
Gut Microbiota as a Source of Uremic Toxins

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Toxins That Can Be Synthesized by the Microbiome
2.2. Toxins with the Least Abundant Synthesizing Human Enzymes
2.3. Bacteria with the Ability to Synthesize or Metabolize Uremic Retention Solutes
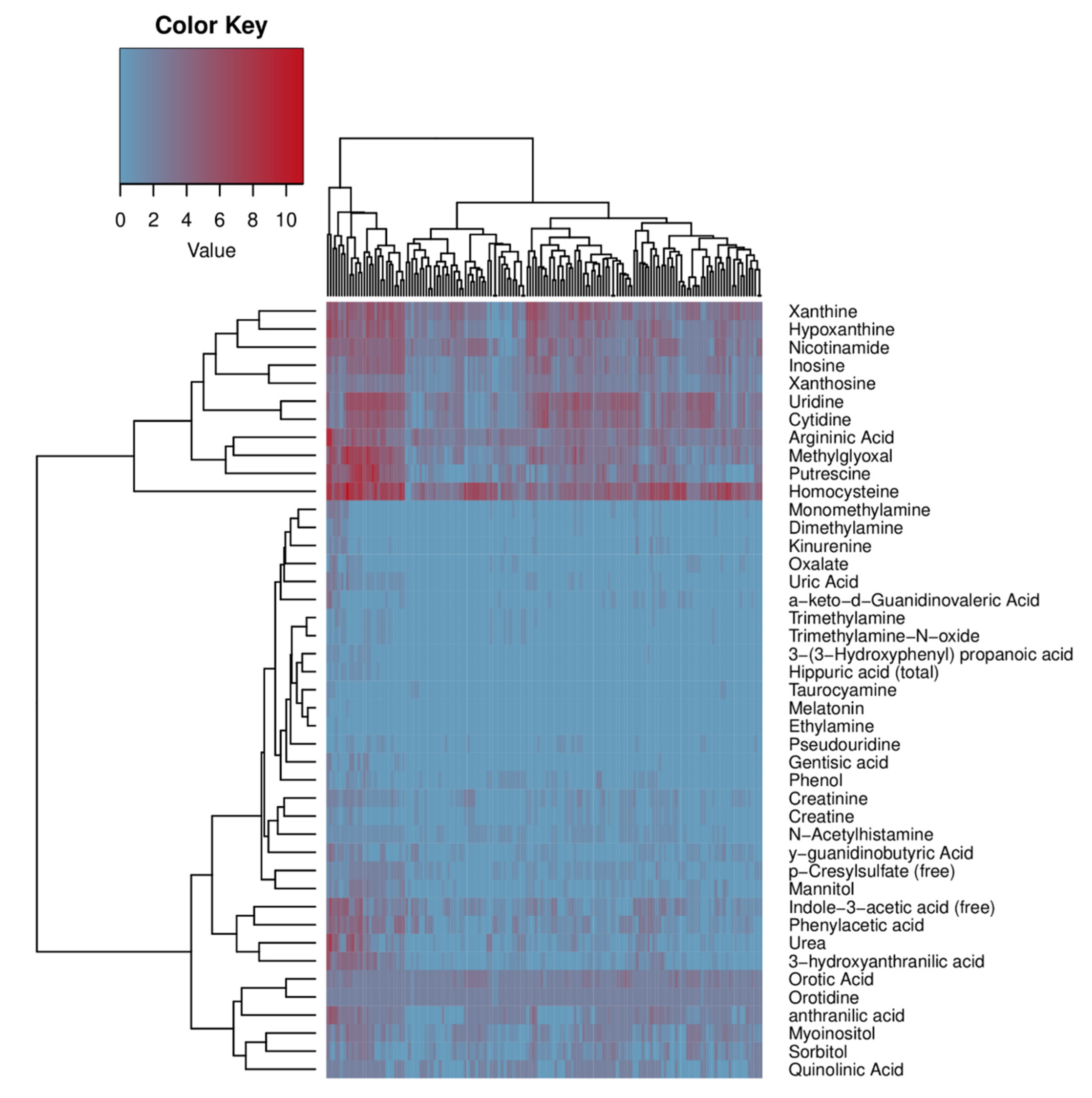
2.4. Human Microbiome Project metadata analysis
3. Discussion
Limitations
4. Materials and Methods
4.1. Database Usage
4.2. Analysis
4.3. Code Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Levin, A.; Tonelli, M.; Bonventre, J.; Coresh, J.; Donner, J.-A.; Fogo, A.B.; Fox, C.S.; Gansevoort, R.T.; Heerspink, H.J.L.; Jardine, M.; et al. Global kidney health 2017 and beyond: A roadmap for closing gaps in care, research, and policy. Lancet 2017, 390, 1888–1917. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, T.W.; Hostetter, T.H. Uremia. N. Engl. J. Med. 2007, 357, 1316–1325. [Google Scholar] [CrossRef]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Glorieux, G.; Poesen, R.; Bakker, S.J.L. Nonextracorporeal methods for decreasing uremic solute concentration: A future way to go? Semin. Nephrol. 2014, 34, 228–243. [Google Scholar] [CrossRef]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J. European uremic toxin work group a bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Meert, N.; Schepers, E.; De Smet, R.; Argiles, A.; Cohen, G.; Deppisch, R.; Drüeke, T.; Massy, Z.; Spasovski, G.; Stegmayr, B.; et al. Inconsistency of reported uremic toxin concentrations. Artif. Organs 2007, 31, 600–611. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. European uremic toxin work group normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Almeras, C.; Argilés, A. The general picture of uremia. Semin. Dial. 2009, 22, 329–333. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einheber, A.; Carter, D. The role of the microbial flora in uremia. I. Survival times of germfree, limited-flora, and conventionalized rats after bilateral nephrectomy and fasting. J. Exp. Med. 1966, 123, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronov, P.A.; Luo, F.J.-G.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mair, R.D.; Sirich, T.L.; Plummer, N.S.; Meyer, T.W. Characteristics of colon-derived uremic solutes. Clin. J. Am. Soc. Nephrol. 2018, 13, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, M.; Ueno, M.; Itoh, Y.; Suda, W.; Hattori, M. Uremic toxin-producing gut microbiota in rats with chronic kidney disease. Nephron 2017, 135, 51–60. [Google Scholar] [CrossRef]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the gut microbiome in uremia: A potential therapeutic target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [Green Version]
- Gryp, T.; De Paepe, K.; Vanholder, R.; Kerckhof, F.-M.; Van Biesen, W.; Van de Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020, 97, 1230–1242. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–6. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–10. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2013, 41, D801–7. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-derived uremic toxins and thrombosis in chronic kidney disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Sirich, T.L.; Meyer, T.W. Uremic solutes produced by colon microbes. Blood Purif. 2015, 40, 306–311. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and quantification of uremic toxin precursor-generating gut bacteria in chronic kidney disease patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef]
- Carter, J.E.; Cimolai, N. Hemolytic-uremic syndrome associated with acute campylobacter upsaliensis gastroenteritis. Nephron 1996, 74, 489. [Google Scholar] [CrossRef]
- Figueras, M.J.; Aldea, M.J.; Fernández, N.; Aspíroz, C.; Alperi, A.; Guarro, J. Aeromonas hemolytic uremic syndrome. A case and a review of the literature. Diagn. Microbiol. Infect. Dis. 2007, 58, 231–234. [Google Scholar] [CrossRef]
- Firouzi, S.; Mohd-Yusof, B.-N.; Majid, H.-A.; Ismail, A.; Kamaruddin, N.-A. Effect of microbial cell preparation on renal profile and liver function among type 2 diabetics: A randomized controlled trial. BMC Complement. Altern. Med. 2015, 15, 433. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Mora, J.; Martínez-Hernández, N.E.; del Campo-López, F.M.; Viramontes-Hörner, D.; Vizmanos-Lamotte, B.; Muñoz-Valle, J.F.; García-García, G.; Parra-Rojas, I.; Castro-Alarcón, N. Effects of a symbiotic on gut microbiota in mexican patients with end-stage renal disease. J. Ren. Nutr. 2014, 24, 330–335. [Google Scholar] [CrossRef]
- Guida, B.; Germanò, R.; Trio, R.; Russo, D.; Memoli, B.; Grumetto, L.; Barbato, F.; Cataldi, M. Effect of short-term synbiotic treatment on plasma p-cresol levels in patients with chronic renal failure: A randomized clinical trial. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1043–1049. [Google Scholar] [CrossRef]
- Fagundes, R.A.B.; Soder, T.F.; Grokoski, K.C.; Benetti, F.; Mendes, R.H. Probiotics in the treatment of chronic kidney disease: A systematic review. J. Bras. Nefrol. 2018, 40, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alatriste, P.V.M.; Arronte, R.U.; Espinosa, C.O.G.; Cuevas, M.d.L.Á.E. Effect of probiotics on human blood urea levels in patients with chronic renal failure. Nutr. Hosp. 2014, 29, 582–590. [Google Scholar]
- Dehghani, H.; Heidari, F.; Mozaffari-Khosravi, H.; Nouri-Majelan, N.; Dehghani, A. Synbiotic supplementations for azotemia in patients with chronic kidney disease: A randomized controlled trial. Iran. J. Kidney Dis. 2016, 10, 351–357. [Google Scholar]
- Sugimoto, M.; Yamaoka, Y. Review of Helicobacter pylori infection and chronic renal failure. Ther. Apher. Dial. 2011, 15, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effect of Prebiotics and/or Probiotics on Uremic Toxins and Inflammation Markers in Peritoneal Dialysis Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03770611 (accessed on 12 July 2021).
