
Non-Alcoholic Steatohepatitis (NASH) and Organokines: What Is Now and What Will Be in the Future

 ,
,  , , and
, , and 
Abstract
:
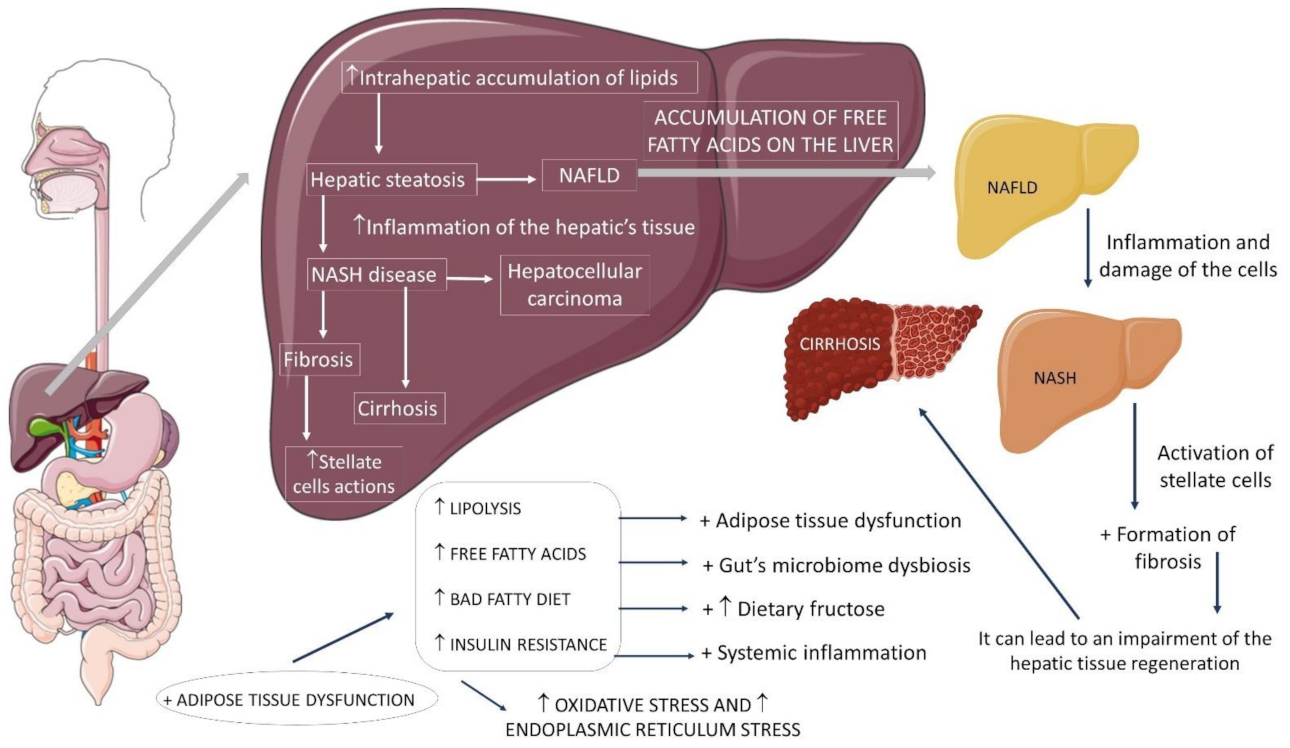
1. Introduction
2. Insulin Resistance, the Role of Fructose, Gut Microbiota and Organokines
2.1. Insulin Resistance, Inflammation, and Mitochondrial Dysfunction
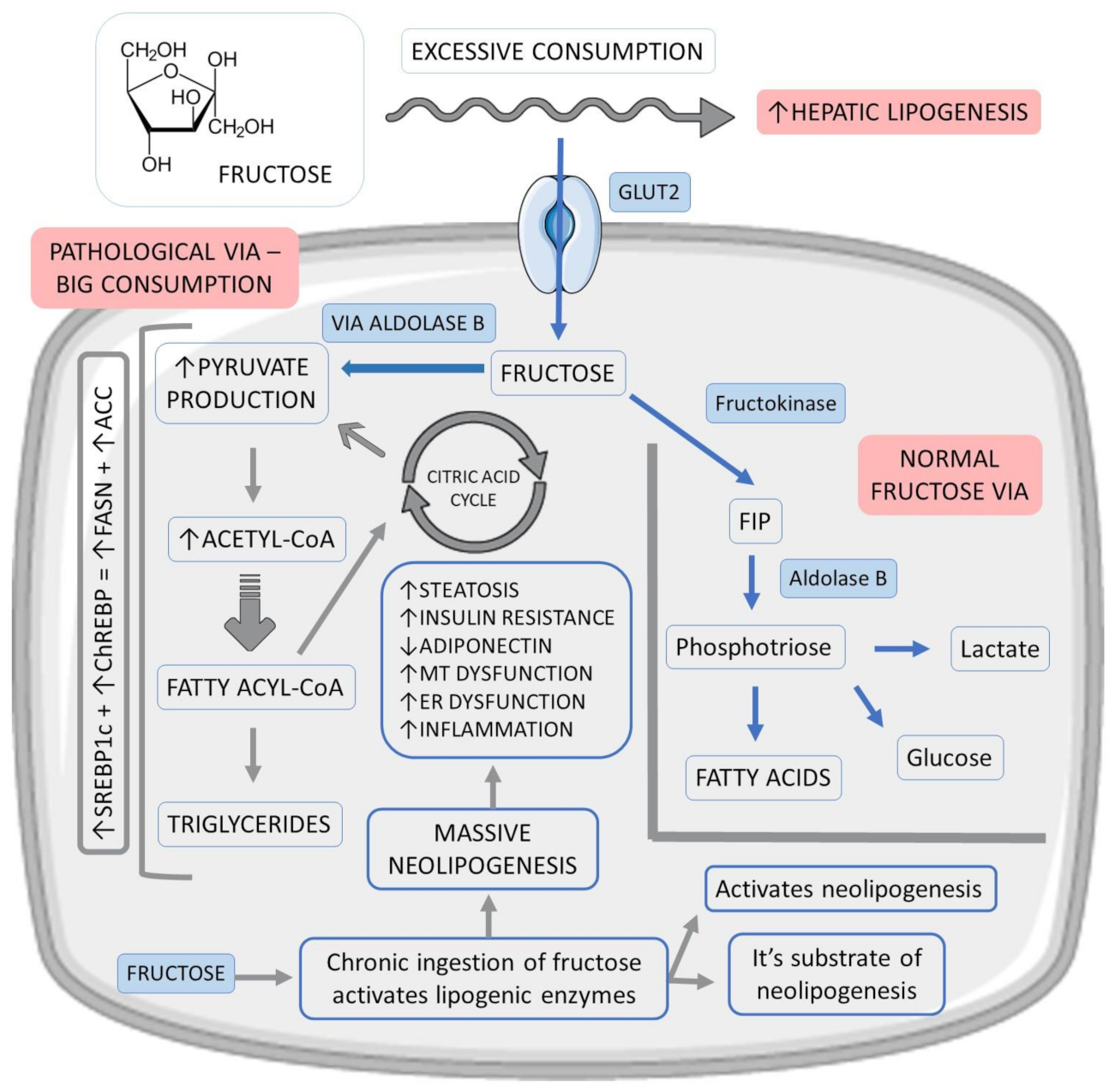
2.2. The Role of Fructose
2.3. Intestinal Microbiota
2.4. The Role of Diacylglycerols
3. Organokines
3.1. Adipokines
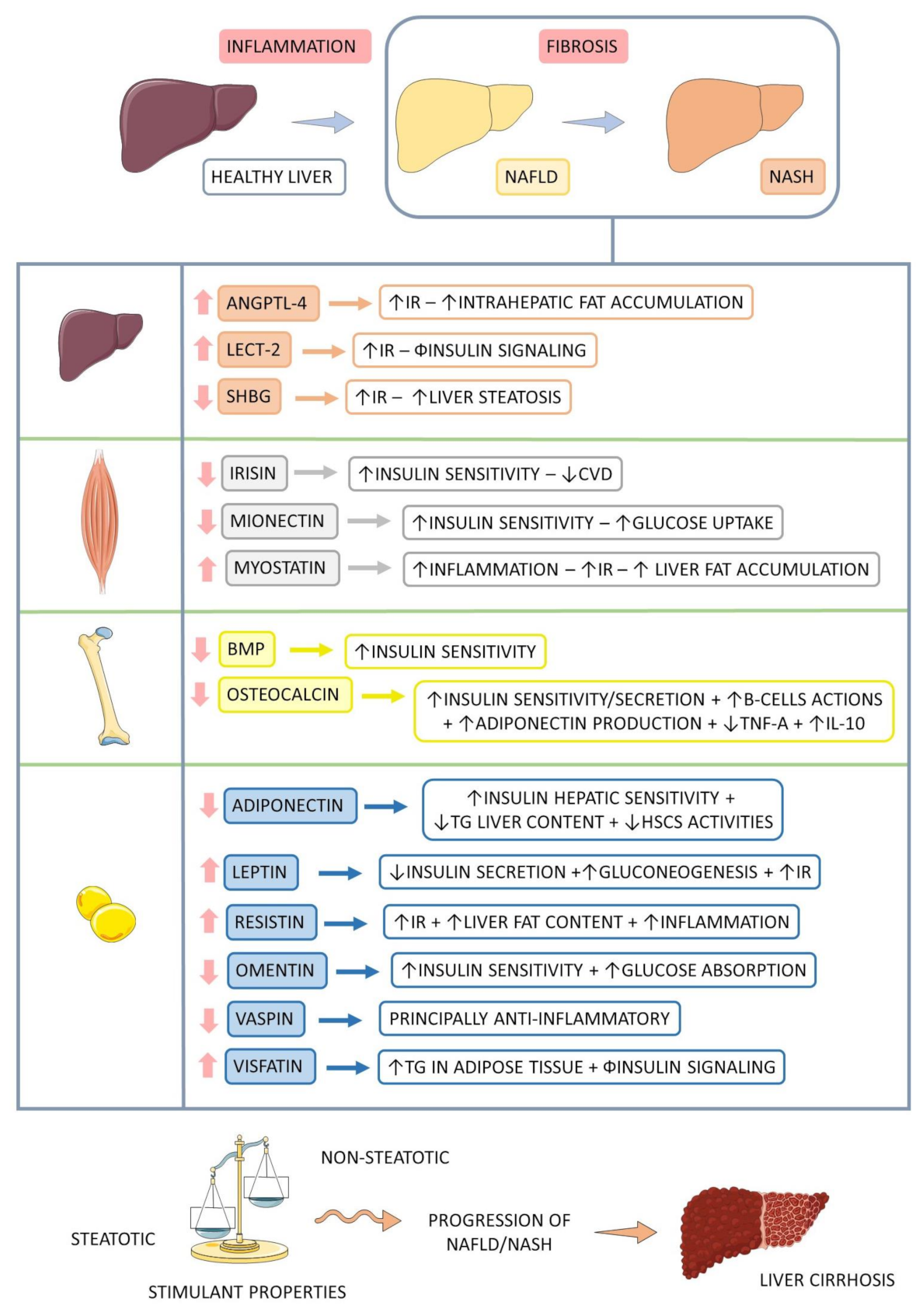
3.1.1. Adiponectin
3.1.2. Leptin
3.1.3. Omentin
3.1.4. Resistin
3.1.5. Vaspin
3.1.6. Visfatin
3.2. Myokines
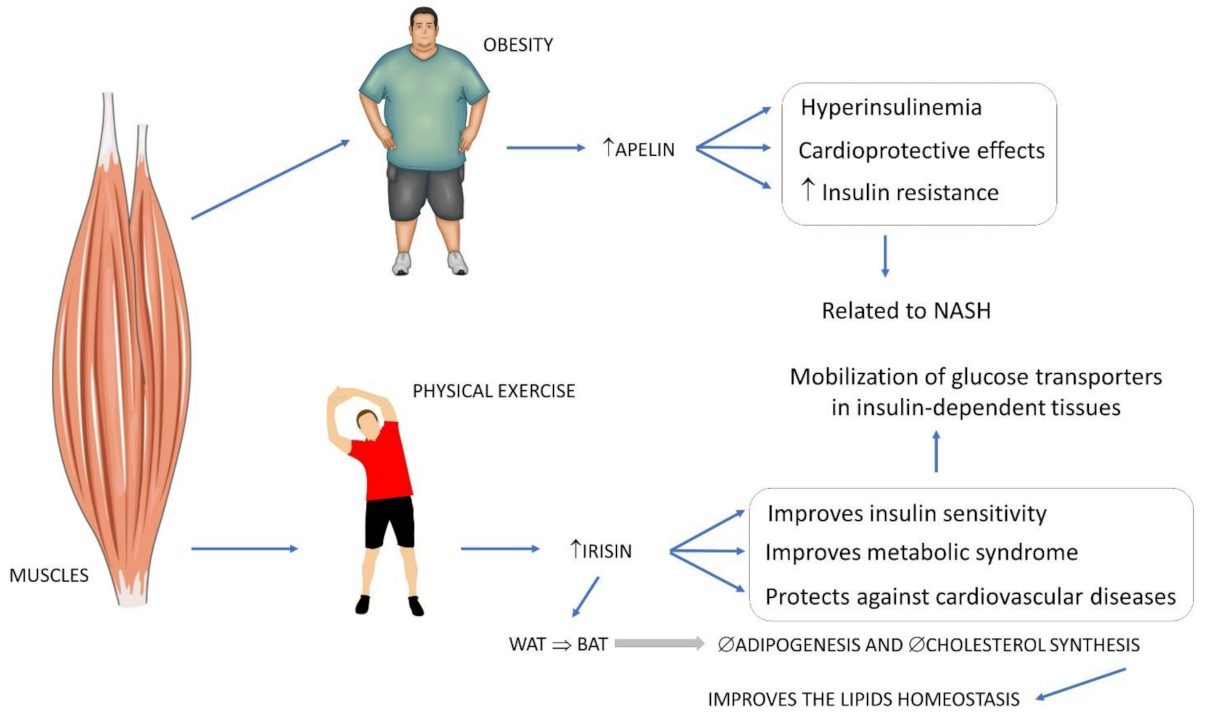
3.2.1. Irisin
3.2.2. Mionectin
3.2.3. Myostatin
3.3. Hepatokines
3.3.1. Angiopoietin-Like 4 (ANGPTL 4)
3.3.2. Leukocyte Cell-Derived Chemotaxin 2 (LECT2)
3.3.3. Sexual Hormone-Binding Globulin (SHBG)
3.4. Osteokines
3.4.1. Bone Morphogenetic Protein (BMP)
3.4.2. Osteocalcin (OCN)
3.5. Organokines: A Cross-Talk
3.5.1. Apelin
3.5.2. Chemerin
3.5.3. Fetuin-A
3.5.4. Fibroblast Growth Factor 21 (FGF-21)
3.5.5. Follistatin
3.5.6. IL-6
3.5.7. Lipocalin 2 (LCN-2)
3.5.8. Osteonectin
3.5.9. Selenoprotein-P (SeP)
3.5.10. TNF-α
3.5.11. TGF-β
4. Paths to Unravel
4.1. Genetics
4.2. Diet and Microbiota
4.3. Exercise
4.4. Sleep, Melatonin and Organokines
4.5. Other Metabolic Conditions
4.6. Intervention, Management, and Therapy
5. General Comments
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ajmera, V.; Loomba, R. Imaging biomarkers of NAFLD, NASH, and fibrosis. Mol. Metab. 2021, 50, 101167. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Jornayvaz, F.R. Pathophysiology of NASH in endocrine diseases. Endocr. Connect. 2021, 10, R52–R65. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Morone, M.V.; Silvestri, C.; Giordano, M.; Salvatore, T.; Sasso, F.C. Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants 2021, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Ayada, I.; van Kleef, L.A.; Alferink, L.J.M.; Li, P.; de Knegt, R.J.; Pan, Q. Systematically comparing epidemiological and clinical features of MAFLD and NAFLD by meta-analysis: Focusing on the non-overlap groups. Liver. Int. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Effenberger, M. From NAFLD to MAFLD: When pathophysiology succeeds. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 387–388. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zheng, J.; Zhang, S.; Wang, B.; Wu, C.; Guo, X. Advances in the Involvement of Gut Microbiota in Pathophysiology of NAFLD. Front. Med. 2020, 7, 361. [Google Scholar] [CrossRef]
- Lonardo, A.; Mantovani, A.; Lugari, S.; Targher, G. Epidemiology and pathophysiology of the association between NAFLD and metabolically healthy or metabolically unhealthy obesity. Ann. Hepatol. 2020, 19, 359–366. [Google Scholar] [CrossRef]
- Baltieri, L.; Chaim, E.; Chaim, F.; Utrini, M.; Gestic, M.; Cazzo, E. Correlation between nonalcoholic fatty liver disease features and levels of adipokines and inflammatory cytokines among morbidly obese individuals. Arq. Gastroenterol. 2018, 55, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- DiStefano, J.K. NAFLD and NASH in Postmenopausal Women: Implications for Diagnosis and Treatment. Endocrinology 2020, 161, bqaa134. [Google Scholar] [CrossRef]
- Zarghamravanbakhsh, P.; Frenkel, M.; Poretsky, L. Metabolic causes and consequences of nonalcoholic fatty liver disease (NAFLD). Metab. Open 2021, 12, 100149. [Google Scholar] [CrossRef]
