT (p.I2949Ffs*4) + c.11781_11782dupGC (p.P3928Rfs*17), c-het | 10 | SIRPT3 +between XPCB & DNAJ, rs | ~20 yrs |
|
| [90] | |
| c.7272C>A (p.C2424X), c.11319_11321del (p.R3774del), c-het | 10 | SIRPT3 +XPCB, rs | ~10 yrs |
|
| [91] | |
| c.11803C>T (p.Q3935X)+ 1.33Mb deletion, c-het | 10 | between SIRPT3 & XPCB | 6 yrs |
|
| China (Chinese) | [92] |
| c.12637 _12638delGA (p.Q4213Rfs*3)+ c.11274_11276delAAC (p.I3758_ TdelinsM), c-het | 10 | between XPCB & DNAJ + XPCB, rs | 10’s |
|
| [93] | |
| c. 8000T>C (p.F2667S), c. 10685_10689del (p.F3562X), c-het | 10 | SIRPT3 + between XPCB & DNAJ + XPCB, rs | early childhood |
|
| [94] | |
| c.5236dupA (p.T1746fs)+ c.13085T/G (p.I4362R), c-het | 10 | SIRPT2 +DNAJ, rs | NA |
|
| [96] | |
| c.9019C>T, p.P3007S and c.10174_10183del, p.H3392fs | 10 | SIRPT2 + between XPCB & DNAJ | early childhood |
|
| [95] | |
| c.1773C>A (p.S578X) + c.8088_8089 insCA (p.M2697Q fs*43), c-het | 8 & 10, rs | SIRPT1 + SIRPT3, rs | 6 yrs |
|
| ||
| c.5692 G>T, p.E1898X; c.12673-12677 del TATCA, p.Y4225D fs*6-c-het | 10 | SIRPT1 +DNAJ, rs | Early childhood |
|
| [97] | |
| c.1773C>A, p.S578X; c.8088-8089 in. CA, p.M2697Q fs*4 | 10 | SIRPT1 + SIRPT3, rs | 6 yrs |
|
| ||
| c.382_383del (p.Q128Sfs*2), hm | 7 | SIRPT1 | 2 yrs |
|
| Thailand (Thai) | [98] |
| c.5824_5827delTACT (p.Y1942Mfs*9), hm | 10 | SIRPT2 | early teens |
|
| Kuwait (Kuwait) | [99] |
| c.429_430delTT (p.W144VfsX39), hm | 7 | SIRPT1 | 3 yrs |
|
| Iran (Iranian) | [100] |
| c.4117_4118delGCinsAG (p.A1373R), hm | 10 | SIRPT2 | early childhood |
|
| [101] | |
| c.14329fs*2725 (p.R707Kfs*6), hm | 10 | SIRPT2SIRPT1 | 9–15 yrs |
|
| India (Indian) | [102] |
| c.11690_11693dupGTGA (p.N3898QfsX2),hm | 10 | XPCB | 4 yrs |
|
| [103] | |
| c.8605delT (p.C2869VfsX15), hm | 10 | SIRPT3 | 14 mths |
|
| [104] | |
| c.8793 delA, hm | 10 | SIRPT1 | early childhood |
|
| [105] | |
| c. 4232T>G + c.8132C>T, c-het | 10 | SIRPT2 + SIRPT3, rs | 3 yrs |
|
| [106] | |
| c.2656C>T (p.Q886*), hm | 10 | SIRPT1 | 11–12 yrs |
|
| Pakistan (Pakistani) | [107] |
| c.4756_4760delAATCA (p.N1586Yfs*3), hm | 10 | SIRPT2 | 9–10 yrs |
|
| ||
| c.9119dupA (p.N3040Kfs*4), hm | 10 | SIRPT3 | 1.5 yrs |
|
| [108] | |
| p.N2380K & p.D3269N, c-het | 10 | SIRPT2 + SIRPT3, rs | 16 mths |
|
| Israel (Ashkenazi Jews) | [109] |
| Mutation | Exon | Domain | AOO | Clinical Symptoms | Neurological Changes | Country (Ethnicity) | Refs |
|---|---|---|---|---|---|---|---|
| c.3484G>T (p.E1162X), & c. 11707C>T (p.R3903X), c-het | 10 | SIRPT1+ between SIRPT3 & XPBC, rs | 2.5–3.5 yrs |
|
| USA (American) | [110] |
| c.11637_11638delAG (p.R3879fs), hm | 10 | SIRPT3 & XPBC, rs | 4 yrs |
|
| [111] | |
| Chr13 duplication | NA | NA | 13 yrs |
|
| [112] | |
| c.4744G>A (p.D1582N) + c.7205_7206delTT (p.L2402Rfs*6), c-het | 10 | SIRPT2 + SIRPT3, rs | 2 yrs |
|
| USA (American or mixed European) | [113] |
| c.11824dup (p.M3942Nfs*4), hm | 10 | XPCB | 11 yrs |
|
| USA (African American) | [114] |
| 11 SACS variants in 39 patients, hm or c-het | 8 or 10 | SIRPT 1-3, XPBC or HEPN | NA |
|
| USA (NA) | [115] |
| NA | NA | NA | early childhood |
|
| Brazil (Brazilian) | [116] |
| c.5150_5151insA | 10 | SIRPT2 | early childhood |
|
| Brazil (German) | [117] |
| Several variants, including p.L393Cfs*17 & p.N2760Mfs*6 | 8 or 10, rs | SIRPT 1-3 or XPBC | 1–44 yrs |
|
| Brazil (Brazilian) | [118] |
| c.8107C>T (p.R2703C) +c.922C>T-(p.L308F), hm | 10 + 8, rs | SIRPT3 + SIRPT1, rs | 8–9 yrs |
|
| [119] | |
| c.7962T>G (p.Y2654X), hm | 10 | SIRPT3 | 20’s |
|
| New Zealand (Maori & English) | [120] |
5. Potential Involvement of SACS in Other Neurodegenerative Diseases
6. ARSACS Diagnosis and Potential Therapeutics
7. Discussion and Future Insights
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takiyama, Y. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuropathology 2006, 26, 368–375. [Google Scholar] [CrossRef]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.-M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Gentil, B.J.; Lai, G.-T.; Menade, M.; Larivière, R.; Minotti, S.; Gehring, K.; Chapple, J.-P.; Brais, B.; Durham, H.D. Sacsin, mutated in the ataxia ARSACS, regulates intermediate filament assembly and dynamics. FASEB J. 2019, 33, 2982–2994. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Soehn, A.S.; Gburek-Augustat, J.; Schicks, J.; Karle, K.N.; Schüle, R.; Haack, T.B.; Schöning, M.; Biskup, S.; Rudnik-Schöneborn, S.; et al. Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): Expanding the genetic, clinical and imaging spectrum. Orphanet J. Rare Dis. 2013, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Engert, J.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.-P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000, 24, 120–125. [Google Scholar] [CrossRef]
- De Braekeleer, M.; Giasson, F.; Mathieu, J.; Roy, M.; Bouchard, J.P.; Morgan, K. Genetic epidemiology of autosomal recessive spastic ataxia of Charlevoix-Saguenay in northeastern Quebec. Genet. Epidemiol. 1993, 10, 17–25. [Google Scholar] [CrossRef]