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Hatch, M.; Freel, R.W.; Vaziri, N.D. Intestinal excretion of oxalate in chronic renal failure. J. Am. Soc. Nephrol. 1994, 5, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.R.; Gabuzda, G.M.; Superdock, K.R.; Kolecki, R.S.; Schaedler, R.W.; Simenhoff, M.L. Induction of creatininase activity in chronic renal failure: Timing of creatinine degradation and effect of antibiotics. Am. J. Kidney Dis. 1997, 29, 72–77. [Google Scholar] [CrossRef]
- Miller, A.W.; Dearing, D. The metabolic and ecological interactions of oxalate-degrading bacteria in the mammalian gut. Pathogens 2013, 2, 636–652. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.-J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T. Uraemic syndrome of chronic kidney disease: Altered remote sensing and signalling. Nat. Rev. Nephrol. 2019, 15, 301–316. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Termén, S.; Tollin, M.; Rodriguez, E.; Sveinsdóttir, S.H.; Jóhannesson, B.; Cederlund, A.; Sjövall, J.; Agerberth, B.; Gudmundsson, G.H. PU.1 and bacterial metabolites regulate the human gene CAMP encoding antimicrobial peptide LL-37 in colon epithelial cells. Mol. Immunol. 2008, 45, 3947–3955. [Google Scholar] [CrossRef]
- Mitch, W.E.; Collier, V.U.; Walser, M. Creatinine metabolism in chronic renal failure. Clin. Sci. 1980, 58, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papoulis, A.; Al-Abed, Y.; Bucala, R. Identification of N2-(1-carboxyethyl)guanine (CEG) as a guanine advanced glycosylation end product. Biochemistry 1995, 34, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. 1994, 269, 32299–32305. Available online: https://www.jbc.org/content/269/51/32299.short (accessed on 24 October 2021). [CrossRef]
- Krymkiewicz, N.; Diéguez, E.; Rekarte, U.D. Properties and mode of action of a bactericidal compound (= methylglyoxal) produced by a mutant of Escherichia coli. J. Bacteriol. 1971, 108, 1338–1347. Available online: https://jb.asm.org/content/108/3/1338.short (accessed on 24 October 2021). [CrossRef] [PubMed] [Green Version]
- Rabie, E.; Serem, J.C.; Oberholzer, H.M.; Gaspar, A.R.M.; Bester, M.J. How methylglyoxal kills bacteria: An ultrastructural study. Ultrastruct. Pathol. 2016, 40, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, I.R.; Ferguson, G.P.; Miller, S.; Li, C.; Gunasekera, B.; Kinghorn, S. Bacterial production of methylglyoxal: A survival strategy or death by misadventure? Biochem. Soc. Trans. 2003, 31, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Chen, G.-C.; Wang, Z.; Usyk, M.; Yu, B.; Baeza, Y.V.; Humphrey, G.; Benitez, R.S.; Li, J.; Williams-Nguyen, J.S.; et al. Dietary factors, gut microbiota, and serum trimethylamine-N-oxide associated with cardiovascular disease in the hispanic community health study/study of latinos. Am. J. Clin. Nutr. 2021, 113, 1503–1514. [Google Scholar] [CrossRef]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and clinical impact of organic uremic retention solutes: A comprehensive update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-oxide: The good, the bad and the unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Keogh, J.; Clifton, P. A Review of potential metabolic etiologies of the observed association between red meat consumption and development of type 2 diabetes mellitus. Metabolism 2015, 64, 768–779. [Google Scholar] [CrossRef]
- Nowiński, A.; Ufnal, M. Trimethylamine N-oxide: A harmful, protective or diagnostic marker in lifestyle diseases? Nutrition 2018, 46, 7–12. [Google Scholar] [CrossRef]
- Wang, D.; Ho, L.; Faith, J.; Ono, K.; Janle, E.M.; Lachcik, P.J.; Cooper, B.R.; Jannasch, A.H.; D’Arcy, B.R.; Williams, B.A.; et al. Role of intestinal microbiota in the generation of polyphenol-derived phenolic acid mediated attenuation of Alzheimer’s disease β-amyloid oligomerization. Mol. Nutr. Food Res. 2015, 59, 1025–1040. [Google Scholar] [CrossRef]
- Shaw, W. Increased urinary excretion of a 3-(3-hydroxyphenyl)-3-hydroxypropionic acid (HPHPA), an abnormal phenylalanine metabolite of Clostridia spp. in the gastrointestinal tract, in urine samples from patients with autism and schizophrenia. Nutr. Neurosci. 2010, 13, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J. Biomed. Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Qian, J.; Li, H.; Luo, H.; Luo, W.; Lin, Z. Renal tubular epithelial cells injury induced by mannitol and its potential mechanism. Ren. Fail. 2018, 40, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downs, J.W.; Wills, B.K. Phenol toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Knight, J.; Madduma-Liyanage, K.; Mobley, J.A.; Assimos, D.G.; Holmes, R.P. Ascorbic acid intake and oxalate synthesis. Urolithiasis 2016, 44, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzica, H.; Breljak, D.; Burckhardt, B.C.; Burckhardt, G.; Sabolić, I. Oxalate: From the environment to kidney stones. Arh. Hig. Rada Toksikol. 2013, 64, 609–630. [Google Scholar] [CrossRef] [Green Version]
- Jonassen, J.A.; Kohjimoto, Y.; Scheid, C.R.; Schmidt, M. Oxalate toxicity in renal cells. Urol. Res. 2005, 33, 329–339. [Google Scholar] [CrossRef]
- Cao, L.-C.; Honeyman, T.W.; Cooney, R.; Kennington, L.; Scheid, C.R.; Jonassen, J.A. Mitochondrial dysfunction is a primary event in renal cell oxalate toxicity. Kidney Int. 2004, 66, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Feher, J. 7.4—Tubular reabsorption and secretion. In Quantitative Human Physiology, 2nd ed.; Feher, J., Ed.; Academic Press: Boston, MA, USA, 2017; pp. 719–729. ISBN 9780128008836. [Google Scholar]
- Fargue, S.; Milliner, D.S.; Knight, J.; Olson, J.B.; Lowther, W.T.; Holmes, R.P. Hydroxyproline metabolism and oxalate synthesis in primary hyperoxaluria. J. Am. Soc. Nephrol. 2018, 29, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Penzo, M.; Guerrieri, A.N.; Zacchini, F.; Treré, D.; Montanaro, L. RNA pseudouridylation in physiology and medicine: For better and for worse. Genes 2017, 8, 301. [Google Scholar] [CrossRef] [Green Version]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Consortium, T.H.M.P. The human microbiome project consortium structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Integrative HMP (iHMP) research network consortium the integrative human microbiome project. Nature 2019, 569, 641–648. [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [Green Version]
- KEGGREST. Available online: https://bioconductor.org/packages/release/bioc/html/KEGGREST.html (accessed on 20 May 2021).
