
Generation of Lasso Peptide-Based ClpP Binders


Abstract
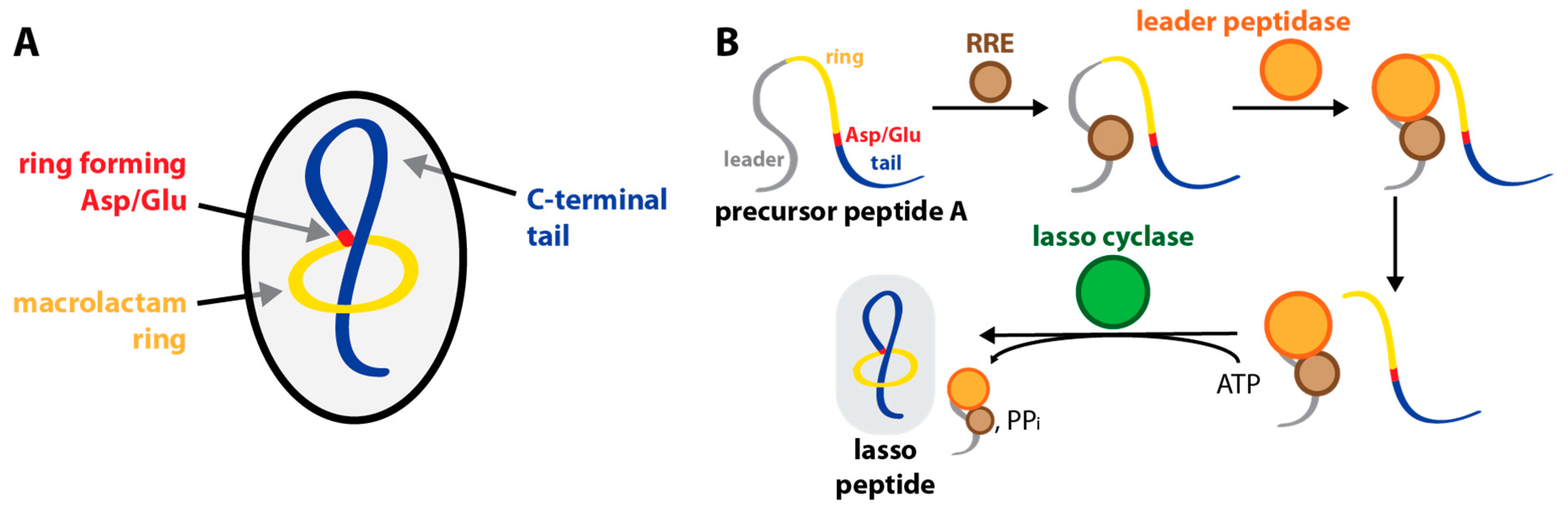
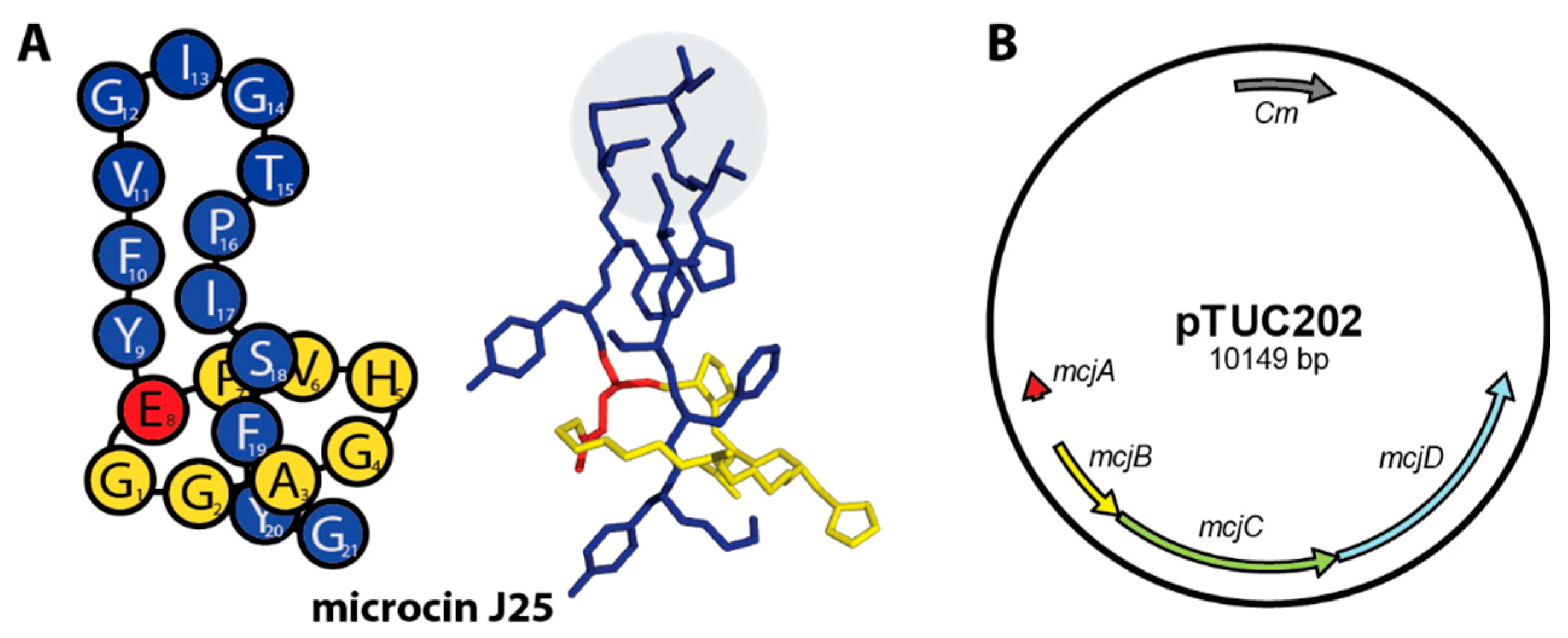
1. Introduction
2. Results
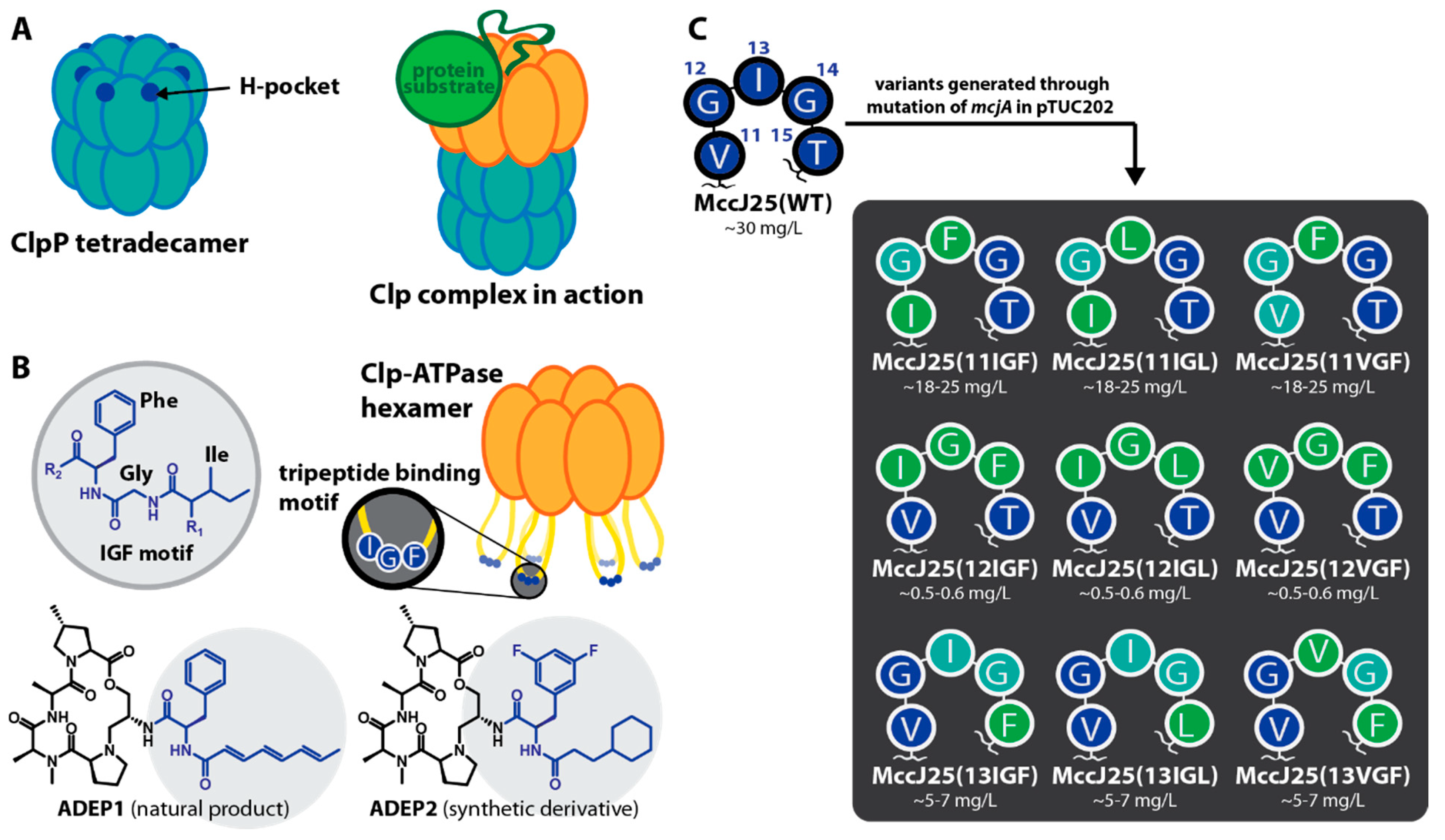
2.1. Construction of MccJ25 Variants with Canonical Clp-ATPase Tripeptide Binding Motifs
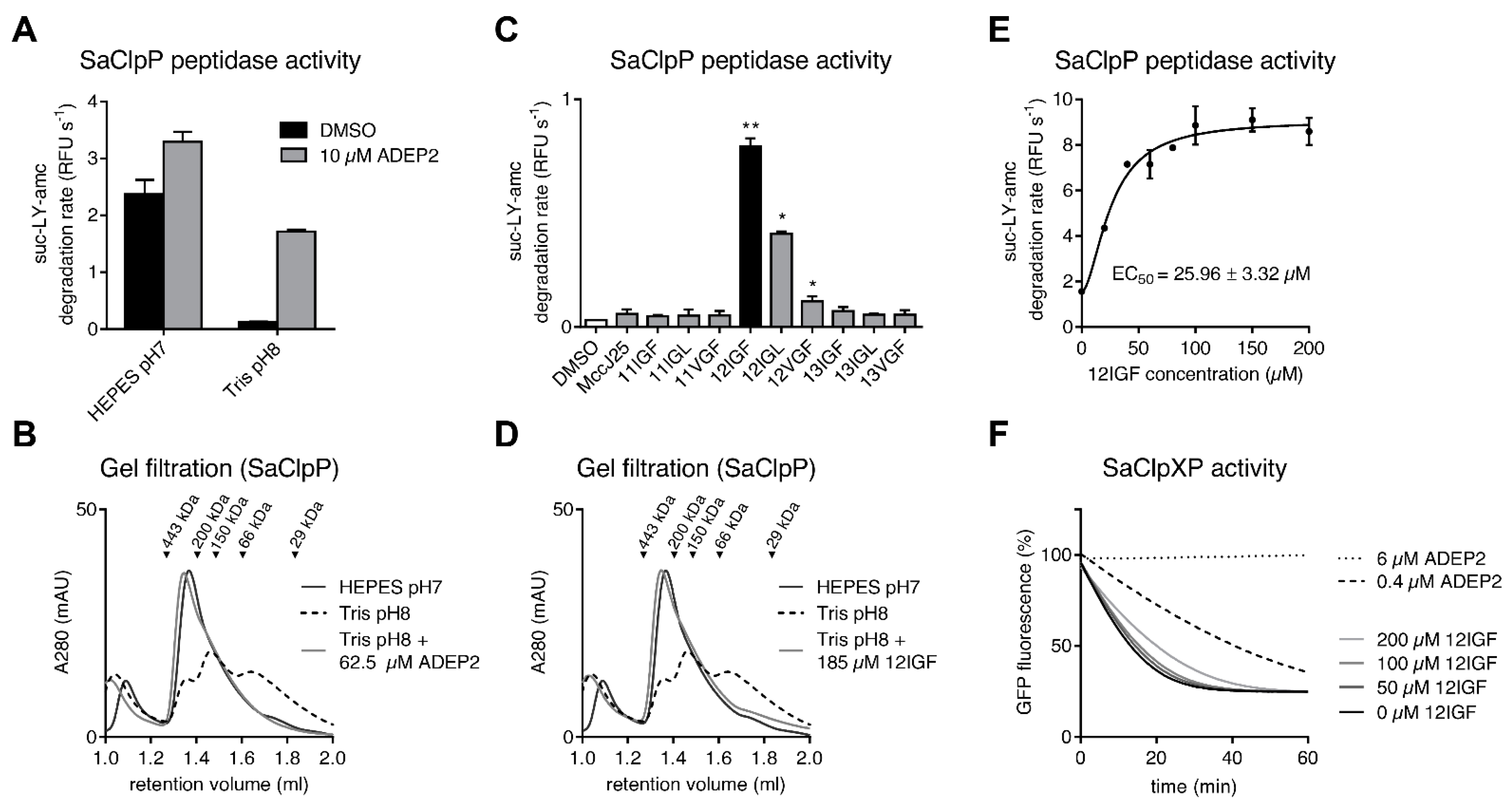
2.2. MccJ25 Variants Stabilize the Oligomeric State of the S. aureus ClpP Barrel
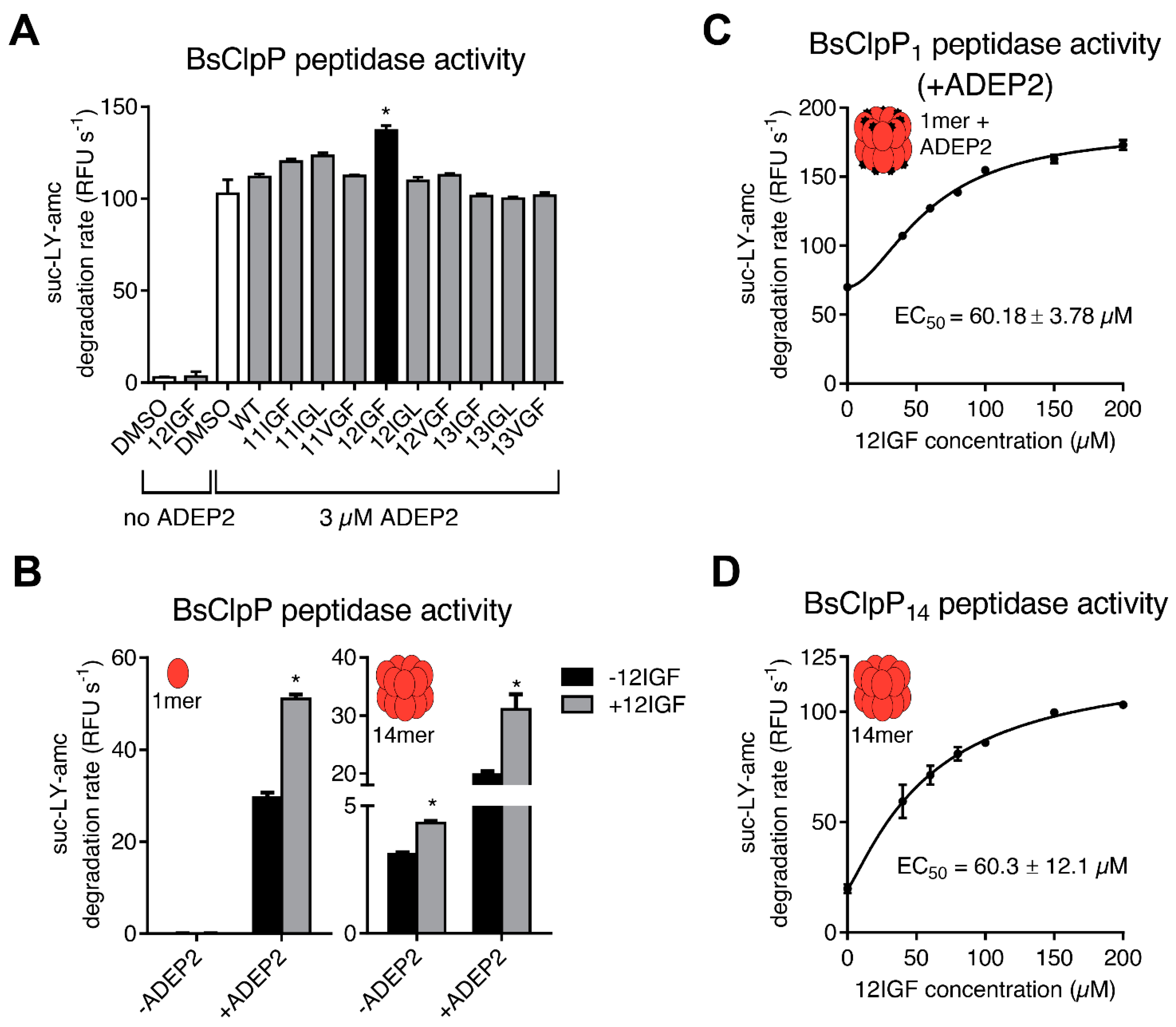
2.3. MccJ25 Variants and ADEPs Synergistically Stimulate B. subtilis ClpP Peptidase Activity
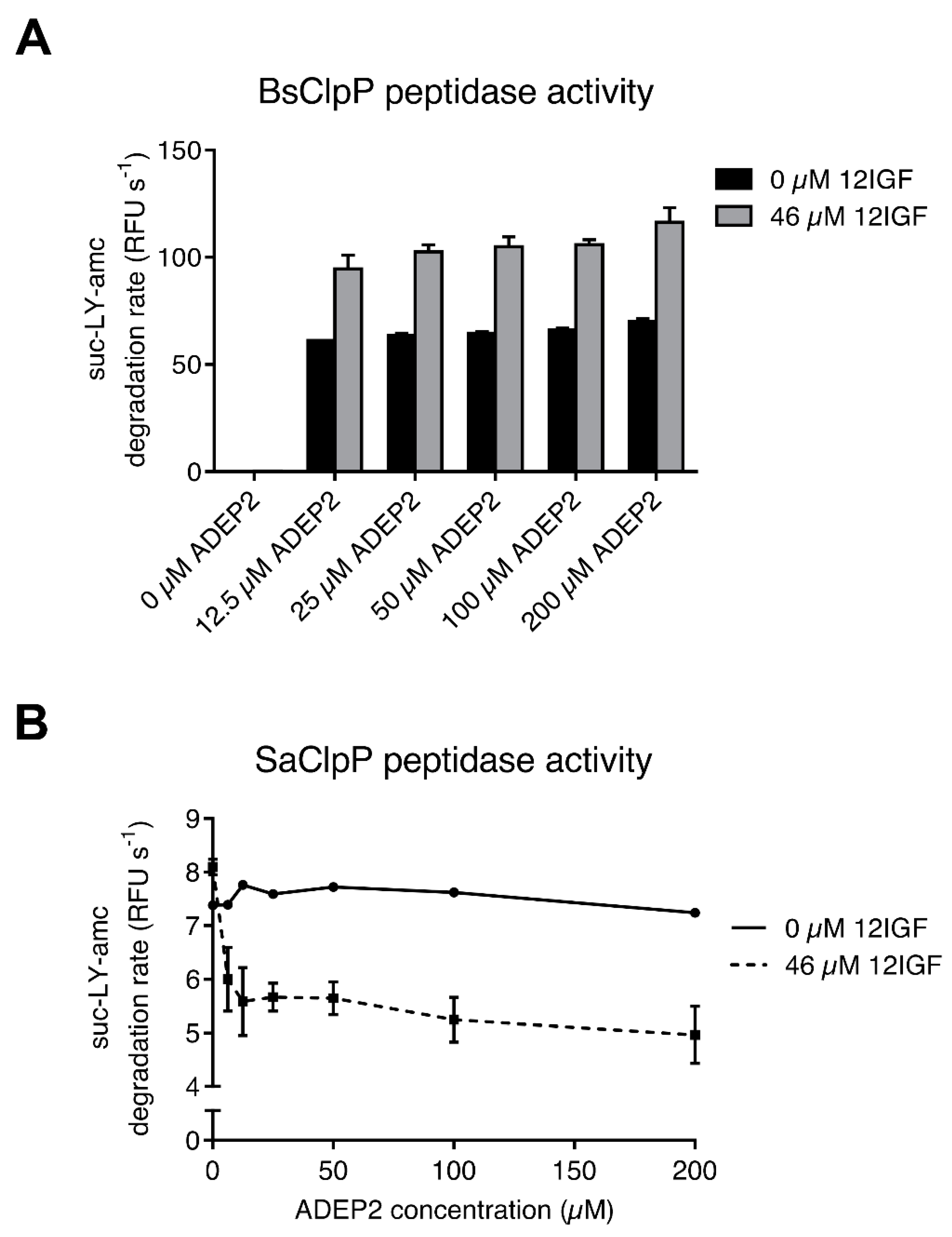
2.4. Lasso Peptide-Mediated Activation Is Not Inhibited by ADEPs
3. Discussion
4. Materials and Methods
4.1. Mutagenesis of mcjA
4.2. Production and Purification of MccJ25 Variants
4.3. Cloning and Protein Purification
4.4. Peptide Degradation Assay
4.5. Casein Degradation Assay
4.6. eGFP Degradation Assay
4.7. Analytical Gel Filtration
4.8. 12IGF Stability Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Montalbán-López, M.; Scott, T.A.; Ramesh, S.; Rahman, I.R.; van Heel, A.J.; Viel, J.H.; Bandarian, V.; Dittmann, E.; Genilloud, O.; Goto, Y.; et al. New devel-opments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 2021, 38, 130–239. [Google Scholar] [CrossRef]
- Hegemann, J.D.; Dit-Foque, K.J.; Xie, X. Comprehensive Natural Products III; Liu, H.-W., Begley, T.P., Eds.; Elsevier: Oxford, UK, 2020; pp. 206–228. [Google Scholar]
- Hegemann, J.; Zimmermann, M.; Xie, X.; Marahiel, M.A. Lasso Peptides: An Intriguing Class of Bacterial Natural Products. Acc. Chem. Res. 2015, 48, 1909–1919. [Google Scholar] [CrossRef]
- Hegemann, J.D. Factors Governing the Thermal Stability of Lasso Peptides. ChemBioChem 2019, 21, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, B.J.; Hudson, G.A.; Dunbar, K.L.; Mitchell, D.A. A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat. Chem. Biol. 2015, 11, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Fage, C.; Hegemann, J.; Mielcarek, A.; Yan, D.; Linne, U.; Marahiel, M.A. The B1 Protein Guides the Biosynthesis of a Lasso Peptide. Sci. Rep. 2016, 6, 35604. [Google Scholar] [CrossRef]
- Hegemann, J.D.; Schwalen, C.J.; Mitchell, D.A.; van der Donk, W.A. Elucidation of the roles of conserved residues in the biosynthesis of the lasso peptide paeninodin. Chem. Commun. 2018, 54, 9007–9010. [Google Scholar] [CrossRef]