- Heyer, E. Genetic consequences of differential demographic behaviour in the Saguenay region, Québec. Am. J. Phys. Anthr. 1995, 98, 1–11. [Google Scholar] [CrossRef]
- Takiyama, Y. Sacsinopathies: Sacsin-related ataxia. Cerebellum 2007, 6, 353–359. [Google Scholar] [CrossRef]
- Bouchard, J.-P.; Richter, A.; Mathieu, J.; Brunet, D.; Hudson, T.J.; Morgan, K.; Melançon, S.B. Autosomal recessive spastic ataxia of Charlevoix–Saguenay. Neuromuscul. Disord. 1998, 8, 474–479. [Google Scholar] [CrossRef]
- Laberge, A.-M.; Michaud, J.; Richter, A.; Lemyre, E.; Lambert, M.; Brais, B.; Mitchell, G. Population history and its impact on medical genetics in Quebec. Clin. Genet. 2005, 68, 287–301. [Google Scholar] [CrossRef]
- Bouhlal, Y.; Amouri, R.; El Euch-Fayeche, G.; Hentati, F. Autosomal recessive spastic ataxia of Charlevoix–Saguenay: An overview. Park. Relat. Disord. 2011, 17, 418–422. [Google Scholar] [CrossRef]
- Larivière, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 2014, 24, 727–739. [Google Scholar] [CrossRef]
- Romano, A.; Tessa, A.; Barca, A.; Fattori, F.; Fulvia de Leva, M.; Terracciano, A.; Storelli, C.; Santorelli, F.M.; Verri, T. Comparative analysis and functional mapping of SACS mutations reveal novel insights into sacsin repeated architecture. Hum. Mutat. 2013, 34, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Ady, V.; Toscano-Márquez, B.; Nath, M.; Chang, P.K.; Hui, J.; Cook, A.; Charron, F.; Larivière, R.; Brais, B.; McKinney, R.A.; et al. Altered synaptic and firing properties of cerebellar Purkinje cells in a mouse model of ARSACS. J. Physiol. 2018, 596, 4253–4267. [Google Scholar] [CrossRef]
- Castro, A.A.; Machuca, C.; Jimenez, F.J.R.; Jendelova, P.; Erceg, S.; Arellano, C.M. Short Review: Investigating ARSACS: Models for understanding cerebellar degeneration. Neuropathol. Appl. Neurobiol. 2019, 45, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Romano, L.E.; Aw, W.Y.; Hixson, K.M.; Novoselova, T.V.; Havener, T.M.; Howell, S.; Hall, C.L.; Xing, L.; Beri, J.; Nethisinghe, S.; et al. The ataxia protein sacsin is required for integrin trafficking and synaptic organization. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef]
- Criscuolo, C.; Procaccini, C.; Meschini, M.C.; Cianflone, A.; Carbone, R.; Doccini, S.; Devos, D.A.V.I.D.; Nesti, C.; Vuillaume, I.; Pellegrino, M.; et al. Powerhouse failure and oxidative damage in autosomal recessive spastic ataxia of Charlevoix-Saguenay. J. Neurol. 2015, 262, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.J.; Larivière, R.; Bradshaw, T.Y.; Longo, F.; Sgarioto, N.; Hayes, M.J.; Romano, L.E.; Nethisinghe, S.; Giunti, P.; Bruntraeger, M.B.; et al. Altered organization of the intermediate filament cytoskeleton and relocalization of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum. Mol. Genet. 2017, 26, 3130–3143. [Google Scholar] [CrossRef]
- Girard, M.; Larivière, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.M.; Duchen, M.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef]
- Mesdom, P.; Colle, R.; Lebigot, E.; Trabado, S.; Deflesselle, E.; Fève, B.; Becquemont, L.; Corruble, E.; Verstuyft, C. Human Dermal Fibroblast: A Promising Cellular Model to Study Biological Mechanisms of Major Depression and Antidepressant Drug Response. Curr. Neuropharmacol. 2020, 18, 301–318. [Google Scholar] [CrossRef]
- Kálmán, S.; Garbett, K.; Janka, Z.; Mirnics, K. Human dermal fibroblasts in psychiatry research. Neuroscience 2016, 320, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Nejaddehbashi, F.; Bayati, V.; Mashali, L.; Hashemitabar, M.; Abbaspour, M.; Moghimipour, E.; Orazizadeh, M. Isolating human dermal fibroblasts using serial explant culture. Stem Cell Investig. 2019, 6, 23. [Google Scholar] [CrossRef]
- Arellano, C.M.; Vilches, A.; Clemente, E.; Pascual, S.I.P.; Bolinches-Amorós, A.; Castro, A.A.; Espinós, C.; Rodriguez, M.L.; Jendelova, P.; Erceg, S. Generation of a human iPSC line from a patient with autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) caused by mutation in SACSIN gene. Stem Cell Res. 2018, 31, 249–252. [Google Scholar] [CrossRef]
- Li, X.; Gehring, K. Structural studies of parkin and sacsin: Mitochondrial dynamics in neurodegenerative diseases. Mov. Disord. 2015, 30, 1610–1619. [Google Scholar] [CrossRef]
- Kozlov, G.; Denisov, A.Y.; Girard, M.; Dicaire, M.-J.; Hamlin, J.; McPherson, P.S.; Brais, B.; Gehring, K. Structural Basis of Defects in the Sacsin HEPN Domain Responsible for Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). J. Biol. Chem. 2011, 286, 20407–20412. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.F.; Siller, E.; Barral, J.M. The Sacsin Repeating Region (SRR): A Novel Hsp90-Related Supra-Domain Associated with Neurodegeneration. J. Mol. Biol. 2010, 400, 665–674. [Google Scholar] [CrossRef]