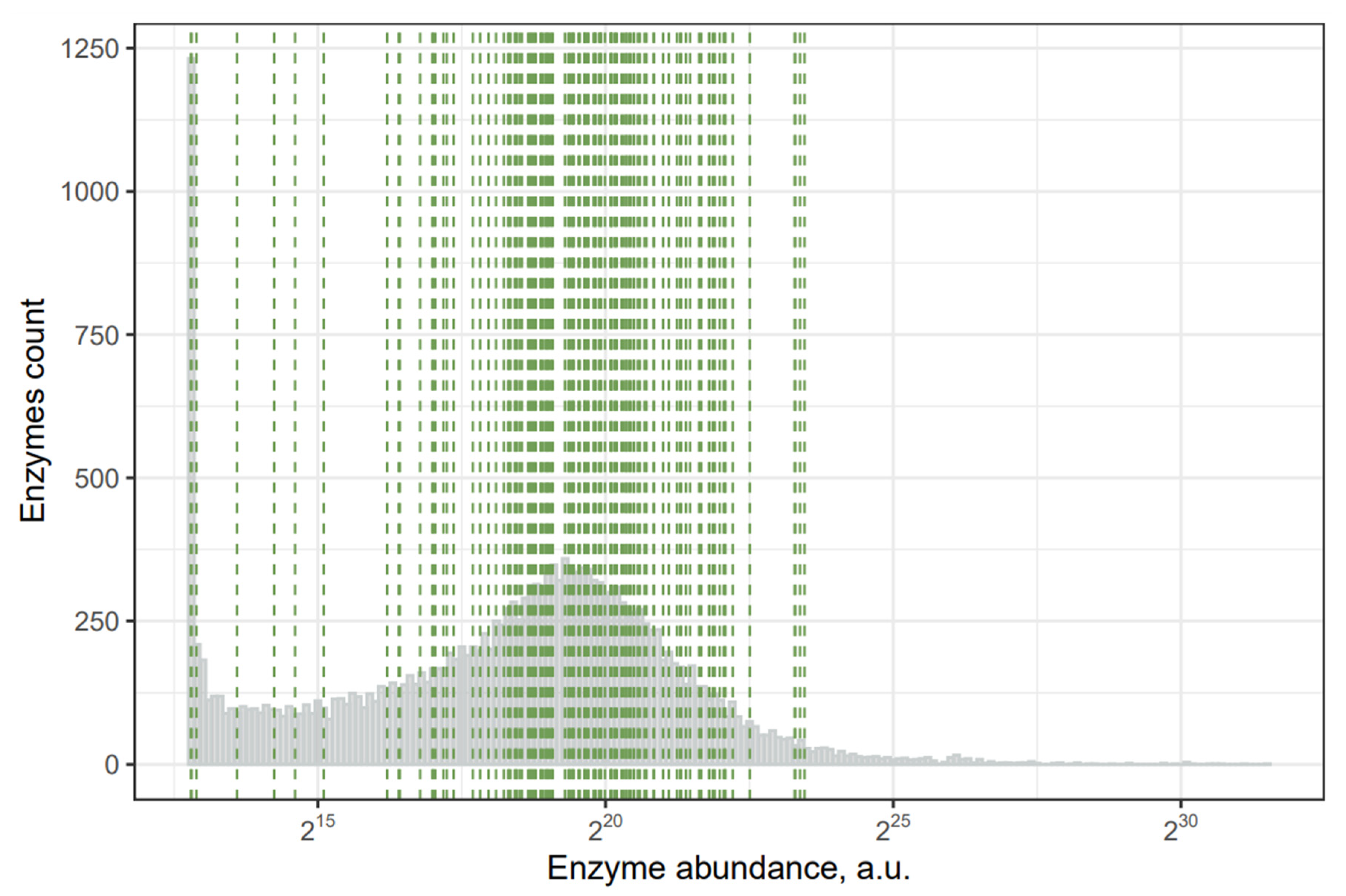

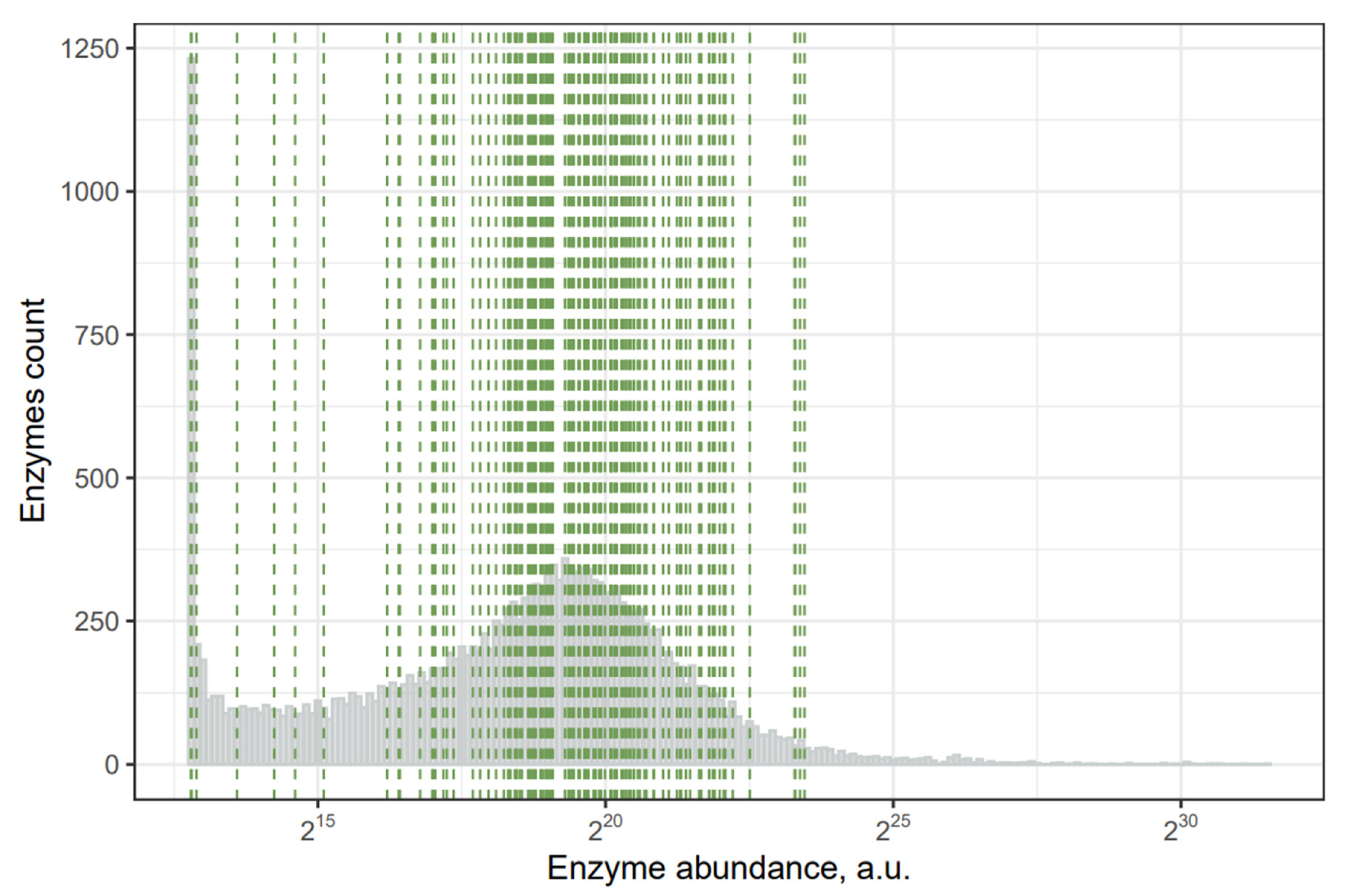{kind=link}
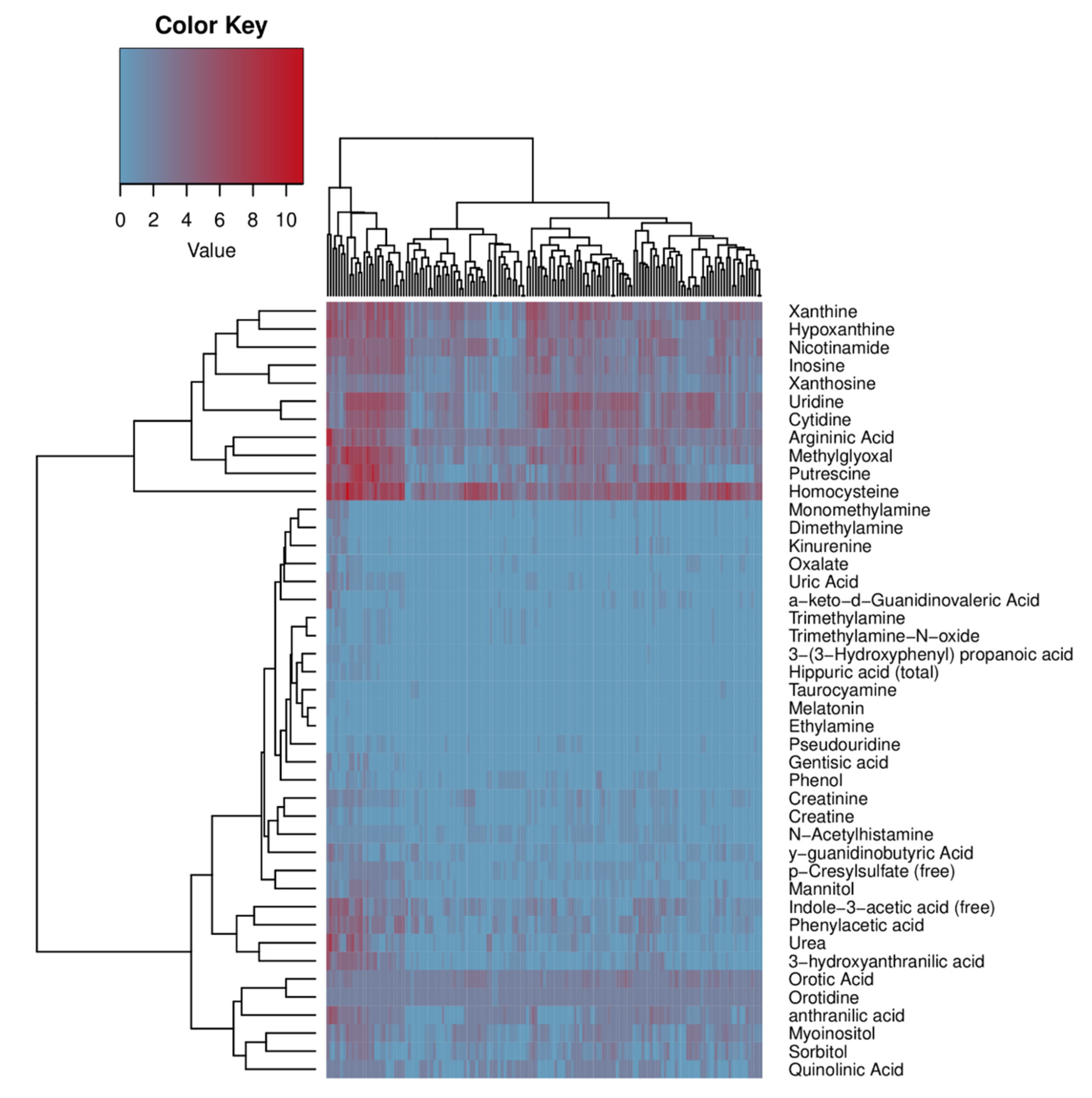{kind=link}
| Toxin | Number of Synthesizing or Metabolizing Bacteria Species | Number of Different Reactions in KEGG | Number of Enzymes in Human |
|---|---|---|---|
| Mannitol | 56 | 4 | 0 |
| Phenol | 37 | 4 | 0 |
| Trimethylamine | 19 | 4 | 0 |
| Oxalate | 20 | 3 | 0 |
| Creatinine | 74 | 2 | 0 |
| Trimethylamine-N-oxide | 16 | 2 | 0 |
| Pseudouridine | 20 | 1 | 0 |
| 3-(3-Hydroxyphenyl) propanoic acid | 13 | 1 | 0 |
| S-Adenosylhomocysteine | 143 | 35 | 1 |
| Homocysteine | 141 | 12 | 1 |
| Argininic Acid | 143 | 11 | 1 |
| Putrescine | 119 | 11 | 1 |
| Methylglyoxal | 138 | 10 | 1 |
| Hypoxanthine | 137 | 9 | 1 |
| Urea | 69 | 9 | 1 |
| Xanthine | 140 | 7 | 1 |
| Nicotinamide | 140 | 7 | 1 |
| Cytidine | 138 | 7 | 1 |
| Uridine | 138 | 7 | 1 |
| anthranilic acid | 108 | 7 | 1 |
| Inosine | 142 | 6 | 1 |
| Indole-3-acetic acid | 117 | 6 | 1 |
| 3-hydroxyanthranilic acid | 70 | 6 | 1 |
| a-keto-d-Guanidinovaleric Acid | 27 | 6 | 1 |
| Myoinositol | 116 | 5 | 1 |
| Phenylacetic acid | 106 | 5 | 1 |
| Sorbitol | 94 | 5 | 1 |
| Dimethylamine | 15 | 5 | 1 |
| Orotic Acid | 143 | 4 | 1 |
| Xanthosine | 142 | 4 | 1 |
| y-guanidinobutyric Acid | 59 | 4 | 1 |
| Monomethylamine | 22 | 4 | 1 |
| p-Cresyl sulfate | 81 | 3 | 1 |
| Uric Acid | 34 | 3 | 1 |
| Kinurenine | 20 | 3 | 1 |
| Orotidine | 143 | 2 | 1 |
| Quinolinic Acid | 89 | 2 | 1 |
| Creatine | 44 | 2 | 1 |
| Gentisic acid | 12 | 2 | 1 |
| N-Acetylhistamine | 82 | 1 | 1 |
| Hippuric acid | 12 | 1 | 1 |
| Taurocyamine | 9 | 1 | 1 |
| Melatonin | 2 | 1 | 1 |
| Ethylamine | 1 | 1 | 1 |
| KEGG + MetaCyc | |
|---|---|
| Synthesis (of More Than Five Toxins) | Decomposition (of More Than Four Toxins) |
| Oxalobacter formigenes Stenotrophomonas sp. Campylobacter coli Brevundimonas sp. Campylobacter upsaliensis Geobacter sp. Escherichia sp. Rhizobium sp. Desulfovibrio sp. Methylobacterium sp. Dialister pneumosintes Helicobacter pylori Desulfovibrio piger Edwardsiella tarda Morganella morganii Gordonibacter pamelaeae Clostridium sp. Eggerthella lenta Coprococcus catus Pediococcus acidilactici Lactobacillus fermentum Corynebacterium ammoniagenes Desulfitobacterium hafniense Paenisporosarcina sp. Paenibacillus sp. Christensenella minuta Aeromonas veronii Propionibacterium sp. Helicobacter bilis Phascolarctobacterium faecium Lachnospiraceae bacterium Helicobacter cinaedi | Escherichia sp. Rhizobium sp. Pediococcus acidilactici Lactobacillus ruminis Stenotrophomonas sp. Lactobacillus fermentum Lactobacillus paracasei Lactobacillus casei Lactobacillus rhamnosus Flavobacteriaceae bacterium Klebsiella sp. Lactobacillus amylolyticus Corynebacterium ammoniagenes Clostridium sporogenes Sphingomonas sp. Hafnia alvei Escherichia coli Providencia alcalifaciens Providencia rustigianii Enterobacter cloacae Listeria grayi Kocuria sp. Lactobacillus acidophilus Pseudomonas sp. Klebsiella oxytoca Lactobacillus helveticus |
| Uremic Retention Solutes | Number of Different Enzymes Where mRNA Detected | Percentage of Patients Who Have at Least One Enzyme’s mRNA for This Uremic Retention Solute | Enzymes |
|---|---|---|---|
| Argininic Acid | 11 | 99.73% | Nitric-oxide synthase (NADPH) (mRNA: 2.59%, Q1, prot: NA); Glycine amidinotransferase (mRNA: 0.68%, Q1, prot: NA); Arginine N-succinyltransferase (mRNA: 5.17%, Q1, prot: NA); Arginine--pyruvate transaminase (mRNA: 0.14%, Q1, prot: NA); Arginine kinase (mRNA: 9.12%, Q2, prot: NA); [Protein ADP-ribosylarginine] hydrolase (mRNA: 2.86%, Q2, prot: NA); Arginase (mRNA: 82.31%, Q2, prot: 0.22%); Arginine deiminase (mRNA: 83.27%, Q2, prot: 8.22%); Arginine decarboxylase (mRNA: 91.29%, Q2, prot: 32%); Argininosuccinate lyase (mRNA: 99.18%, Q4, prot: 14%); Arginine--tRNA ligase (mRNA: 99.59%, Q4, prot: 42%) |