- Sumida, T.; Dubiley, S.; Wilcox, B.; Severinov, K.; Tagami, S. Structural basis of leader peptide recognition in lasso peptide bio-synthesis pathway. ACS Chem. Biol. 2019, 14, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- DiCaprio, A.J.; Firouzbakht, A.; Hudson, G.A.; Mitchell, D.A. Enzymatic Reconstitution and Biosynthetic Investigation of the Lasso Peptide Fusilassin. J. Am. Chem. Soc. 2018, 141, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Koos, J.D.; Link, A.J. Heterologous and in Vitro Reconstitution of Fuscanodin, a Lasso Peptide from Thermobifida fusca. J. Am. Chem. Soc. 2018, 141, 928–935. [Google Scholar] [CrossRef]
- Martin-Gómez, H.; Linne, U.; Albericio, F.; Tulla-Puche, J.; Hegemann, J.D. Investigation of the Biosynthesis of the Lasso Peptide Chaxapeptin Using an E. coli-Based Production System. J. Nat. Prod. 2018, 81, 2050–2056. [Google Scholar] [CrossRef]
- Hegemann, J.D.; Fouque, K.J.D.; Santos-Fernandez, M.; Fernandez-Lima, F. A Bifunctional Leader Peptidase/ABC Transporter Protein Is Involved in the Maturation of the Lasso Peptide Cochonodin I from Streptococcus suis. J. Nat. Prod. 2021, 84, 2683–2691. [Google Scholar] [CrossRef]
- Salomón, R.A.; Farías, R.N. Microcin 25, a novel antimicrobial peptide produced by Escherichia coli. J. Bacteriol. 1992, 174, 7428–7435. [Google Scholar] [CrossRef]
- Braffman, N.R.; Piscotta, F.J.; Hauver, J.; Campbell, E.A.; Link, A.J.; Darst, S.A. Structural mechanism of transcription inhi-bition by lasso peptides microcin J25 and capistruin. Proc. Natl. Acad. Sci. USA 2019, 116, 1273–1278. [Google Scholar] [CrossRef]
- Mukhopadhyay, J.; Sineva, E.; Knight, J.; Levy, R.M.; Ebright, R.H. Antibacterial Peptide Microcin J25 Inhibits Transcription by Binding within and Obstructing the RNA Polymerase Secondary Channel. Mol. Cell 2004, 14, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Adelman, K.; Yuzenkova, J.; La Porta, A.; Zenkin, N.; Lee, J.; Lis, J.T.; Borukhov, S.; Wang, M.D.; Severinov, K. Molecular Mechanism of Transcription Inhibition by Peptide Antibiotic Microcin J25. Mol. Cell 2004, 14, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Yuzenkova, Y.; Delgado, M.; Nechaev, S.; Savalia, D.; Epshtein, V.; Artsimovitch, I.; Mooney, R.A.; Landick, R.; Farias, R.N.; Salomon, R.; et al. Mutations of Bacterial RNA Polymerase Leading to Resistance to Microcin J25. J. Biol. Chem. 2002, 277, 50867–50875. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.A.; Rintoul, M.R.; Farías, R.N.; Salomón, R.A. Escherichia coli RNA Polymerase Is the Target of the Cyclopeptide Antibiotic Microcin J25. J. Bacteriol. 2001, 183, 4543–4550. [Google Scholar] [CrossRef]
- Mathavan, I.; Zirah, S.; Mehmood, S.; Choudhury, H.G.; Goulard, C.; Li, Y.; Robinson, C.V.; Rebuffat, S.; Beis, K. Structural basis for hijacking siderophore receptors by antimicrobial lasso peptides. Nat. Chem. Biol. 2014, 10, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Salomón, R.A.; Farías, R.N. The FhuA protein is involved in microcin 25 uptake. J. Bacteriol. 1993, 175, 7741–7742. [Google Scholar] [CrossRef][Green Version]
- Joshi, S.A.; Hersch, G.L.; Baker, T.A.; Sauer, R.T. Genetic analysis of plasmid determinants for microcin J25 production and immunity. J. Bacteriol. 1996, 178, 3661–3663. [Google Scholar] [CrossRef]
- Hegemann, J.D.; De Simone, M.; Zimmermann, M.; Knappe, T.A.; Xie, X.; Di Leva, F.S.; Marinelli, L.; Novellino, E.; Zahler, S.; Kessler, H.; et al. Rational Improvement of the Affinity and Selectivity of Integrin Binding of Grafted Lasso Peptides. J. Med. Chem. 2014, 57, 5829–5834. [Google Scholar] [CrossRef]
- Romano, M.; Fusco, G.; Choudhury, H.G.; Mehmood, S.; Robinson, C.V.; Zirah, S.; Hegemann, J.; Lescop, E.; Marahiel, M.A.; Rebuffat, S.; et al. Structural Basis for Natural Product Selection and Export by Bacterial ABC Transporters. ACS Chem. Biol. 2018, 13, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Bountra, K.; Hagelueken, G.; Choudhury, H.G.; Corradi, V.; El Omari, K.; Wagner, A.; Mathavan, I.; Zirah, S.; Wahlgren, W.Y.; Tieleman, D.P.; et al. Structural basis for antibacterial peptide self-immunity by the bacterial ABC transporter McjD. EMBO J. 2017, 36, 3062–3079. [Google Scholar] [CrossRef]
- Choudhury, H.G.; Tong, Z.; Mathavan, I.; Li, Y.; Iwata, S.; Zirah, S.; Rebuffat, S.; Van Veen, H.W.; Beis, K. Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. USA 2014, 111, 9145–9150. [Google Scholar] [CrossRef]