- Ménade, M.; Kozlov, G.; Trempe, J.-F.; Pande, H.; Shenker, S.; Wickremasinghe, S.; Li, X.; Hojjat, H.; Dicaire, M.-J.; Brais, B.; et al. Structures of ubiquitin-like (Ubl) and Hsp90-like domains of sacsin provide insight into pathological mutations. J. Biol. Chem. 2018, 293, 12832–12842. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Hanayama, R.; Bloodgood, B.; Mardinly, A.; Lipton, D.M.; Flavell, S.; Kim, T.-K.; Griffith, E.; Waldon, Z.; Maehr, R.; et al. The Angelman Syndrome Protein Ube3A Regulates Synapse Development by Ubiquitinating Arc. Cell 2010, 140, 704–716. [Google Scholar] [CrossRef]
- Dabbaghizadeh, A.; Paré, A.; Cheng-Boivin, Z.; Dagher, R.; Minotti, S.; Dicaire, M.J.; Bernard, B.; Young, J.C.; Durham, H.D.; Gentil, B.J. The J domain of sacsin disrupts intermediate filament assembly. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, X.; Ménade, M.; Kozlov, G.; Hu, Z.; Dai, Z.; McPherson, P.S.; Brais, B.; Gehring, K. High-Throughput Screening for Ligands of the HEPN Domain of Sacsin. PLoS ONE 2015, 10, e0137298. [Google Scholar] [CrossRef]
- Grynberg, M.; Erlandsen, H.; Godzik, A. HEPN: A common domain in bacterial drug resistance and human neurodegenerative proteins. Trends Biochem. Sci. 2003, 28, 224–226. [Google Scholar] [CrossRef]
- Bradshaw, T.Y.; Romano, L.E.L.; Duncan, E.J.; Nethisinghe, S.; Abeti, R.; Michael, G.J.; Giunti, P.; Vermeer, S.; Chapple, J.P. A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 2016, 25, 3232–3244. [Google Scholar] [CrossRef] [PubMed]
- Pilliod, J.; Moutton, S.; Lavie, J.; Maurat, E.; Hubert, C.; Bellance, N.; Anheim, M.; Forlani, S.; Mochel, F.; N’Guyen, K.; et al. New practical definitions for the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Ann. Neurol. 2015, 78, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Morani, F.; Doccini, S.; Chiorino, G.; Fattori, F.; Galatolo, D.; Sciarrillo, E.; Gemignani, F.; Züchner, S.; Bertini, E.S.; Santorelli, F.M. Functional Network Profiles in ARSACS Disclosed by Aptamer-Based Proteomic Technology. Front. Neurol. 2020, 11, 603774. [Google Scholar] [CrossRef] [PubMed]
- Bchetnia, M.; Bouchard, L.; Mathieu, J.; Campeau, P.M.; Morin, C.; Brisson, D.; Laberge, A.M.; Vézina, H.; Gaudet, D.; Laprise, C. Genetic burden linked to founder effects in Saguenay-Lac-Saint-Jean illustrates the importance of genetic screening test availability. J. Med. Genet. 2021, 58, 653–665. [Google Scholar] [CrossRef]
- Longo, F.; De Ritis, D.; Miluzio, A.; Fraticelli, D.; Baets, J.; Scarlato, M.; Santorelli, F.M.; Biffo, S.; Maltecca, F. Assessment of Sacsin Turnover in Patients With ARSACS: Implications for Molecular Diagnosis and Pathogenesis. Neurology 2021, 97, e2315–e2327. [Google Scholar] [CrossRef]
- Thiffault, I.; Dicaire, M.; Tetreault, M.; Huang, K.; Demers-Lamarche, J.; Bernard, G.; Duquette, A.; Larivière, R.; Gehring, K.; Montpetit, A.; et al. Diversity of ARSACS Mutations in French-Canadians. Can. J. Neurol. Sci./J. Can. Sci. Neurol. 2013, 40, 61–66. [Google Scholar] [CrossRef]
- Bouchard, J.P.; Barbeau, A.; Bouchard, R.W. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci./J. Can. des Sci. Neurol. 1978, 5, 61–69. [Google Scholar] [CrossRef]
- McKenzie, E.; Sharma, P.; Parboosingh, J.; Suchowersky, O. Forge Canada Consortium Novel SACS Mutation Deviates from the French Canadian ARSACS Phenotype. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2014, 41, 88–89. [Google Scholar] [CrossRef][Green Version]
- Richter, A.; Rioux, J.D.; Bouchard, J.-P.; Mercier, J.; Mathieu, J.; Ge, B.; Poirier, J.; Julien, D.; Gyapay, G.; Weissenbach, J.; et al. Location Score and Haplotype Analyses of the Locus for Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay, in Chromosome Region 13q11. Am. J. Hum. Genet. 1999, 64, 768–775. [Google Scholar] [CrossRef]
- El Euch-Fayache, G.; Lalani, I.; Amouri, R.; Turki, I.; Ouahchi, K.; Hung, W.-Y.; Belal, S.; Siddique, T.; Hentati, F. Phenotypic Features and Genetic Findings in Sacsin-Related Autosomal Recessive Ataxia in Tunisia. Arch. Neurol. 2003, 60, 982–988. [Google Scholar] [CrossRef]
- Bouhlal, Y.; El Euch-Fayeche, G.; Hentati, F.; Amouri, R. A Novel SACS Gene Mutation in a Tunisian Family. J. Mol. Neurosci. 2009, 39, 333–336. [Google Scholar] [CrossRef]
- Hammer, M.B.; Eleuch-Fayache, G.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Sassi, C.; Bouhlal, Y.; Hentati, F.; Amouri, R.; Singleton, A.B. Exome sequencing: An efficient diagnostic tool for complex neurodegenerative disorders. Eur. J. Neurol. 2013, 20, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.M. ARSACS goes global. Neurology 2004, 62, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Grieco, G.S.; Malandrini, A.; Comanducci, G.; Leuzzi, V.; Valoppi, M.; Tessa, A.; Palmeri, S.; Benedetti, L.; Pierallini, A.; Gambelli, S.; et al. Novel SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay type. Neurology 2004, 62, 103–106. [Google Scholar] [CrossRef]