- Kim, H.; Lee, D.S.; An, T.H.; Park, H.J.; Kim, W.K.; Bae, K.H.; Oh, K.J. Metabolic Spectrum of Liver Failure in Type 2 Diabetes and Obesity: From NAFLD to NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4495. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Choi, K.M. Chapter Six—Organokines in disease. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 94, pp. 261–321. [Google Scholar]
- Kolb, H.; Kempf, K.; Röhling, M.; Martin, S. Insulin: Too much of a good thing is bad. BMC Med. 2020, 18, 224. [Google Scholar] [CrossRef]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef] [PubMed]
- Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Attia, S.L.; Softic, S.; Mouzaki, M. Evolving Role for Pharmacotherapy in NAFLD/NASH. Clin. Transl. Sci. 2021, 14, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants. Biomolecules 2020, 10, 1702. [Google Scholar] [CrossRef]
- Kimura, T.; Singh, S.; Tanaka, N.; Umemura, T. Role of G Protein-Coupled Receptors in Hepatic Stellate Cells and Approaches to Anti-Fibrotic Treatment of Non-Alcoholic Fatty Liver Disease. Front. in Endocrinol. 2021, 12, 773432. [Google Scholar] [CrossRef]
- Fujii, H.; Kawada, N.; Japan Study Group of NAFLD. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef]
- Secor, J.D.; Fligor, S.C.; Tsikis, S.T.; Yu, L.J.; Puder, M. Free Fatty Acid Receptors as Mediators and Therapeutic Targets in Liver Disease. Front. Physiol. 2021, 12, 656441. [Google Scholar] [CrossRef]
- Albhaisi, S.; Noureddin, M. Current and Potential Therapies Targeting Inflammation in NASH. Front. Endocrinol. 2021, 12, 767314. [Google Scholar] [CrossRef]
- Xiao, X.; Hu, Q.; Deng, X.; Shi, K.; Zhang, W.; Jiang, Y.; Ma, X.; Zeng, J.; Wang, X. Old wine in new bottles: Kaempferol is a promising agent for treating the trilogy of liver diseases. Pharmacol. Res. 2021, 175, 106005. [Google Scholar] [CrossRef]
- Farrell, G.C.; Haczeyni, F.; Chitturi, S. Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. Adv. Exp. Med. Biol. 2018, 1061, 19–44. [Google Scholar] [CrossRef]
- Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4173. [Google Scholar] [CrossRef]
- Bates, J.; Vijayakumar, A.; Ghoshal, S.; Marchand, B.; Yi, S.; Kornyeyev, D.; Zagorska, A.; Hollenback, D.; Walker, K.; Liu, K.; et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J. Hepatol. 2020, 73, 896–905. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef]
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e734. [Google Scholar] [CrossRef]
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxid. Med. Cell. Longev. 2020, 2020, 1617805. [Google Scholar] [CrossRef]
- Dong, J.; Viswanathan, S.; Adami, E.; Singh, B.K.; Chothani, S.P.; Ng, B.; Lim, W.W.; Zhou, J.; Tripathi, M.; Ko, N.S.J.; et al. Hepatocyte-specific IL11 cis-signaling drives lipotoxicity and underlies the transition from NAFLD to NASH. Nat. Commun. 2021, 12, 66. [Google Scholar] [CrossRef]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeb, E.; Weiskirchen, R. Fructose and Non-Alcoholic Steatohepatitis. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Neuenschwander, M.; Hoffmann, G.; Buyken, A.E.; Schlesinger, S. Dietary sugars and cardiometabolic risk factors: A network meta-analysis on isocaloric substitution interventions. Am. J. Clin. Nutr. 2020, 111, 187–196. [Google Scholar] [CrossRef]
- Jegatheesan, P.; De Bandt, J.-P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [Green Version]
- DiStefano, J.K. Fructose-mediated effects on gene expression and epigenetic mechanisms associated with NAFLD pathogenesis. Cell. Mol. Life Sci. 2020, 77, 2079–2090. [Google Scholar] [CrossRef]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Muriel, P.; López-Sánchez, P.; Ramos-Tovar, E. Fructose and the Liver. Int. J. Mol. Sci. 2021, 22, 6969. [Google Scholar] [CrossRef]