- Rosengren, K.J.; Clark, R.J.; Daly, N.L.; Göransson, U.; Jones, A.; Craik, D.J. Microcin J25 Has a Threaded Sidechain-to-Backbone Ring Structure and Not a Head-to-Tail Cyclized Backbone. J. Am. Chem. Soc. 2003, 125, 12464–12474. [Google Scholar] [CrossRef]
- Wilson, K.-A.; Kalkum, M.; Ottesen, J.; Yuzenkova, J.; Chait, B.T.; Landick, R.; Muir, T.; Severinov, A.K.; Darst, S.A. Structure of Microcin J25, a Peptide Inhibitor of Bacterial RNA Polymerase, is a Lassoed Tail. J. Am. Chem. Soc. 2003, 125, 12475–12483. [Google Scholar] [CrossRef]
- Rosengren, K.J.; Blond, A.; Afonso, C.; Tabet, J.-C.; Rebuffat, S.; Craik, D.J. Structure of Thermolysin Cleaved Microcin J25: Extreme Stability of a Two-Chain Antimicrobial Peptide Devoid of Covalent Links. Biochemistry 2004, 43, 4696–4702. [Google Scholar] [CrossRef] [PubMed]
- Fouque, K.J.D.; Lavanant, H.; Zirah, S.; Hegemann, J.D.; Fage, C.D.; Marahiel, M.A.; Rebuffat, S.; Afonso, C. General rules of fragmentation evidencing lasso structures in CID and ETD. Analyst 2018, 143, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Genet. 2015, 14, 33–44. [Google Scholar] [CrossRef]
- Illigmann, A.; Thoma, Y.; Pan, S.; Reinhardt, L.; Brötz-Oesterhelt, H. Contribution of the Clp Protease to Bacterial Survival and Mitochondrial Homoeostasis. Microb. Physiol. 2021, 31, 260–279. [Google Scholar] [CrossRef]
- Frees, D.; Gerth, U.; Ingmer, H. Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. Int. J. Med. Microbiol. 2014, 304, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Y.H.; Houry, W.A. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007, 581, 3749–3757. [Google Scholar] [CrossRef]
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. eLife 2020, 9. [Google Scholar] [CrossRef]
- Gatsogiannis, C.; Balogh, D.; Merino, F.; Sieber, S.A.; Raunser, S. Cryo-EM structure of the ClpXP protein degradation ma-chinery. Nat. Struct. Mol. Biol. 2019, 26, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.; Lee, H.S.; Maurizi, M.R.; Steven, A.C. Alternating translocation of protein substrates from both ends of ClpXP protease. EMBO J. 2002, 21, 4938–4949. [Google Scholar] [CrossRef]
- Thompson, M.; Singh, S.; Maurizi, M. Processive degradation of proteins by the ATP-dependent Clp protease from Escherichia coli. Requirement for the multiple array of active sites in ClpP but not ATP hydrolysis. J. Biol. Chem. 1994, 269, 18209–18215. [Google Scholar] [CrossRef]