- Criscuolo, C.; Banfi, S.; Orio, M.; Gasparini, P.; Monticelli, A.; Scarano, V.; Santorelli, F.M.; Perretti, A.; Santoro, L.; De Michele, G.; et al. A novel mutation in SACS gene in a family from southern Italy. Neurology 2004, 62, 100–102. [Google Scholar] [CrossRef]
- Ricca, I.; Morani, F.; Bacci, G.M.; Nesti, C.; Caputo, R.; Tessa, A.; Santorelli, F.M. Clinical and molecular studies in two new cases of ARSACS. Neurogenetics 2019, 20, 45–49. [Google Scholar] [CrossRef]
- Prodi, E.; Grisoli, M.; Panzeri, M.; Minati, L.; Fattori, F.; Erbetta, A.; Uziel, G.; D’Arrigo, S.; Tessa, A.; Ciano, C.; et al. Supratentorial and pontine MRI abnormalities characterize recessive spastic ataxia of Charlevoix-Saguenay. A comprehensive study of an Italian series. Eur. J. Neurol. 2013, 20, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Pensabene, M.C.; Melis, M.; De Corato, L.; Di Stefano, C.; Pizzicannella, G.; Mondillo, M.; Amico, A.; Tatulli, D.; Floris, R. Autosomal recessive spastic ataxia of charlevoix-saguenay: Findings from MRI in two adult Italian siblings. Radiol. Case Rep. 2020, 15, 507–510. [Google Scholar] [CrossRef]
- Anesi, L.; De Gemmis, P.; Pandolfo, M.; Hladnik, U. Two Novel Homozygous SACS Mutations in Unrelated Patients Including the First Reported Case of Paternal UPD as an Etiologic Cause of ARSACS. J. Mol. Neurosci. 2011, 43, 346–349. [Google Scholar] [CrossRef]
- Terracciano, A.; Casali, C.; Grieco, G.S.; Orteschi, D.; Di Giandomenico, S.; Seminara, L.; Di Fabio, R.; Carrozzo, R.; Simonati, A.; Stevanin, G.; et al. An inherited large-scale rearrangement in SACS associated with spastic ataxia and hearing loss. Neurogenetics 2009, 10, 151–155. [Google Scholar] [CrossRef]
- Masciullo, M.; Modoni, A.; Fattori, F.; Santoro, M.; Denora, P.S.; Tonali, P.; Santorelli, F.M.; Silvestri, G. A novel mutation in the SACS gene associated with a complicated form of spastic ataxia. J. Neurol. 2008, 255, 1429–1431. [Google Scholar] [CrossRef] [PubMed]
- Gücüyener, K.; Özgül, K.; Paternotte, C.; Erdem, H.; Prud’Homme, J.F.; Özgüç, M.; Topaloğlu, H. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay in Two Unrelated Turkish Families. Neuropediatrics 2001, 32, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Ozgul, R.K.; Poisson, V.C.; Topaloglu, H. Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) families from Turkey. Neurogenetics 2004, 5, 165–170. [Google Scholar] [CrossRef]
- Oguz, K.; Haliloglu, G.; Temucin, C.; Gocmen, R.; Has, A.; Doerschner, K.; Dolgun, A.B.; Alikasifoglu, M. Assessment of Whole-Brain White Matter by DTI in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Am. J. Neuroradiol. 2013, 34, 1952–1957. [Google Scholar] [CrossRef] [PubMed]
- Kurt, S.; Kartal, E.; Aksoy, D.; Cevik, B.; Eken, A.G.; Sahbaz, I.; Basak, A.N. Coexistence of autosomal recessive spastic ataxia of Charlevoix Saguenay and spondyloepiphyseal dysplasia in a Turkish patient. J. Neurol. Sci. 2015, 357, 290–291. [Google Scholar] [CrossRef]
- Incecik, F.; Hergüner, O.M.; Bisgin, A. Autosomal-Recessive Spastic Ataxia of Charlevoix-Saguenay: A Turkish Child. J. Pediatr. Neurosci. 2018, 13, 355–357. [Google Scholar] [CrossRef]
- Samanci, B.; Gokalp, E.E.; Bilgic, B.; Gurvit, H.; Artan, S.; Hanagasi, H.A. A novel SACS p.Pro4154GlnfsTer20 mutation in a family with autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neurol. Sci. 2021, 42, 1–5. [Google Scholar] [CrossRef]
- Vermeer, S.; Meijer, R.P.P.; Pijl, B.J.; Timmermans, J.; Cruysberg, J.R.M.; Bos, M.M.; Schelhaas, H.J.; Van De Warrenburg, B.P.C.; Knoers, N.V.A.M.; Scheffer, H.; et al. ARSACS in the Dutch population: A frequent cause of early-onset cerebellar ataxia. Neurogenetics 2008, 9, 207–214. [Google Scholar] [CrossRef]
- Ouyang, Y.; Segers, K.; Bouquiaux, O.; Wang, F.; Janin, N.; Andris, C.; Shimazaki, H.; Sakoe, K.; Nakano, I.; Takiyama, Y. Novel SACS mutation in a Belgian family with sacsin-related ataxia. J. Neurol. Sci. 2008, 264, 73–76. [Google Scholar] [CrossRef]
- Baets, J.; Deconinck, T.; Smets, K.; Goossens, D.; Bergh, P.V.D.; Dahan, K.; Schmedding, E.; Santens, P.; Rasic, V.M.; Van Damme, P.; et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 2010, 75, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, C.; Saccà, F.; De Michele, G.; Mancini, P.; Combarros, O.; Infante, J.; Garcia, A.; Banfi, S.; Filla, A.; Berciano, J. Novel mutation of SACS gene in a Spanish family with autosomal recessive spastic ataxia. Mov. Disord. 2005, 20, 1358–1361. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.G.; Pérez, J.E.; Pérez, M.R.; Redondo, A.G. Novel SACS mutation in autosomal recessive spastic ataxia of Charlevoix-Saguenay. J. Neurol. Sci. 2015, 358, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Xiromerisiou, G.; Dadouli, K.; Marogianni, C.; Provatas, A.; Ntellas, P.; Rikos, D.; Stathis, P.; Georgouli, D.; Loules, G.; Zamanakou, M.; et al. A novel homozygous SACS mutation identified by whole exome sequencing-genotype phenotype correlations of all published cases. J. Mol. Neurosci. 2020, 70, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Krygier, M.; Konkel, A.; Schinwelski, M.; Rydzanicz, M.; Walczak, A.; Sildatke-Bauer, M.; Płoski, R.; Slawek, J. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS)—A Polish family with novel SACS mutations. Neurol. Neurochir. Polska 2017, 51, 481–485. [Google Scholar] [CrossRef]