- Powell, E.S.; Smith-Taillie, L.P.; Popkin, B.M. Added Sugars Intake Across the Distribution of US Children and Adult Consumers: 1977–2012. J. Acad. Nutr. Diet. 2016, 116, 1543–1550.e1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievenpiper, J.L.; de Souza, R.J.; Cozma, A.I.; Chiavaroli, L.; Ha, V.; Mirrahimi, A. Fructose vs. glucose and metabolism: Do the metabolic differences matter? Curr. Opin. Lipidol. 2014, 25, 8–19. [Google Scholar] [CrossRef]
- Chan, A.M.L.; Ng, A.M.H.; Mohd Yunus, M.H.; Idrus, R.B.H.; Law, J.X.; Yazid, M.D.; Chin, K.-Y.; Shamsuddin, S.A.; Lokanathan, Y. Recent Developments in Rodent Models of High-Fructose Diet-Induced Metabolic Syndrome: A Systematic Review. Nutrients 2021, 13, 2497. [Google Scholar] [CrossRef]
- Eshraghian, A.; Nikeghbalian, S.; Geramizadeh, B.; Kazemi, K.; Shamsaeefar, A.; Malek-Hosseini, S.A. Characterization of biopsy proven non-alcoholic fatty liver disease in healthy non-obese and lean population of living liver donors: The impact of uric acid. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 572–578. [Google Scholar] [CrossRef]
- Brennan, P.; Clare, K.; George, J.; Dillon, J.F. Determining the role for uric acid in non-alcoholic steatohepatitis development and the utility of urate metabolites in diagnosis: An opinion review. World J. Gastroenterol. 2020, 26, 1683–1690. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, J.; Shi, H.; Hu, W.; Song, L.; Zhao, Q. Serum uric acid is positively associated with the prevalence of nonalcoholic fatty liver in non-obese type 2 diabetes patients in a Chinese population. J. Diabetes Complicat. 2021, 35, 107874. [Google Scholar] [CrossRef]
- Federico, A.; Rosato, V.; Masarone, M.; Torre, P.; Dallio, M.; Romeo, M.; Persico, M. The Role of Fructose in Non-Alcoholic Steatohepatitis: Old Relationship and New Insights. Nutrients 2021, 13, 1314. [Google Scholar] [CrossRef]
- Pérez-Montes de Oca, A.; Julián, M.T.; Ramos, A.; Puig-Domingo, M.; Alonso, N. Microbiota, Fiber, and NAFLD: Is There Any Connection? Nutrients 2020, 12, 3100. [Google Scholar] [CrossRef]
- Carter, J.K.; Bhattacharya, D.; Borgerding, J.N.; Fiel, M.I.; Faith, J.J.; Friedman, S.L. Modeling dysbiosis of human NASH in mice: Loss of gut microbiome diversity and overgrowth of Erysipelotrichales. PLoS ONE 2021, 16, e0244763. [Google Scholar] [CrossRef]
- Martinez, K.B.; Leone, V.; Chang, E.B. Western diets, gut dysbiosis, and metabolic diseases: Are they linked? Gut Microb. 2017, 8, 130–142. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-C.; Liu, Y.-Y.; Lin, C.-C.; Wang, C.-C.; Wu, Y.-J.; Yong, C.-C.; Chen, K.-D.; Chuah, S.-K.; Yao, C.-C.; Huang, P.-Y.; et al. Gut Microbiota Dysbiosis in Patients with Biopsy-Proven Nonalcoholic Fatty Liver Disease: A Cross-Sectional Study in Taiwan. Nutrients 2020, 12, 820. [Google Scholar] [CrossRef] [Green Version]
- Ferro, D.; Baratta, F.; Pastori, D.; Cocomello, N.; Colantoni, A.; Angelico, F.; Del Ben, M. New Insights into the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Gut-Derived Lipopolysaccharides and Oxidative Stress. Nutrients 2020, 12, 2762. [Google Scholar] [CrossRef]
- Vasques-Monteiro, I.M.L.; Silva-Veiga, F.M.; Miranda, C.S.; de Andrade Gonçalves, É.C.B.; Daleprane, J.B.; Souza-Mello, V. A rise in Proteobacteria is an indicator of gut-liver axis-mediated nonalcoholic fatty liver disease in high-fructose-fed adult mice. Nutr. Res. 2021, 91, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Semmler, G.; Datz, C.; Reiberger, T.; Trauner, M. Diet and exercise in NAFLD/NASH: Beyond the obvious. Liver. Int. 2021, 41, 2249–2268. [Google Scholar] [CrossRef] [PubMed]