- Joshi, S.A.; Hersch, G.L.; Baker, T.A.; Sauer, R.T. Communication between ClpX and ClpP during substrate processing and degradation. Nat. Struct. Mol. Biol. 2004, 11, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Baker, T.A.; Sauer, R.T. Distinct Static and Dynamic Interactions Control ATPase-Peptidase Communication in a AAA+ Protease. Mol. Cell 2007, 27, 41–52. [Google Scholar] [CrossRef]
- Kim, Y.I.; Levchenko, I.; Fraczkowska, K.; Woodruff, R.V.; Sauer, R.T.; Baker, T.A. Molecular determinants of complex for-mation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat. Struct. Biol. 2001, 8, 230–233. [Google Scholar] [CrossRef]
- Brötz-Oesterhelt, H.; Beyer, D.; Kroll, H.-P.; Endermann, R.; Ladel, C.; Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 2005, 11, 1082–1087. [Google Scholar] [CrossRef]
- Malik, I.T.; Brötz-Oesterhelt, H. Conformational control of the bacterial Clp protease by natural product antibiotics. Nat. Prod. Rep. 2017, 34, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.; Famulla, K.; Dahmen, M.; Gobl, C.; Malik, I.; Richter, K.; Korotkov, V.S.; Sass, P.; Rübsamen-Schaeff, H.; Madl, T.; et al. AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat. Commun. 2015, 6, 6320. [Google Scholar] [CrossRef]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Sass, P.; Josten, M.; Famulla, K.; Schiffer, G.; Sahl, H.-G.; Hamoen, L.; Brötz-Oesterhelt, H. Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc. Natl. Acad. Sci. USA 2011, 108, 17474–17479. [Google Scholar] [CrossRef]
- Li, D.H.; Chung, Y.S.; Gloyd, M.; Joseph, E.; Ghirlando, R.; Wright, G.D.; Cheng, Y.Q.; Maurizi, M.R.; Guarné, A.; Ortega, J. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem. Biol. 2010, 17, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-G.; Park, E.Y.; Lee, K.-E.; Jeon, H.; Sung, K.H.; Paulsen, H.; Rübsamen-Schaeff, H.; Brötz-Oesterhelt, H.; Song, H.K. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat. Struct. Mol. Biol. 2010, 17, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Famulla, K.; Sass, P.; Malik, I.; Akopian, T.; Kandror, O.; Alber, M.; Hinzen, B.; Ruebsamen-Schaeff, H.; Kalscheuer, R.; Goldberg, A.L.; et al. Acyldepsipeptide antibiotics kill mycobacteria by preventing the physiological functions of the ClpP1P2 protease. Mol. Microbiol. 2016, 101, 194–209. [Google Scholar] [CrossRef]