| S-Adenosylhomocysteine | 64 | 99.59% | Homocysteine S-methyltransferase (mRNA: 4.49%, Q1, prot: NA); Protein-S-isoprenylcysteine O-methyltransferase (mRNA: 0%, QNA, prot: NA); Caffeoyl-CoA O-methyltransferase (mRNA: 8.98%, Q1, prot: NA); Uroporphyrinogen-III C-methyltransferase (mRNA: 52.65%, Q1, prot: 1.33%); Site-specific DNA-methyltransferase (cytosine-N(4)-specific) (mRNA: 2.45%, Q1, prot: NA); Precorrin-2 C(20)-methyltransferase (mRNA: 57.96%, Q1, prot: NA); Precorrin-3B C(17)-methyltransferase (mRNA: 78.91%, Q2, prot: NA); Precorrin-6B C(5,15)-methyltransferase (decarboxylating) (mRNA: 86.94%, Q2, prot: 0.22%); Precorrin-4 C(11)-methyltransferase (mRNA: 71.56%, Q1, prot: NA); Trans-aconitate 2-methyltransferase (mRNA: 8.98%, Q1, prot: 0.22%)... (see all 64 enzymes in ST 4) |
| Orotidine | 2 | 99.59% | Orotate phosphoribosyltransferase (mRNA: 99.05%, Q4, prot: 35.78%); Orotidine-5’-phosphate decarboxylase (mRNA: 99.32%, Q4, prot: 63.78%) |
| Uridine | 7 | 99.46% | Pyrimidine-nucleoside phosphorylase (mRNA: 50.88%, Q1, prot: 0.22%); Uridine phosphorylase (mRNA: 91.84%, Q2, prot: 4.67%); Uridine kinase (mRNA: 99.05%, Q4, prot: 2.89%); 5’-nucleotidase (mRNA: 98.64%, Q4, prot: 1.56%); 3’-nucleotidase (mRNA: 19.46%, Q1, prot: 0.89%); Uridine nucleosidase (mRNA: 0.54%, Q1, prot: NA); Cytidine deaminase (mRNA: 87.62%, Q2, prot: 11.56%) |
| Hypoxanthine | 8 | 99.32% | Xanthine dehydrogenase (mRNA: 80%, Q2, prot: 0.89%); Purine-nucleoside phosphorylase (mRNA: 99.18%, Q4, prot: 62.67%); Thymidine phosphorylase (mRNA: 58.5%, Q1, prot: 0.89%); S-methyl-5’-thioinosine phosphorylase (mRNA: 0.41%, Q1, prot: NA); Hypoxanthine phosphoribosyltransferase (mRNA: 97.69%, Q3, prot: 6.89%); Purine nucleosidase (mRNA: 28.44%, Q1, prot: NA); Ribosylpyrimidine nucleosidase (mRNA: 3.67%, Q1, prot: NA); Adenine deaminase (mRNA: 96.46%, Q2, prot: 0.22%) |
| Cytidine | 6 | 99.32% | Pyrimidine-nucleoside phosphorylase (mRNA: 50.88%, Q1, prot: 0.22%); Uridine kinase (mRNA: 99.05%, Q4, prot: 2.89%); 5’-nucleotidase (mRNA: 98.64%, Q4, prot: 1.56%); 3’-nucleotidase (mRNA: 19.46%, Q1, prot: 0.89%); Ribosylpyrimidine nucleosidase (mRNA: 3.67%, Q1, prot: NA); Cytidine deaminase (mRNA: 87.62%, Q2, prot: 11.56%) |
| Orotic Acid | 4 | 99.32% | Dihydroorotate dehydrogenase (NAD(+)) (mRNA: 91.43%, Q3, prot: 14.44%); Dihydroorotate dehydrogenase (quinone) (mRNA: 93.47%, Q3, prot: 0.44%); Dihydroorotate oxidase (fumarate) (mRNA: 2.86%, Q1, prot: NA); Orotate phosphoribosyltransferase (mRNA: 99.05%, Q4, prot: 35.78%) |
| Nicotinamide | 7 | 99.18% | Purine-nucleoside phosphorylase (mRNA: 99.18%, Q4, prot: 62.67%); Nicotinamide phosphoribosyltransferase (mRNA: 0.41%, Q1, prot: NA); NAD(+) ADP-ribosyltransferase (mRNA: 0.41%, Q2, prot: 0%); Purine nucleosidase (mRNA: 28.44%, Q1, prot: NA); Uridine nucleosidase (mRNA: 0.54%, Q1, prot: NA); ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase (mRNA: 0%, QNA, prot: NA); Nicotinamidase (mRNA: 19.32%, Q1, prot: NA) |
| Xanthine | 6 | 99.18% | Xanthine dehydrogenase (mRNA: 80%, Q2, prot: 0.89%); Purine-nucleoside phosphorylase (mRNA: 99.18%, Q4, prot: 62.67%); Xanthine phosphoribosyltransferase (mRNA: 98.37%, Q4, prot: 16.22%); Hypoxanthine phosphoribosyltransferase (mRNA: 97.69%, Q3, prot: 6.89%); Purine nucleosidase (mRNA: 28.44%, Q1, prot: NA); Guanine deaminase (mRNA: 42.04%, Q1, prot: 0%) |
| Inosine | 6 | 99.18% | Purine-nucleoside phosphorylase (mRNA: 99.18%, Q4, prot: 62.67%); Inosine kinase (mRNA: 17.69%, Q1, prot: NA); 5’-nucleotidase (mRNA: 98.64%, Q4, prot: 1.56%); Purine nucleosidase (mRNA: 28.44%, Q1, prot: NA); Ribosylpyrimidine nucleosidase (mRNA: 3.67%, Q1, prot: NA); Adenosine deaminase (mRNA: 36.46%, Q1, prot: 0.22%) |