- Lyu, K.; Zhang, Y.; Zhang, D.; Kahn, M.; Ter Horst, K.W.; Rodrigues, M.R.S.; Gaspar, R.C.; Hirabara, S.M.; Luukkonen, P.K.; Lee, S.; et al. A Membrane-Bound Diacylglycerol Species Induces PKCϵ-Mediated Hepatic Insulin Resistance. Cell Metab. 2020, 32, 654–664.e655. [Google Scholar] [CrossRef]
- Marušić, M.; Paić, M.; Knobloch, M.; Liberati Pršo, A.-M. NAFLD, Insulin Resistance, and Diabetes Mellitus Type 2. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6613827. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Morgan, E.; Watts, L.; Xia, S.; Hannan, L.A.; Geary, R.S.; Baker, B.F.; Bhanot, S. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: A multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 829–838. [Google Scholar] [CrossRef]
- Kirk, B.; Feehan, J.; Lombardi, G.; Duque, G. Muscle, Bone, and Fat Crosstalk: The Biological Role of Myokines, Osteokines, and Adipokines. Curr. Osteoporos Rep. 2020, 18, 388–400. [Google Scholar] [CrossRef]
- Kucukoglu, O.; Sowa, J.P.; Mazzolini, G.D.; Syn, W.K.; Canbay, A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J. Hepatol. 2021, 74, 442–457. [Google Scholar] [CrossRef]
- Choi, K.M. The Impact of Organokines on Insulin Resistance, Inflammation, and Atherosclerosis. Endocrinol. Metab. 2016, 31, 1–6. [Google Scholar] [CrossRef]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharm. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef] [Green Version]
- Albhaisi, S.; Sanyal, A. Recent advances in understanding and managing non-alcoholic fatty liver disease. F1000Research 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Petta, S.; Gastaldelli, A.; Rebelos, E.; Bugianesi, E.; Messa, P.; Miele, L.; Svegliati-Baroni, G.; Valenti, L.; Bonino, F. Pathophysiology of Non Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 2082. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira dos Santos, A.R.; de Oliveira Zanuso, B.; Miola, V.F.B.; Barbalho, S.M.; Santos Bueno, P.C.; Flato, U.A.P.; Detregiachi, C.R.P.; Buchaim, D.V.; Buchaim, R.L.; Tofano, R.J.; et al. Adipokines, Myokines, and Hepatokines: Crosstalk and Metabolic Repercussions. Int. J. Mol. Sci. 2021, 22, 2639. [Google Scholar] [CrossRef] [PubMed]
- Panera, N.; Della Corte, C.; Crudele, A.; Stronati, L.; Nobili, V.; Alisi, A. Recent advances in understanding the role of adipocytokines during non-alcoholic fatty liver disease pathogenesis and their link with hepatokines. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 393–403. [Google Scholar] [CrossRef]
- Marques, V.; Afonso, M.B.; Bierig, N.; Duarte-Ramos, F.; Santos-Laso, Á.; Jimenez-Agüero, R.; Eizaguirre, E.; Bujanda, L.; Pareja, M.J.; Luís, R.; et al. Adiponectin, Leptin, and IGF-1 Are Useful Diagnostic and Stratification Biomarkers of NAFLD. Front. Med. 2021, 8, 811. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Cortegana, C.; García-Galey, A.; Tami, M.; del Pino, P.; Carmona, I.; López, S.; Alba, G.; Sánchez-Margalet, V. Role of Leptin in Non-Alcoholic Fatty Liver Disease. Biomedicines 2021, 9, 762. [Google Scholar] [CrossRef]
- Zorena, K.; Jachimowicz-Duda, O.; Ślęzak, D.; Robakowska, M.; Mrugacz, M. Adipokines and Obesity. Potential Link to Metabolic Disorders and Chronic Complications. Int. J. Mol. Sci. 2020, 21, 3570. [Google Scholar] [CrossRef] [PubMed]
- Parrettini, S.; Cavallo, M.; Gaggia, F.; Calafiore, R.; Luca, G. Adipokines: A Rainbow of Proteins with Metabolic and Endocrine Functions. Protein Pept. Lett. 2020, 27, 1204–1230. [Google Scholar] [CrossRef]
- Han, D.; Chen, J.; Liu, S.; Zhang, Z.; Zhao, Z.; Jin, W.; Xin, Y. Serum Resistin Levels in Adult Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. J. Clin. Transl. Hepatol. 2021, 9, 484–493. [Google Scholar] [CrossRef]