- Kirstein, J.; Hoffmann, A.; Lilie, H.; Schmidt, R.; Rübsamen-Waigmann, H.; Brötz-Oesterhelt, H.; Mogk, A.; Turgay, K. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med. 2009, 1, 37–49. [Google Scholar] [CrossRef]
- Brötz-Oesterhelt, H.; Vorbach, A. Reprogramming of the Caseinolytic Protease by ADEP Antibiotics: Molecular Mechanism, Cellular Consequences, Therapeutic Potential. Front. Mol. Biosci. 2021, 8, 690902. [Google Scholar] [CrossRef]
- Thomy, D.; Culp, E.; Adamek, M.; Cheng, E.Y.; Ziemert, N.; Wright, G.D.; Sass, P.; Brötz-Oesterhelt, H. The ADEP Biosynthetic Gene Cluster in Streptomyces hawaiiensis NRRL 15010 Reveals an Accessory clpP Gene as a Novel Antibiotic Resistance Factor. Appl. Environ. Microbiol. 2019, 85, e01292-19. [Google Scholar] [CrossRef]
- Carney, D.W.; Compton, C.L.; Schmitz, K.R.; Stevens, J.P.; Sauer, R.; Sello, J.K. A simple fragment of cyclic acyldepsipeptides is necessary and sufficient for ClpP activation and antibacterial activity. ChemBioChem 2014, 15, 2216–2220. [Google Scholar] [CrossRef]
- Pavlova, O.; Mukhopadhyay, J.; Sineva, E.; Ebright, R.H.; Severinov, K. Systematic Structure-Activity Analysis of Microcin J25. J. Biol. Chem. 2008, 283, 25589–25595. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.T.; Pereira, R.; Vielberg, M.; Mayer, C.; Straetener, J.; Thomy, D.; Famulla, K.; Castro, H.C.; Sass, P.; Groll, M.; et al. Functional Characterisation of ClpP Mutations Conferring Resistance to Acyldepsipeptide Antibiotics in Firmicutes. ChemBioChem 2020, 21, 1997–2012. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kim, K.K. Crystal Structure of ClpX Molecular Chaperone from Helicobacter pylori. J. Biol. Chem. 2003, 278, 50664–50670. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Hegemann, J.D.; Bobeica, S.C.; Walker, M.C.; Bothwell, I.R.; van der Donk, W.A. Assessing the Flexibility of the Prochlorosin 2.8 Scaffold for Bioengineering Applications. ACS Synth. Biol. 2019, 8, 1204–1214. [Google Scholar] [CrossRef]
- Hetrick, K.J.; Walker, M.C.; van der Donk, W.A. Development and Application of Yeast and Phage Display of Diverse Lan-thipeptides. ACS Cent. Sci. 2018, 4, 458–467. [Google Scholar] [CrossRef]