| Myoinositol | 5 | 99.18% | Inositol 2-dehydrogenase (mRNA: 16.33%, Q1, prot: 0.89%); Galactinol--raffinose galactosyltransferase (mRNA: 0.27%, Q2, prot: NA); CDP-diacylglycerol--inositol 3-phosphatidyltransferase (mRNA: 3.54%, Q2, prot: NA); Inositol-phosphate phosphatase (mRNA: 87.48%, Q2, prot: 0.22%); Alpha-galactosidase (mRNA: 99.05%, Q3, prot: 11.56%) |
| Xanthosine | 3 | 99.18% | Purine-nucleoside phosphorylase (mRNA: 99.18%, Q4, prot: 62.67%); 5’-nucleotidase (mRNA: 98.64%, Q4, prot: 1.56%); Purine nucleosidase (mRNA: 28.44%, Q1, prot: NA) |
| Methylglyoxal | 8 | 99.05% | Aldehyde reductase (mRNA: 11.43%, Q1, prot: NA); Methylglyoxal reductase (NADPH) (mRNA: 5.71%, Q2, prot: NA); Glycerol dehydrogenase (mRNA: 54.01%, Q1, prot: 7.11%); Glyoxylate reductase (NADP(+)) (mRNA: 17.28%, Q1, prot: NA); Lactaldehyde dehydrogenase (mRNA: 24.76%, Q1, prot: 0.89%); D-lactate dehydratase (mRNA: 15.78%, Q1, prot: 0.22%); Methylglyoxal synthase (mRNA: 98.91%, Q4, prot: 58.22%); Lactoylglutathione lyase (mRNA: 66.8%, Q2, prot: 0.67%) |
| Homocysteine | 8 | 99.05% | Homocysteine S-methyltransferase (mRNA: 4.49%, Q1, prot: NA); Methionine synthase (mRNA: 82.45%, Q2, prot: 0.44%); 5-methyltetrahydropteroyltriglutamate--homocysteine S-methyltransferase (mRNA: 38.23%, Q1, prot: 18.67%); Cystathionine gamma-synthase (mRNA: 29.52%, Q1, prot: 0.67%); O-acetylhomoserine aminocarboxypropyltransferase (mRNA: 94.01%, Q2, prot: 21.56%); Adenosylhomocysteinase (mRNA: 97.82%, Q3, prot: 16.22%); Cystathionine beta-synthase (mRNA: 4.35%, Q1, prot: NA); S-ribosylhomocysteine lyase (mRNA: 97.82%, Q3, prot: 42.89%) |
| Sorbitol | 4 | 99.05% | L-iditol 2-dehydrogenase (mRNA: 63.27%, Q1, prot: 4.89%); Aldehyde reductase (mRNA: 11.43%, Q1, prot: NA); Hexokinase (mRNA: 33.88%, Q1, prot: NA); Alpha-galactosidase (mRNA: 99.05%, Q3, prot: 11.56%) |
| Phenylacetic acid | 6 | 98.37% | Phenylacetaldehyde dehydrogenase (mRNA: 4.08%, Q1, prot: NA); Aldehyde dehydrogenase (NAD(P)(+)) (mRNA: 5.85%, Q1, prot: NA); Penicillin amidase (mRNA: 62.99%, Q1, prot: 0.44%); Amidase (mRNA: 1.09%, Q1, prot: NA); Nitrilase (mRNA: 0.27%, Q1, prot: NA); Phenylacetate--CoA ligase (mRNA: 98.23%, Q4, prot: 12.44%) |
| Putrescine | 11 | 98.1% | Non-specific polyamine oxidase (mRNA: 0.41%, Q2, prot: NA); Putrescine carbamoyltransferase (mRNA: 1.5%, Q1, prot: NA); Diamine N-acetyltransferase (mRNA: 83.4%, Q2, prot: NA); Spermidine synthase (mRNA: 76.87%, Q2, prot: 0.44%); Homospermidine synthase (mRNA: 0.54%, Q1, prot: NA); Diamine transaminase (mRNA: 0.14%, Q1, prot: NA); Putrescine aminotransferase (mRNA: 16.19%, Q1, prot: NA); N-carbamoylputrescine amidase (mRNA: 28.84%, Q1, prot: NA); Agmatinase (mRNA: 74.29%, Q2, prot: 1.33%); Ornithine decarboxylase (mRNA: 48.71%, Q2, prot: 2.89%); Glutamate--putrescine ligase (mRNA: 6.8%, Q1, prot: NA) |
| Quinolinic Acid | 2 | 98.1% | Nicotinate-nucleotide diphosphorylase (carboxylating) (mRNA: 77.55%, Q2, prot: 8.89%); Quinolinate synthase (mRNA: 97.82%, Q3, prot: 4.67%) |
| 3-hydroxyanthranilic acid | 4 | 97.82% | Catalase peroxidase (mRNA: 4.9%, Q2, prot: 5.11%); Catalase (mRNA: 97.69%, Q3, prot: 22.22%); 3-hydroxyanthranilate 3,4-dioxygenase (mRNA: 0.27%, Q2, prot: NA); Kynureninase (mRNA: 0.27%, Q2, prot: NA) |
| Urea | 8 | 96.87% | Urease (mRNA: 58.78%, Q2, prot: 0.67%); Arginase (mRNA: 82.31%, Q2, prot: 0.22%); Agmatinase (mRNA: 74.29%, Q2, prot: 1.33%); Creatinase (mRNA: 0.95%, Q2, prot: NA); Allantoicase (mRNA: 0.68%, Q2, prot: NA); Guanidinobutyrase (mRNA: 0.27%, Q1, prot: NA); Ureidoglycolate lyase (mRNA: 5.44%, Q1, prot: 0.22%); Urea carboxylase (mRNA: 27.76%, Q1, prot: NA) |
| Anthranilic acid | 5 | 96.33% | Anthranilate phosphoribosyltransferase (mRNA: 94.29%, Q2, prot: 2%); Arylformamidase (mRNA: 0.41%, Q1, prot: NA); Kynureninase (mRNA: 0.27%, Q2, prot: NA); Anthranilate synthase (mRNA: 55.1%, Q1, prot: 0.89%); Anthranilate--CoA ligase (mRNA: 0.27%, Q1, prot: NA) |
| Phenol | 3 | 89.66% | Arylesterase (mRNA: 16.73%, Q1, prot: NA); 4-hydroxybenzoate decarboxylase (mRNA: 3.95%, Q2, prot: NA); Tyrosine phenol-lyase (mRNA: 86.67%, Q3, prot: NA) |