- Deb, A.; Deshmukh, B.; Ramteke, P.; Bhati, F.K.; Bhat, M.K. Resistin: A journey from metabolism to cancer. Transl. Oncol. 2021, 14, 101178. [Google Scholar] [CrossRef]
- Díaz, B.B.; González, D.A.; Gannar, F.; Pérez, M.C.R.; de León, A.C. Myokines, physical activity, insulin resistance and autoimmune diseases. Immunol. Lett. 2018, 203, 1–5. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Anastasilakis, A.D. Irisin in nonalcoholic fatty liver disease: Need for an updated meta-analysis. Metabolism 2021, 121, 154818. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Anastasilakis, A.D.; Geladari, E.V.; Mantzoros, C.S. Irisin in patients with nonalcoholic fatty liver disease. Metabolism 2014, 63, 207–217. [Google Scholar] [CrossRef]
- Gonzalez-Gil, A.M.; Elizondo-Montemayor, L. The Role of Exercise in the Interplay between Myokines, Hepatokines, Osteokines, Adipokines, and Modulation of Inflammation for Energy Substrate Redistribution and Fat Mass Loss: A Review. Nutrients 2020, 12, 1899. [Google Scholar] [CrossRef]
- Delogu, W.; Caligiuri, A.; Provenzano, A.; Rosso, C.; Bugianesi, E.; Coratti, A.; Macias-Barragan, J.; Galastri, S.; Di Maira, G.; Marra, F. Myostatin regulates the fibrogenic phenotype of hepatic stellate cells via c-jun N-terminal kinase activation. Dig. Liver Dis. 2019, 51, 1400–1408. [Google Scholar] [CrossRef]
- Ke, Y.; Xu, C.; Lin, J.; Li, Y. Role of Hepatokines in Non-alcoholic Fatty Liver Disease. J. Transl. Int. Med. 2019, 7, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Lebensztejn, D.M.; Flisiak-Jackiewicz, M.; Białokoz-Kalinowska, I.; Bobrus-Chociej, A.; Kowalska, I. Hepatokines and non-alcoholic fatty liver disease. Acta Biochim. Pol. 2016, 63, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Barja-Fernández, S.; Folgueira, C.; Castelao, C.; Pena-León, V.; González-Saenz, P.; Vázquez-Cobela, R.; Aguilera, C.M.; Gil-Campos, M.; Bueno, G.; Gil, Á.; et al. ANGPTL-4 is Associated with Obesity and Lipid Profile in Children and Adolescents. Nutrients 2019, 11, 1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, N.; Ishii, K.-a.; Takayama, H.; Nagashimada, M.; Kamoshita, K.; Tanaka, T.; Kikuchi, A.; Takeshita, Y.; Matsumoto, Y.; Ota, T.; et al. LECT2 as a hepatokine links liver steatosis to inflammation via activating tissue macrophages in NASH. Sci. Rep. 2021, 11, 555. [Google Scholar] [CrossRef] [PubMed]
- Sáez-López, C.; Salcedo-Allende, M.T.; Hernandez, C.; Simó-Servat, O.; Simó, R.; Selva, D.M. Sex Hormone-Binding Globulin Expression Correlates With Acetyl-Coenzyme A Carboxylase and Triglyceride Content in Human Liver. J. Clin. Endocrinol. Metab. 2019, 104, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Ho, H.N. Hepatic manifestations of women with polycystic ovary syndrome. Best Pr. Res. Clin. Obs. Gynaecol. 2016, 37, 119–128. [Google Scholar] [CrossRef]
- Baboota, R.K.; Blüher, M.; Smith, U. Emerging Role of Bone Morphogenetic Protein 4 in Metabolic Disorders. Diabetes 2021, 70, 303–312. [Google Scholar] [CrossRef]
- Colaianni, G.; Storlino, G.; Sanesi, L.; Colucci, S.; Grano, M. Myokines and Osteokines in the Pathogenesis of Muscle and Bone Diseases. Curr. Osteoporos. Rep. 2020, 18, 401–407. [Google Scholar] [CrossRef]
- Oury, F.; Khrimian, L.; Denny, C.A.; Gardin, A.; Chamouni, A.; Goeden, N.; Huang, Y.Y.; Lee, H.; Srinivas, P.; Gao, X.B.; et al. Maternal and offspring pools of osteocalcin influence brain development and functions. Cell 2013, 155, 228–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.M.; Singh, P.; Khrimian, L.; Morgan, D.A.; Chowdhury, S.; Arteaga-Solis, E.; Horvath, T.L.; Domingos, A.I.; Marsland, A.L.; Yadav, V.K.; et al. Mediation of the Acute Stress Response by the Skeleton. Cell Metab. 2019, 30, 890–902.e898. [Google Scholar] [CrossRef]