- Rudenskaya, G.E.; Kadnikova, V.A.; Ryzhkova, O.P. Spastic ataxia of Charlevoix-Saguenay: The first Russian case report and literature review. Zhurnal Nevrol. Psikhiatrii Im. SS Korsakova 2020, 120, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Johansson, S.; Haukanes, B.I.; Boman, H.; Knappskog, P.M.; Bindoff, L.A. Novel SACS Mutations Identified by Whole Exome Sequencing in a Norwegian Family with Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. PLoS ONE 2013, 8, e66145. [Google Scholar] [CrossRef] [PubMed]
- Vill, K.; Müller-Felber, W.; Gläser, D.; Kuhn, M.; Teusch, V.; Schreiber, H.; Weis, J.; Klepper, J.; Schirmacher, A.; Blaschek, A.; et al. SACS variants are a relevant cause of autosomal recessive hereditary motor and sensory neuropathy. Qual. Life Res. 2018, 137, 911–919. [Google Scholar] [CrossRef]
- Pyle, A.; Griffin, H.R.; Yu-Wai-Man, P.; Duff, J.; Eglon, G.; Pickering-Brown, S.; Santibanez-Korev, M.; Horvath, R.; Chinnery, P.F. Prominent Sensorimotor Neuropathy Due to SACS Mutations Revealed by Whole-Exome Sequencing. Arch. Neurol. 2012, 69, 1351–1354. [Google Scholar] [CrossRef]
- Parkinson, M.H.; Bartmann, A.P.; Clayton, L.M.S.; Nethisinghe, S.; Pfundt, R.; Chapple, J.P.; Reilly, M.M.; Manji, H.; Wood, N.; Bremner, F.; et al. Optical coherence tomography in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Brain 2018, 141, 989–999. [Google Scholar] [CrossRef]
- Miressi, F.; Faye, P.-A.; Pyromali, I.; Bourthoumieux, S.; Derouault, P.; Husson, M.; Favreau, F.; Sturtz, F.; Magdelaine, C.; Lia, A.-S. A mutation can hide another one: Think Structural Variants! Comput. Struct. Biotechnol. J. 2020, 18, 2095–2099. [Google Scholar] [CrossRef] [PubMed]
- Palmio, J.; Kärppä, M.; Baumann, P.; Penttilä, S.; Moilanen, J.S.; Udd, B. Novel compound heterozygous mutation in SACS gene leads to a milder autosomal recessive spastic ataxia of Charlevoix-Saguenay, ARSACS, in a Finnish family. Clin. Case Rep. 2016, 4, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Petrov, I. Novel Mutation in SACS Gene in a Patient with Autosomal Recessive Spastic Ataxia Charlevoix-Saguenay. Mov. Disord. Clin. Pract. 2021, 8, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Breckpot, J.; Takiyama, Y.; Thienpont, B.; Van Vooren, S.; Vermeesch, J.R.; Ortibus, E.; Devriendt, K. A novel genomic disorder: A deletion of the SACS gene leading to Spastic Ataxia of Charlevoix–Saguenay. Eur. J. Hum. Genet. 2008, 16, 1050–1054. [Google Scholar] [CrossRef]
- Pascual-Castroviejo, I.; I Pascual-Pascual, S.; Viaño, J.; Martínez, V. Carlevoix-Saguenay type recessive spastic ataxia. A report of a Spanish case. Rev. Neurol. 2000, 31, 36–38. [Google Scholar]
- Okawa, S.; Sugawara, M.; Watanabe, S.; Toyoshima, I.; Imota, T. A novel sacsin mutation in a Japanese woman showing clinical uniformity of autosomal recessive spastic ataxia of Charlevoix-Saguenay. J. Neurol. Neurosurg. Psychiatry 2006, 77, 280–282. [Google Scholar] [CrossRef][Green Version]
- Yamamoto, Y.; Hiraoka, K.; Araki, M.; Nagano, S.; Shimazaki, H.; Takiyama, Y.; Sakoda, S. Novel compound heterozygous mutations in sacsin-related ataxia. J. Neurol. Sci. 2005, 239, 101–104. [Google Scholar] [CrossRef]
- Ouyang, Y.; Takiyama, Y.; Sakoe, K.; Shimazaki, H.; Ogawa, T.; Nagano, S.; Yamamoto, Y.; Nakano, I. Sacsin-related ataxia (ARSACS): Expanding the genotype upstream from the gigantic exon. Neurology 2006, 66, 1103–1104. [Google Scholar] [CrossRef]
- Hara, K.; Onodera, O.; Endo, M.; Kondo, H.; Shiota, H.; Miki, K.; Tanimoto, N.; Kimura, T.; Nishizawa, M. Sacsin-related autosomal recessive ataxia without prominent retinal myelinated fibers in Japan. Mov. Disord. 2005, 20, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, H.; Sakoe, K.; Niijima, K.; Nakano, I.; Takiyama, Y. An unusual case of a spasticity-lacking phenotype with a novel SACS mutation. J. Neurol. Sci. 2007, 255, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, H.; Takiyama, Y.; Sakoe, K.; Ando, Y.; Nakano, I. A phenotype without spasticity in sacsin-related ataxia. Neurology 2005, 64, 2129–2131. [Google Scholar] [CrossRef]
- Hara, K.; Shimbo, J.; Nozaki, H.; Kikugawa, K.; Onodera, O.; Nishizawa, M. Sacsin-related ataxia with neither retinal hypermyelination nor spasticity. Mov. Disord. 2007, 22, 1362–1363. [Google Scholar] [CrossRef] [PubMed]
- Kamada, S.; Okawa, S.; Imota, T.; Sugawara, M.; Toyoshima, I. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): Novel compound heterozygous mutations in the SACS gene. J. Neurol. 2008, 255, 803–806. [Google Scholar] [CrossRef]