| Uric Acid | 3 | 80.00% | FAD-dependent urate hydroxylase (mRNA: 0.68%, Q1, prot: NA); Xanthine dehydrogenase (mRNA: 80%, Q2, prot: 0.89%); 8-oxoguanine deaminase (mRNA: 0%, QNA, prot: NA) |
| Trimethylamine | 3 | 78.23% | Betaine reductase (mRNA: 75.65%, Q3, prot: 0.44%); Trimethylamine dehydrogenase (mRNA: 0.27%, Q1, prot: NA); Trimethylamine-N-oxide reductase (cytochrome c) (mRNA: 10.2%, Q1, prot: 0.44%) |
| Hippuric acid | 1 | 44.63% | Hippurate hydrolase (mRNA: 44.63%, Q1, prot: NA) |
| Monomethylamine | 1 | 42.99% | Sarcosine reductase (mRNA: 42.99%, Q1, prot: 0.44%) |
| Creatine | 3 | 28.44% | Creatine kinase (mRNA: 1.5%, Q1, prot: 0%); Creatininase (mRNA: 26.39%, Q1, prot: 0.67%); Creatinase (mRNA: 0.95%, Q2, prot: NA) |
| Creatinine | 1 | 26.39% | Creatininase (mRNA: 26.39%, Q1, prot: 0.67%) |
| Oxalate | 3 | 22.72% | Formyl-CoA transferase (mRNA: 20.54%, Q1, prot: 1.33%); CoA:oxalate CoA-transferase (mRNA: 3.27%, Q1, prot: 0.22%); Oxalate--CoA ligase (mRNA: 0.14%, Q1, prot: NA) |
| Mannitol | 3 | 21.9% | D-arabinitol 4-dehydrogenase (mRNA: 0.68%, Q1, prot: NA); Mannitol dehydrogenase (mRNA: 0.41%, Q1, prot: NA); Mannitol 2-dehydrogenase (mRNA: 21.22%, Q1, prot: NA) |
| Phenol sulfate | 2 | 20.27% | Aryl-sulfate sulfotransferase (mRNA: 5.99%, Q1, prot: NA); Arylsulfatase (mRNA: 15.37%, Q1, prot: NA) |
| Arab(in)itol | 1 | 11.43% | Aldehyde reductase (mRNA: 11.43%, Q1, prot: NA) |
| Trimethylamine-N-oxide | 1 | 10.2% | Trimethylamine-N-oxide reductase (cytochrome c) (mRNA: 10.2%, Q1, prot: 0.44%) |
| Pseudouridine | 1 | 9.66% | Pseudouridine kinase (mRNA: 9.66%, Q1, prot: NA) |
| p-Cresyl sulfate | 2 | 9.12% | 4-hydroxyphenylacetate decarboxylase (mRNA: 0.95%, Q2, prot: NA); 2-iminoacetate synthase (mRNA: 8.71%, Q1, prot: NA) |
| 3-(3-Hydroxyphenyl) propanoic acid | 1 | 9.12% | 3-(3-hydroxy-phenyl)propanoic acid hydroxylase (mRNA: 9.12%, Q1, prot: NA) |
| a-keto-d-Guanidinovaleric Acid | 2 | 5.85% | D-amino-acid transaminase (mRNA: 5.71%, Q1, prot: NA); Arginine--pyruvate transaminase (mRNA: 0.14%, Q1, prot: NA) |
| Indole-3-acetic acid | 3 | 3.95% | Aldehyde dehydrogenase (NAD(+)) (mRNA: 2.72%, Q1, prot: NA); Amidase (mRNA: 1.09%, Q1, prot: NA); Nitrilase (mRNA: 0.27%, Q1, prot: NA) |
| y-guanidinobutyric Acid | 3 | 1.9% | Glycine amidinotransferase (mRNA: 0.68%, Q1, prot: NA); Amidase (mRNA: 1.09%, Q1, prot: NA); Guanidinobutyrase (mRNA: 0.27%, Q1, prot: NA) |
| Hyaluronic acid (Hyaluronan) | 1 | 1.9% | Hyaluronan synthase (mRNA: 1.9%, Q1, prot: 0.22%) |
| Gentisic acid | 1 | 1.22% | 3-hydroxybenzoate 6-monooxygenase (mRNA: 1.22%, Q1, prot: 0.22%) |
| Kinurenine | 4 | 0.95% | Kynurenine 3-monooxygenase (mRNA: 0%, QNA, prot: NA); Kynurenine--oxoglutarate transaminase (mRNA: 0.82%, Q2, prot: 0%); Arylformamidase (mRNA: 0.41%, Q1, prot: NA); Kynureninase (mRNA: 0.27%, Q2, prot: NA) |
| Dimethylamine | 2 | 0.95% | Trimethylamine dehydrogenase (mRNA: 0.27%, Q1, prot: NA); Dimethylargininase (mRNA: 0.68%, Q2, prot: NA) |
| Kynurenic Acid | 1 | 0.82% | Kynurenine--oxoglutarate transaminase (mRNA: 0.82%, Q2, prot: 0%) |
| Taurocyamine | 1 | 0.68% | Glycine amidinotransferase (mRNA: 0.68%, Q1, prot: NA) |
| Asymmetric Dimethylarginine (ADMA) | 1 | 0.68% | Dimethylargininase (mRNA: 0.68%, Q2, prot: NA) |
| Enzyme Name | Number of Bacteria That Express mRNA for This Enzyme | Percentage of Patients with Detected mRNA for This Enzyme | Quartile of mRNA Abundance | Percentage of Patients with Indicated Enzyme Detected in the Proteome | A Toxin That Could Be Potentially Synthesised by the Indicated Enzyme |
|---|---|---|---|---|---|
| Orotidine-5’-phosphate decarboxylase | 205 | 99.32% | 2 | 63.78% | Orotidine |
| Purine-nucleoside phosphorylase | 187 | 99.18% | 2 | 62.67% | Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine |
| Methylglyoxal synthase | 124 | 98.91% | 3 | 58.22% | Methylglyoxal |
| S-ribosylhomocysteine lyase | 129 | 97.82% | 3 | 42.89% | Homocysteine |
| Arginine--tRNA ligase | 221 | 99.59% | 2 | 42% | Argininic Acid |
| Orotate phosphoribosyltransferase | 199 | 99.05% | 3 | 35.78% | Orotic Acid; Orotidine |
| Arginine decarboxylase | 39 | 91.29% | 2 | 32% | Argininic Acid |
| Catalase | 56 | 97.69% | 3 | 22.22% | 3-hydroxyanthranilic acid |
| O-acetylhomoserine aminocarboxypropyl-transferase | 46 | 94.01% | 2 | 21.56% | Homocysteine |
| 5-methyltetrahydropteroyltriglutamate-homocysteine S-methyltransferase | 31 | 38.23% | 1 | 18.67% | Homocysteine |
| Xanthine phosphoribosyltransferase | 161 | 98.37% | 2 | 16.22% | Xanthine |
| Adenosylhomocysteinase | 68 | 97.82% | 2 | 16.22% | Homocysteine; S-Adenosylhomocysteine |
| Dihydroorotate dehydrogenase (NAD(+)) | 28 | 91.43% | 2 | 14.44% | Orotic Acid |
| Argininosuccinate lyase | 192 | 99.18% | 2 | 14% | Argininic Acid |
| Phenylacetate--CoA ligase | 84 | 98.23% | 2 | 12.44% | Phenylacetic acid |
| tRNA-2-methylthio-N(6)-dimethylallyladenosine synthase | 185 | 99.05% | 2 | 11.56% | S-Adenosylhomocysteine |
| Alpha-galactosidase | 71 | 99.05% | 2 | 11.56% | Myoinositol; Sorbitol |
| Cytidine deaminase | 48 | 87.62% | 3 | 11.56% | Cytidine; Uridine |
| Bacteria | Percentage of Patients with Detected mRNA for Indicated Bacteria | Number of Uremic Retention Solutes for Which mRNA Was Detected of at Least One of the Toxin-Producing Enzymes | Number of Different Enzymes Detected for Indicated Bacteria | Uremic Retention Solutes That Could Be Potentially Synthesized by Indicated Bacteria |
|---|---|---|---|---|
| Ruminococcus torques | 93.61% | 22 | 45 | Sorbitol; Arab(in)itol; Methylglyoxal; Hypoxanthine; Uric Acid; Xanthine; S-Adenosylhomocysteine; Homocysteine; Inosine; Nicotinamide; Xanthosine; Orotic Acid; Orotidine; anthranilic acid; Uridine; Putrescine; Quinolinic Acid; Cytidine; Myoinositol; Urea; Argininic Acid; Phenylacetic acid |
| Faecalibacterium prausnitzii | 93.33% | 17 | 34 | Sorbitol; Homocysteine; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; Putrescine; Cytidine; Uridine; Myoinositol; Urea; Argininic Acid; Methylglyoxal |
| Eubacterium rectale | 81.77% | 17 | 27 | Sorbitol; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; anthranilic acid; Uridine; Putrescine; Homocysteine; Quinolinic Acid; Cytidine; Methylglyoxal; Argininic Acid |
| Bacteroides uniformis | 79.05% | 18 | 20 | S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; anthranilic acid; Quinolinic Acid; Cytidine; Uridine; Myoinositol; Sorbitol; Methylglyoxal; Argininic Acid; Homocysteine; Phenylacetic acid |
| Bacteroides ovatus | 78.37% | 21 | 34 | Sorbitol; Mannitol; 3-hydroxyanthranilic acid; Orotic Acid; S-Adenosylhomocysteine; Putrescine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotidine; anthranilic acid; Homocysteine; Quinolinic Acid; Cytidine; Uridine; Myoinositol; Phenylacetic acid; Methylglyoxal; Argininic Acid |
| Bacteroides vulgatus | 77.14% | 17 | 24 | Orotic Acid; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotidine; anthranilic acid; Cytidine; Uridine; Myoinositol; Sorbitol; Methylglyoxal; Argininic Acid; Homocysteine; Phenylacetic acid |
| Ruminococcus obeum | 75.78% | 23 | 41 | Sorbitol; Arab(in)itol; Methylglyoxal; Hypoxanthine; Uric Acid; Xanthine; S-Adenosylhomocysteine; Inosine; Nicotinamide; Xanthosine; Orotic Acid; Orotidine; anthranilic acid; Quinolinic Acid; Uridine; Putrescine; Homocysteine; Cytidine; Phenol sulfate; Phenol; Urea; Argininic Acid; Phenylacetic acid |
| Lachnospiraceae bacterium | 68.98% | 13 | 16 | S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; Cytidine; Uridine; Homocysteine; Argininic Acid; Urea |
| Roseburia inulinivorans | 62.04% | 15 | 21 | S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; Uridine; Putrescine; Quinolinic Acid; Cytidine; Methylglyoxal; Argininic Acid; Homocysteine |