- Urban, J.H.; Moosmeier, M.A.; Aumüller, T.; Thein, M.; Bosma, T.; Rink, R.; Groth, K.; Zulley, M.; Siegers, K.; Tissot, K.; et al. Phage display and selection of lanthipeptides on the carboxy-terminus of the gene-3 minor coat protein. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-G.; Kim, M.K.; Song, H.K. Structural insights into the conformational diversity of ClpP from Bacillus subtilis. Mol. Cells 2011, 32, 589–595. [Google Scholar] [CrossRef]
- Geiger, S.R.; Böttcher, T.; Sieber, S.A.; Cramer, P. A Conformational Switch Underlies ClpP Protease Function. Angew. Chem. Int. Ed. 2011, 50, 5749–5752. [Google Scholar] [CrossRef]
- Chiu, J.; Tillett, D.; Dawes, I.W.; March, P.E. Site-directed, Ligase-Independent Mutagenesis (SLIM) for highly efficient muta-genesis of plasmids greater than 8kb. J. Microbiol. Methods 2008, 73, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.; March, P.E.; Lee, R.; Tillett, D. Site-directed, Ligase-Independent Mutagenesis (SLIM): A single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 2004, 32, e174. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.; List, A.; Groll, M.; Sieber, S.A. Insights into structural network responsible for oligomerization and activity of bac-terial virulence regulator caseinolytic protease P (ClpP) protein. J. Biol. Chem. 2012, 287, 9484–9494. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| mcjA_SLIM_FP | TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_RP | CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_11IGL_Tail_FP | TAT TTT ATT GGG CTG GGT ACA CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_11IGL_Tail_RP | AGA TAT AGG TGT ACC CAG CCC AAT AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_11IGF_Tail_FP | TAT TTT ATT GGG TTT GGT ACA CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_11IGF_Tail_RP | AGA TAT AGG TGT ACC AAA CCC AAT AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_11VGF_Tail_FP | TAT TTT GTG GGG TTT GGT ACA CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_11VGF_Tail_RP | AGA TAT AGG TGT ACC AAA CCC CAC AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_12IGL_Tail_FP | TAT TTT GTG ATT GGC CTG ACA CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_12IGL_Tail_RP | AGA TAT AGG TGT CAG GCC AAT CAC AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_12IGF_Tail_FP | TAT TTT GTG ATT GGC TTT ACA CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_12IGF_Tail_RP | AGA TAT AGG TGT AAA GCC AAT CAC AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_12VGF_Tail_FP | TAT TTT GTG GTG GGC TTT ACA CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_12VGF_Tail_RP | AGA TAT AGG TGT AAA GCC CAC CAC AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_13IGL_Tail_FP | TAT TTT GTG GGG ATT GGT CTG CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_13IGL_Tail_RP | AGA TAT AGG CAG ACC AAT CCC CAC AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_13IGF_Tail_FP | TAT TTT GTG GGG ATT GGT TTT CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_13IGF_Tail_RP | AGA TAT AGG AAA ACC AAT CCC CAC AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| mcjA_SLIM_13VGF_Tail_FP | TAT TTT GTG GGG GTG GGT TTT CCT ATA TCT TTC TAT GGC TGA TAT TCT GAA AGA AGA ACT CTG |
| mcjA_SLIM_13VGF_Tail_RP | AGA TAT AGG AAA ACC CAC CCC CAC AAA ATA CTC AGG CAC ATG TCC TGC ACC ACC |
| Component | Amount |
|---|---|
| choline chloride | 1.0 g |
| folic acid | 1.0 g |
| pantothenic acid | 1.0 g |
| Nicotinamide | 1.0 g |
| myo-inositol | 2.0 g |
| pyridoxal hydrochloride | 1.0 g |
| Thiamine | 1.0 g |
| Riboflavin | 0.1 g |
| disodium adenosine 5′-triphosphate | 0.3 g |
| Biotin | 0.2 g |
| add 300 mL ddH2O * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, I.T.; Hegemann, J.D.; Brötz-Oesterhelt, H. Generation of Lasso Peptide-Based ClpP Binders. Int. J. Mol. Sci. 2022, 23, 465. https://doi.org/10.3390/ijms23010465
Malik IT, Hegemann JD, Brötz-Oesterhelt H. Generation of Lasso Peptide-Based ClpP Binders. International Journal of Molecular Sciences. 2022; 23(1):465. https://doi.org/10.3390/ijms23010465
Chicago/Turabian StyleMalik, Imran T., Julian D. Hegemann, and Heike Brötz-Oesterhelt. 2022. "Generation of Lasso Peptide-Based ClpP Binders" International Journal of Molecular Sciences 23, no. 1: 465. https://doi.org/10.3390/ijms23010465
APA StyleMalik, I. T., Hegemann, J. D., & Brötz-Oesterhelt, H. (2022). Generation of Lasso Peptide-Based ClpP Binders. International Journal of Molecular Sciences, 23(1), 465. https://doi.org/10.3390/ijms23010465