- Shimazaki, H.; Takiyama, Y.; Honda, J.; Sakoe, K.; Namekawa, M.; Tsugawa, J.; Tsuboi, Y.; Suzuki, C.; Baba, M.; Nakano, I. Middle Cerebellar Peduncles and Pontine T2 Hypointensities in ARSACS. J. Neuroimaging 2013, 23, 82–85. [Google Scholar] [CrossRef]
- Aida, I.; Ozawa, T.; Fujinaka, H.; Goto, K.; Ohta, K.; Nakajima, T. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay without Spasticity. Intern. Med. 2021, 7401–7421. [Google Scholar] [CrossRef]
- Haga, R.; Miki, Y.; Funamizu, Y.; Kon, T.; Suzuki, C.; Ueno, T.; Nishijima, H.; Arai, A.; Tomiyama, M.; Shimazaki, H.; et al. Novel compound heterozygous mutations of the SACS gene in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Clin. Neurol. Neurosurg. 2012, 114, 746–747. [Google Scholar] [CrossRef]
- Kwon, K.-Y.; Huh, K.; Eun, B.-L.; Yoo, H.-W.; Kamsteeg, E.-J.; Scheffer, H.; Koh, S.-B. A Probable Korean Case of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci./J. Can. Sci. Neurol. 2015, 42, 271–273. [Google Scholar] [CrossRef]
- Bong, J.B.; Kim, S.W.; Lee, S.T.; Choi, J.R.; Shin, H.Y. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. J. Korean Neurol. Assoc. 2019, 37, 69–72. [Google Scholar] [CrossRef]
- Cho, H.; Lyoo, C.H.; Park, S.E.; Seo, Y.; Han, S.-H.; Han, J. Optical Coherence Tomography Findings Facilitate the Diagnosis of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Korean J. Ophthalmol. 2021, 35, 330–331. [Google Scholar] [CrossRef]
- Liu, L.; Li, X.; Zi, X.; Shen, L.; Hu, Z.; Huang, S.; Yu, D.; Li, H.; Xia, K.; Tang, B.; et al. A novel hemizygous SACS mutation identified by whole exome sequencing and SNP array analysis in a Chinese ARSACS patient. J. Neurol. Sci. 2016, 362, 111–114. [Google Scholar] [CrossRef]
- Sun, W.; Meng, Y.U.; Zhuo, Y.; Wang, Z.; Yuan, Y. Novel spastic ataxia of Charlevoix-Saguenay gene compound heterozygous mutations in late onset autosomal recessive spastic ataxia of Charlevoix-Saguenay. Chin. J. Neurol. 2017, 50, 831–836. [Google Scholar]
- Luo, W.; Chen, Y.; Cen, Z.; Zheng, X.; Chen, S.; Xie, F. Novel Compound Heterozygous SACS Mutations in a Case with a Spasticity-Lacking Phenotype of Sacsin-Related Ataxia. Neurol. India 2021, 69, 219. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Shang, L.; Tian, W.T.; Cao, L.; Zhang, X.; Liu, Q. Complicated paroxysmal kinesigenic dyskinesia associated with SACS mutations. Ann. Transl. Med. 2020, 8, 8. [Google Scholar] [CrossRef]
- Li, S.; Chen, Y.; Yuan, X.; Wei, Q.; Ou, R.; Gu, X.; Shang, H. Identification of compound heterozygous mutations of SACS gene in two patients from a pedigree with spastic ataxia of Charlevoix-Saguenay. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2018, 35, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Song, Y.; Wang, X.; Li, X.; Xu, F.; Si, L.; Dong, Y.; Yao, T.; Zhu, J.; Lai, H.; et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay caused by novel mutations in SACS gene: A report of two Chinese families. Neurosci. Lett. 2021, 752, 135831. [Google Scholar] [CrossRef] [PubMed]
- Srikajon, J.; Pitakpatapee, Y.; Limwongse, C.; Chirapapaisan, N.; Srivanitchapoom, P. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) in a Thai Patient: The Classic Clinical Manifestations, Funduscopic Feature, and Brain Imaging Findings with a Novel Mutation in the SACS Gene. Tremor Other Hyperkinetic Mov. 2020, 10, 1. [Google Scholar] [CrossRef]
- Al-Ajmi, A.; Shamsah, S.; Janicijevic, A.; Williams, M.; Al-Mulla, F. Novel frameshift mutation in the SACS gene causing spastic ataxia of charlevoix-saguenay in a consanguineous family from the Arabian Peninsula: A case report and review of literature. World J. Clin. Cases 2020, 8, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Habibzadeh, P.; Tabatabaei, Z.; Inaloo, S.; Nashatizadeh, M.M.; Synofzik, M.; Ostovan, V.R.; Faghihi, M.A. Case Report: Expanding the Genetic and Phenotypic Spectrum of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Front. Genet. 2020, 11, 585136. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, Z.; Taheri, M.; Fallah, M.-S.; Abiri, M.; Golnabi, F.; Bagherian, H.; Zeinali, R.; Farahzadi, H.; Alborji, M.; Tehrani, P.G.; et al. Comprehensive Mutation Analysis and Report of 12 Novel Mutations in a Cohort of Patients with Spinal Muscular Atrophy in Iran. J. Mol. Neurosci. 2021, 71, 2281–2298. [Google Scholar] [CrossRef]
- Faruq, M.; Narang, A.; Kumari, R.; Pandey, R.; Garg, A.; Behari, M.; Dash, D.; Srivastava, A.; Mukerji, M. Novel mutations in typical and atypical genetic loci through exome sequencing in autosomal recessive cerebellar ataxia families. Clin. Genet. 2014, 86, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.A.; Ate-Upasani, P.; Ramprasad, V.L. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS)-First Report of Clinical and Imaging Features from India, and a Novel SACS Gene Duplication. Mov. Disord. Clin. Pract. 2017, 4, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, R.A.H.; Mir, Y.R.; Zeng, X.; Hassan, A.; Musarrat, J.; Parwez, I.; Kernstock, C.; Traschütz, A.; Synofzik, M. ARSACS as a Worldwide Disease: Novel SACS Mutations Identified in a Consanguineous Family from the Remote Tribal Jammu and Kashmir Region in India. Cerebellum 2019, 18, 807–812. [Google Scholar] [CrossRef]