| Bacteroides thetaiotaomicron | 61.77% | 19 | 30 | 3-hydroxyanthranilic acid; Orotic Acid; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotidine; anthranilic acid; Homocysteine; Quinolinic Acid; Cytidine; Uridine; Myoinositol; Sorbitol; Methylglyoxal; Argininic Acid; Phenylacetic acid |
| Clostridium bolteae | 61.5% | 16 | 22 | Sorbitol; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; anthranilic acid; Uridine; Quinolinic Acid; Methylglyoxal; Argininic Acid; Homocysteine; Phenylacetic acid |
| Bacteroides caccae | 60.95% | 18 | 26 | Orotic Acid; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotidine; anthranilic acid; Homocysteine; Quinolinic Acid; Cytidine; Uridine; Myoinositol; Sorbitol; Methylglyoxal; Argininic Acid; Phenylacetic acid |
| Bacteroides xylanisolvens | 60.14% | 21 | 37 | Sorbitol; Mannitol; Orotic Acid; S-Adenosylhomocysteine; Putrescine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotidine; anthranilic acid; Homocysteine; Quinolinic Acid; Cytidine; Uridine; Myoinositol; Phenol sulfate; Phenylacetic acid; Methylglyoxal; Argininic Acid |
| Dorea longicatena | 59.32% | 16 | 25 | Sorbitol; Methylglyoxal; Homocysteine; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; Uridine; Quinolinic Acid; Myoinositol; Argininic Acid; Phenylacetic acid |
| Roseburia hominis | 59.32% | 16 | 21 | S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; anthranilic acid; Putrescine; Quinolinic Acid; Cytidine; Uridine; Methylglyoxal; Argininic Acid; Homocysteine |
| Roseburia intestinalis | 57.14% | 20 | 42 | Sorbitol; Hypoxanthine; Uric Acid; Xanthine; S-Adenosylhomocysteine; Inosine; Nicotinamide; Xanthosine; Orotic Acid; Orotidine; anthranilic acid; Quinolinic Acid; Cytidine; Uridine; Putrescine; Homocysteine; Phenol; Myoinositol; Argininic Acid; Methylglyoxal |
| Parabacteroides merdae | 56.05% | 17 | 23 | Orotic Acid; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotidine; anthranilic acid; Quinolinic Acid; Cytidine; Uridine; Homocysteine; Argininic Acid; Urea; Methylglyoxal; Phenylacetic acid |
| Bifidobacterium longum | 55.1% | 16 | 21 | S-Adenosylhomocysteine; Orotic Acid; Orotidine; anthranilic acid; Xanthine; Hypoxanthine; Putrescine; Homocysteine; Cytidine; Inosine; Uridine; Xanthosine; Myoinositol; Sorbitol; Nicotinamide; Argininic Acid |
| Flavonifractor plautii | 54.83% | 13 | 17 | S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; Cytidine; Uridine; Homocysteine; Argininic Acid; Urea |
| Alistipes putredinis | 54.56% | 15 | 19 | 3-hydroxyanthranilic acid; Homocysteine; S-Adenosylhomocysteine; Hypoxanthine; Inosine; Nicotinamide; Xanthine; Xanthosine; Orotic Acid; Orotidine; Quinolinic Acid; Cytidine; Uridine; Methylglyoxal; Argininic Acid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popkov, V.A.; Zharikova, A.A.; Demchenko, E.A.; Andrianova, N.V.; Zorov, D.B.; Plotnikov, E.Y. Gut Microbiota as a Source of Uremic Toxins. Int. J. Mol. Sci. 2022, 23, 483. https://doi.org/10.3390/ijms23010483
Popkov VA, Zharikova AA, Demchenko EA, Andrianova NV, Zorov DB, Plotnikov EY. Gut Microbiota as a Source of Uremic Toxins. International Journal of Molecular Sciences. 2022; 23(1):483. https://doi.org/10.3390/ijms23010483
Chicago/Turabian StylePopkov, Vasily A., Anastasia A. Zharikova, Evgenia A. Demchenko, Nadezda V. Andrianova, Dmitry B. Zorov, and Egor Y. Plotnikov. 2022. "Gut Microbiota as a Source of Uremic Toxins" International Journal of Molecular Sciences 23, no. 1: 483. https://doi.org/10.3390/ijms23010483
APA StylePopkov, V. A., Zharikova, A. A., Demchenko, E. A., Andrianova, N. V., Zorov, D. B., & Plotnikov, E. Y. (2022). Gut Microbiota as a Source of Uremic Toxins. International Journal of Molecular Sciences, 23(1), 483. https://doi.org/10.3390/ijms23010483