- Xia, M.; Rong, S.; Zhu, X.; Yan, H.; Chang, X.; Sun, X.; Zeng, H.; Li, X.; Zhang, L.; Chen, L.; et al. Osteocalcin and Non-Alcoholic Fatty Liver Disease: Lessons From Two Population-Based Cohorts and Animal Models. J. Bone Miner. Res. 2021, 36, 712–728. [Google Scholar] [CrossRef] [PubMed]
- Barbalho, S.M.; Flato, U.A.P.; Tofano, R.J.; Goulart, R.d.A.; Guiguer, E.L.; Detregiachi, C.R.P.; Buchaim, D.V.; Araújo, A.C.; Buchaim, R.L.; Reina, F.T.R.; et al. Physical Exercise and Myokines: Relationships with Sarcopenia and Cardiovascular Complications. Int. J. Mol. Sci. 2020, 21, 3607. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Chen, Q. Adipokines: New Therapeutic Target for Osteoarthritis? Curr. Rheumatol. Rep. 2019, 21, 71. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Liu, J.; Li, Y.; Dou, Z.; Li, N.; Suo, Y.; Ma, Y.; Sun, M.; Tian, Z.; Xu, L. Chemerin/CMKLR1 ameliorates nonalcoholic steatohepatitis by promoting autophagy and alleviating oxidative stress through the JAK2-STAT3 pathway. Peptides 2021, 135, 170422. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Yang, K.; Zang, S.; Lin, Z.; Chau, H.T.; Wang, Y.; Zhang, J.; Shi, J.; Xu, A.; Lin, S.; et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 2016, 65, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef]
- Mazzolini, G.; Atorrasagasti, C.; Onorato, A.; Peixoto, E.; Schlattjan, M.; Sowa, J.-P.; Sydor, S.; Gerken, G.; Canbay, A. SPARC expression is associated with hepatic injury in rodents and humans with non-alcoholic fatty liver disease. Sci. Rep. 2018, 8, 725. [Google Scholar] [CrossRef] [Green Version]
- Onorato, A.M.; Fiore, E.; Bayo, J.; Casali, C.; Fernandez-Tomé, M.; Rodríguez, M.; Domínguez, L.; Argemi, J.; Hidalgo, F.; Favre, C.; et al. SPARC inhibition accelerates NAFLD-associated hepatocellular carcinoma development by dysregulating hepatic lipid metabolism. Liver. Int. 2021, 41, 1677–1693. [Google Scholar] [CrossRef]
- Flisiak-Jackiewicz, M.; Bobrus-Chociej, A.; Wasilewska, N.; Tarasow, E.; Wojtkowska, M.; Lebensztejn, D.M. Can hepatokines be regarded as novel non-invasive serum biomarkers of intrahepatic lipid content in obese children? Adv. Med. Sci. 2019, 64, 280–284. [Google Scholar] [CrossRef]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (NAFLD)—Pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Ezquerro, S.; Mocha, F.; Frühbeck, G.; Guzmán-Ruiz, R.; Valentí, V.; Mugueta, C.; Becerril, S.; Catalán, V.; Gómez-Ambrosi, J.; Silva, C.; et al. Ghrelin Reduces TNF-α-Induced Human Hepatocyte Apoptosis, Autophagy, and Pyroptosis: Role in Obesity-Associated NAFLD. J Clin. Endocrinol. Metab. 2019, 104, 21–37. [Google Scholar] [CrossRef]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014, 59, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Janzi, S.; González-Padilla, E.; Najafi, K.; Ramne, S.; Ahlqvist, E.; Borné, Y.; Sonestedt, E. Single Nucleotide Polymorphisms in Close Proximity to the Fibroblast Growth Factor 21 (FGF21) Gene Found to be Associated with Sugar Intake in a Swedish Population. Nutrients 2021, 13, 3954. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef]
- Safari, Z.; Gérard, P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell. Mol. Life Sci. CMLS 2019, 76, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Tsujimoto, T.; Kim, B.; Uchida, F.; Suzuki, H.; Iizumi, S.; Isobe, T.; Sakae, T.; Tanaka, K.; Shoda, J. Weight-loss-independent benefits of exercise on liver steatosis and stiffness in Japanese men with NAFLD. JHEP Rep. Innov. Hepatol. 2021, 3, 100253. [Google Scholar] [CrossRef] [PubMed]