- Sheetal, S.; Kumar, S.A.; Byju, P. SACS Mutation-Positive Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay (ARSACS) from Kerala. Ann. Indian Acad. Neurol. 2020, 23, 374–376. [Google Scholar] [PubMed]
- Qavi, A.; Agarwal, A.; Garg, D.; Kharat, A. Autosomal recessive spastic ataxia of charlevoix-saguenay (ARSACS): Case report of a novel nonsense mutation in the SACS gene. Ann. Indian Acad. Neurol. 2020, 23, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Klar, J.; Jameel, M.; Khan, K.; Fatima, A.; Raininko, R.; Baig, S.; Dahl, N. Novel SACS mutations associated with intellectual disability, epilepsy and widespread supratentorial abnormalities. J. Neurol. Sci. 2016, 371, 105–111. [Google Scholar] [CrossRef]
- Manzoor, H.; Brüggemann, N.; Hussain, H.M.J.; Bäumer, T.; Hinrichs, F.; Wajid, M.; Münchau, A.; Naz, S.; Lohmann, K. Novel homozygous variants in ATCAY, MCOLN1, and SACS in complex neurological disorders. Park. Relat. Disord. 2018, 51, 91–95. [Google Scholar] [CrossRef]
- Blumkin, L.; Bradshaw, T.; Michelson, M.; Kopler, T.; Dahari, D.; Lerman-Sagie, T.; Lev, D.; Chapple, J.P.; Leshinsky-Silver, E. Molecular and functional studies of retinal degeneration as a clinical presentation of SACS-related disorder. Eur. J. Paediatr. Neurol. 2015, 19, 472–476. [Google Scholar] [CrossRef]
- Narayanan, V.; Rice, S.G.; Olfers, S.S.; Sivakumar, K. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: Compound heterozygotes for nonsense mutations of the SACS gene. J. Child. Neurol. 2011, 26, 1585–1589. [Google Scholar] [CrossRef]
- Liew, W.K.; Ben-Omran, T.; Darras, B.T.; Prabhu, S.P.; Darryl, C.; Vatta, M.; Yang, Y.; Eng, C.M.; Chung, W.K. Clinical application of whole-exome sequencing: A novel autosomal recessive spastic ataxia of Charlevoix-Saguenay sequence variation in a child with ataxia. JAMA Neurol. 2013, 70, 788–791. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McElroy, J.P.; Krupp, L.; Johnson, B.; McCauley, J.L.; Qi, Z.; Caillier, S.J.; Gourraud, P.-A.; Yu, J.; Nathanson, L.; Belman, A.L.; et al. Copy number variation in pediatric multiple sclerosis. Mult. Scler. J. 2013, 19, 1014–1021. [Google Scholar] [CrossRef]
- Wagner, F.; Titelbaum, D.S.; Engisch, R.; Coskun, E.K.; Waugh, J.L. Subtle Imaging Findings Aid the Diagnosis of Adolescent Hereditary Spastic Paraplegia and Ataxia. Clin. Neuroradiol. 2019, 29, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, S.C.; Harper, A.; Al Saif, H.; Vorona, G.; Haines, S.R. A Chromosomal Deletion and New Frameshift Mutation Cause ARSACS in an African-American. Front. Neurol. 2018, 9, 956. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Lee, J.Y.; Lane, J.; Bs, A.W.; Chan, S.; Huang, A.; Bs, G.E.O.; Klein, E.; Mamah, C.; Perlman, S.; et al. Mutations in rare ataxia genes are uncommon causes of sporadic cerebellar ataxia. Mov. Disord. 2012, 27, 442–446. [Google Scholar] [CrossRef]
- Pedroso, J.L.; Braga-Neto, P.; Abrahao, A.; Rivero, R.L.M.; Abdalla, C.; Abdala, N.; Barsottini, O.G.P. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): Typical clinical and neuroimaging features in a Brazilian family. Arq. Neuro-Psiquiatr. 2011, 69, 288–291. [Google Scholar] [CrossRef]
- Burguêz, D.; De Oliveira, C.M.; Rockenbach, M.A.B.C.; Fussiger, H.; Vedolin, L.M.; Winckler, P.B.; Maestri, M.K.; Finkelsztejn, A.; Santorelli, F.M.; Jardim, L.B.; et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: A family report from South Brazil. Arq. Neuro-Psiquiatr. 2017, 75, 339–344. [Google Scholar] [CrossRef]
- Filho, F.M.R.; Parkinson, M.H.; Pedroso, J.L.; Poh, R.; Faber, I.; Lourenço, C.M.; Júnior, W.M.; Junior, M.C.F.; Kok, F.; Sallum, J.; et al. Clinical, ophthalmological, imaging and genetic features in Brazilian patients with ARSACS. Park. Relat. Disord. 2019, 62, 148–155. [Google Scholar] [CrossRef]
- Souza, P.V.S.; Bortholin, T.; Naylor, F.G.M.; Pinto, W.B.V.D.R.; Oliveira, A.S.B. Early-onset axonal Charcot-Marie-Tooth disease due to SACS mutation. Neuromuscul. Disord. 2018, 28, 169–172. [Google Scholar] [CrossRef]
- Nickerson, S.L.; Marquis-Nicholson, R.; Claxton, K.; Ashton, F.; Leong, I.U.S.; Prosser, D.O.; Love, J.M.; George, A.M.; Taylor, G.; Wilson, C.; et al. SNP Analysis and Whole Exome Sequencing: Their Application in the Analysis of a Consanguineous Pedigree Segregating Ataxia. Microarrays 2015, 4, 490–502. [Google Scholar] [CrossRef]
- Burnett, B.; Li, F.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205. [Google Scholar] [CrossRef]
- Feng, S.-T.; Wang, Z.-Z.; Yuan, Y.-H.; Wang, X.-L.; Sun, H.-M.; Chen, N.-H.; Zhang, Y. Dynamin-related protein 1: A protein critical for mitochondrial fission, mitophagy, and neuronal death in Parkinson’s disease. Pharmacol. Res. 2020, 151, 104553. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.; Reddy, P.H. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019, 8, 961. [Google Scholar] [CrossRef]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.F.; Siller, E.; Barral, J.M. The Neurodegenerative-Disease-Related Protein Sacsin Is a Molecular Chaperone. J. Mol. Biol. 2011, 411, 870–880. [Google Scholar] [CrossRef]
- Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Armijo, E.; Morales, R.; Kramm, C.; Flores, A.; Tiwari, A.; Bian, J.; Telling, G.C.; Pandita, T.K.; Hunt, C.R.; et al. Prion disease is accelerated in mice lacking stress-induced heat shock protein 70 (HSP70). J. Biol. Chem. 2019, 294, 13619–13628. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Berthet, A.; Citron, Y.R.; Tsiolaki, P.L.; Stanley, R.; Gestwicki, J.E.; Agard, D.A.; McConlogue, L. Hsp70 chaperone blocks α-synuclein oligomer formation via a novel engagement mechanism. J. Biol. Chem. 2021, 296, 100613. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Tang, J.-G.; Yang, Y.-F.; Tan, Z.-P.; Tan, J.-Q. A Novel Homozygous SACS Mutation Identified by Whole-Exome Sequencing in a Consanguineous Family with Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Cytogenet. Genome Res. 2017, 152, 16–21. [Google Scholar] [CrossRef]
- Rezende Filho, F.M.; Bremner, F.; Pedroso, J.L.; de Andrade, J.B.C.; Marianelli, B.F.; Lourenço, C.M.; Marques-Júnior, W.; França, M.C.; Kok, F.; Sallum, J.M.; et al. Retinal architecture in autosomal recessive spastic ataxia of charlevoix-saguenay (ARSACS): Insights into disease pathogenesis and biomarkers. Mov. Disord. 2021, 36, 2027–2035. [Google Scholar] [CrossRef]
- Lessard, I.; Lavoie, C.; Côté, I.; Mathieu, J.; Brais, B.; Gagnon, C. Validity and reliability of the LEMOCOT in the adult ARSACS population: A measure of lower limb coordination. J. Neurol. Sci. 2017, 377, 193–1966. [Google Scholar] [CrossRef]
- Bui, H.T.; Gagnon, C.; Audet, O.; Mathieu, J.; Leone, M. Measurement properties of a new wireless electrogoniometer for quantifying spasticity during the pendulum test in ARSACS patients. J. Neurol. Sci. 2017, 375, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Audet, O.; Bui, H.T.; Allisse, M.; Comtois, A.-S.; Leone, M. Assessment of the impact of an exercise program on the physical and functional capacity in patients with autosomal recessive spastic ataxia of Charlevoix-Saguenay: An exploratory study. Intractable Rare Dis. Res. 2018, 7, 164–171. [Google Scholar] [CrossRef]
- Vogel, A.P.; Stoll, L.H.; Oettinger, A.; Rommel, N.; Kraus, E.-M.; Timmann, D.; Scott, D.; Atay, C.; Storey, E.; Schöls, L.; et al. Speech treatment improves dysarthria in multisystemic ataxia: A rater-blinded, controlled pilot-study in ARSACS. J. Neurol. 2019, 266, 1260–1266. [Google Scholar] [CrossRef]
- Martinelli, C.; Battaglini, M.; Pucci, C.; Gioi, S.; Caracci, C.; Macaluso, G.; Doccini, S.; Santorelli, F.M.; Ciofani, G. Development of Nanostructured Lipid Carriers for the Delivery of Idebenone in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. ACS Omega 2020, 5, 12451–12466. [Google Scholar] [CrossRef] [PubMed]
- Ricca, I.; Tessa, A.; Trovato, R.; Bacci, G.M.; Santorelli, F.M. Docosahexaenoic acid in ARSACS: Observations in two patients. BMC Neurol. 2020, 20, 215. [Google Scholar] [CrossRef] [PubMed]
- Nethisinghe, S.; Abeti, R.; Kesavan, M.; Wigley, W.C.; Giunti, P. Hsp90 Inhibition: A Promising Therapeutic Approach for ARSACS. Int. J. Mol. Sci. 2021, 22, 11722. [Google Scholar] [CrossRef]
- Zuehlke, A.D.; Moses, M.A.; Neckers, L. Heat shock protein 90: Its inhibition and function. Philos. Trans. R. Soc. B Biol. Sci. 2017, 373, 20160527. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagaria, J.; Bagyinszky, E.; An, S.S.A. Genetics of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) and Role of Sacsin in Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 552. https://doi.org/10.3390/ijms23010552
Bagaria J, Bagyinszky E, An SSA. Genetics of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) and Role of Sacsin in Neurodegeneration. International Journal of Molecular Sciences. 2022; 23(1):552. https://doi.org/10.3390/ijms23010552
Chicago/Turabian StyleBagaria, Jaya, Eva Bagyinszky, and Seong Soo A. An. 2022. "Genetics of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) and Role of Sacsin in Neurodegeneration" International Journal of Molecular Sciences 23, no. 1: 552. https://doi.org/10.3390/ijms23010552
APA StyleBagaria, J., Bagyinszky, E., & An, S. S. A. (2022). Genetics of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) and Role of Sacsin in Neurodegeneration. International Journal of Molecular Sciences, 23(1), 552. https://doi.org/10.3390/ijms23010552