- Marjot, T.; Ray, D.W.; Williams, F.R.; Tomlinson, J.W.; Armstrong, M.J. Sleep and liver disease: A bidirectional relationship. Lancet. Gastroenterol. Hepatol. 2021, 6, 850–863. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Favero, G.; Rodella, L.F. Impact of Melatonin on Skeletal Muscle and Exercise. Cells 2020, 9, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
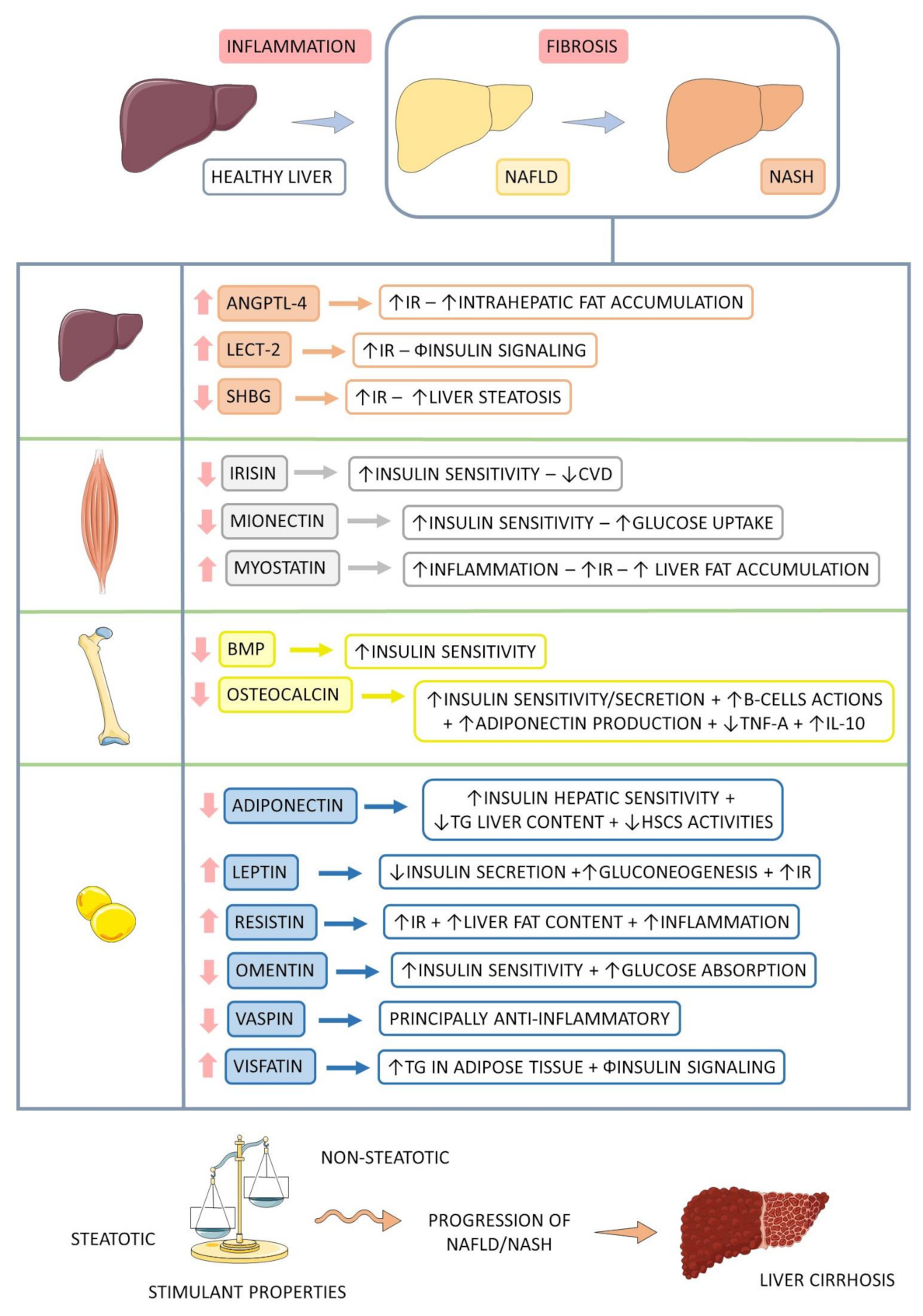
| Release | Organokine | Expression | General Function | Role in NASH | Reference |
|---|---|---|---|---|---|
| ADIPOKINE | Adiponectin | ↓ | Anti-inflammatory; Increases fatty acid oxidation; Increases glucose uptake in skeletal muscle; Inhibition of NFκβ and TNF-α by secreting IL-10; IR-related plasma reduction and glucose intolerance. | Hepatic insulin-sensitizing effect; Prevention of TG accumulation in the liver; Maintenance of HSCs in their quiescent state. | [57,61,63,64,65,66] |
| Leptin | ↑ | Weight loss increased energy expenditure and FFA oxidation; Increases glucose and FFA absorption in skeletal muscle; Stimulates production of IL-6 and TNF-α; Reduction of appetite and TAG synthesis; Neutralization of lipogenic action of insulin; Pro-inflammatory action; NO release and increases endothelin. | Reduction of insulin secretion; Failure in the antisteatotic effect; Increase of gluconeogenesis and IR. | [57,63,64,67] | |
| Omentin | ↓ | Anti-atherosclerotic effects. | Improvement of insulin sensitivity and glucose absorption. | [64,68,69] | |
| Resistin | ↑ | Pro-inflammatory effects. | Induction of inflammation, IR, angiogenesis, and smooth muscle cell proliferation; Reduction of mitochondria content and increases fat accumulation. | [9,58,64,65,70] | |
| Vaspin | ↓ | Anti-inflammatory effects. | Inhibition of kallikrein 7; Reduction of insulin sensitivity; Reduce the synthesis of pro-inflammatory cytokines. | [60,64] | |
| Visfatin | ↑ | Dysfunction of pancreatic β cells, Activation of leukocytes; Production of pro-inflammatory cytokines in adipose tissue. | Stimulation of the production and storage of triacylglycerols in adipose tissue; Impair insulin signaling. | [64,65,68] | |
| HEPATOKI-NE | ANGPTL-4 | ↑ | Induction of IR in the liver, skeletal muscle, and adipose tissue. | Accumulation of liver lipids. | [16,75,79] |
| LECT-2 | ↑ | Promotion of IR in skeletal muscle by activating JNK. | Impairment of insulin signaling. | [16,64] | |
| SHBG | ↓ | Association with systemic IR. | Suppression of lipogenesis in the liver, exacerbating steatosis. | [16,64] | |
| MYOKINE | Irisin | ↓ | Energy homeostasis and interactions between skeletal muscle and other tissues; Darkening of the WAT; Suppression of adipogenesis and cholesterol synthesis; Increase in lipid oxidation. | Increase of insulin sensitivity and improvement of Metabolic Syndrome and CVD. | [57,63,64,73] |
| Mionectin | ↓ | Increase of lipid uptake by adipose tissue and liver. | Insulin sensitivity. | [64] | |
| Myostatin | ↑ | Pro-inflammatory effects; Reduction of muscle mass, increasing metabolic disorders. | Increase of IR and liver fat deposition. | [64,76] | |
| OSTEOKINE | BMP | ↓ | Darkening of the WAT and increase of thermogenesis; Stimulation of insulin secretion. | Improvement of insulin sensitivity. | [57,83] |
| Osteocalcin | ↓ | Stimulation of glucose uptake and increase in insulin sensitivity; It increases the glucose and FFA uptake by muscles; Muscle hypertrophy and strength. | Increase of sensitivity and insulin secretion; Survival and functioning of pancreatic β cells; Stimulates the production of adiponectin and IL-10; Reduces TNF-α levels in adipocytes | [57,75,84,87] |
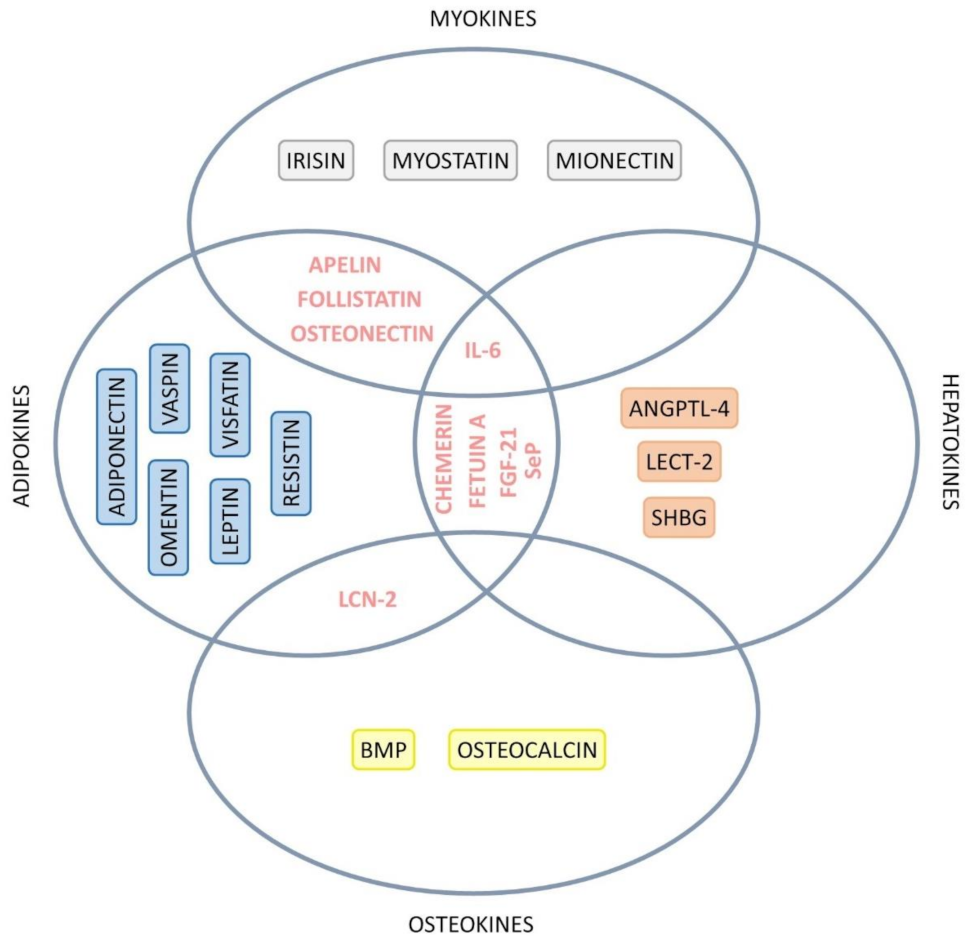
| Organokine | Classification | Expression | General Actions | Role in NASH | Reference |
|---|---|---|---|---|---|
| Apelin | Adipokine and myokine | ↓ | Control of cardiac muscle; Control of the cycle and cell death; Muscle regeneration. | Anti-inflammatory. | [64,88] |
| Chemerin | Adipokine and hepatokine | ↑ | Increase in glucose tolerance and hinders insulin signaling. | Impairment of glucose homeostasis. | [64,90] |
| Fetuin A | Adipokine and hepatokine | ↑ | Inhibition of insulin receptors in the liver and skeletal muscle; Pro-adipogenic; Suppression of adiponectin; IR in the liver via ER stress and JNK activation. | Association with NAFLD severity; IR and accelerates atherogenesis; Association with fatty liver. | [58,63,64,77,78] |
| FGF-21 | Adipokine and hepatokine | ↑ | Lipolysis; Stimulates adiponectin, insulin sensitivity, fatty acid oxidation in the liver, and glucose retention in adipocytes; Decrease of glucose production; Insulin secretion. | Modulates oxidative and ER stress, mitochondrial dysfunction, and low-grade inflammation to improve NAFLD; Increase of hepatic fat oxidation; Decrease of adipose tissue lipolysis. | [20,57,58,64,77,78] |
| Follistatin like-1 | Adipokine and myokine | ↑ | Stimulation of bone regeneration. | Stimulation of IL-1β production; Stimulation of fibrosis development. | [57] |
| IL-6 | Adipokine, hepatokine, and myokine | From adipocytes: pro-inflammatory effects; From myocytes: anti-inflammatory actions. | Adipocytes: RI; Myocytes: insulin sensitization. | [57,64] | |
| LCN-2 | Adipokyne and osteokyne | ↓ | Satiety and stimulation of energy expenditure. | Production of pro-inflammatory chemokines. | [57,64] |
| Osteonectin | Adipokine and myokine | ↑ | Modulates expression of pro-inflammatory cytokines. | IR; Adipogenesis. | [64] |
| SeP | Adipokine and hepatokine | ↑ | Transportation of selenium from the liver to the rest of the body; Contribution to IR, impairing insulin signaling in hepatocytes. | IR | [16,63,64,77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.P.M.d.; Maio, M.C.d.; Lemes, M.A.; Laurindo, L.F.; Haber, J.F.d.S.; Bechara, M.D.; Prado, P.S.d., Jr.; Rauen, E.C.; Costa, F.; Pereira, B.C.d.A.; et al. Non-Alcoholic Steatohepatitis (NASH) and Organokines: What Is Now and What Will Be in the Future. Int. J. Mol. Sci. 2022, 23, 498. https://doi.org/10.3390/ijms23010498
Santos JPMd, Maio MCd, Lemes MA, Laurindo LF, Haber JFdS, Bechara MD, Prado PSd Jr., Rauen EC, Costa F, Pereira BCdA, et al. Non-Alcoholic Steatohepatitis (NASH) and Organokines: What Is Now and What Will Be in the Future. International Journal of Molecular Sciences. 2022; 23(1):498. https://doi.org/10.3390/ijms23010498
Chicago/Turabian StyleSantos, João Paulo Margiotti dos, Mariana Canevari de Maio, Monike Alves Lemes, Lucas Fornari Laurindo, Jesselina Francisco dos Santos Haber, Marcelo Dib Bechara, Pedro Sidnei do Prado, Jr., Eduardo Costa Rauen, Fernando Costa, Barbara Cristina de Abreu Pereira, and et al. 2022. "Non-Alcoholic Steatohepatitis (NASH) and Organokines: What Is Now and What Will Be in the Future" International Journal of Molecular Sciences 23, no. 1: 498. https://doi.org/10.3390/ijms23010498
APA StyleSantos, J. P. M. d., Maio, M. C. d., Lemes, M. A., Laurindo, L. F., Haber, J. F. d. S., Bechara, M. D., Prado, P. S. d., Jr., Rauen, E. C., Costa, F., Pereira, B. C. d. A., Flato, U. A. P., Goulart, R. d. A., Chagas, E. F. B., & Barbalho, S. M. (2022). Non-Alcoholic Steatohepatitis (NASH) and Organokines: What Is Now and What Will Be in the Future. International Journal of Molecular Sciences, 23(1), 498. https://doi.org/10.3390/ijms23010498