
Cancer-Homing CAR-T Cells and Endogenous Immune Population Dynamics


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
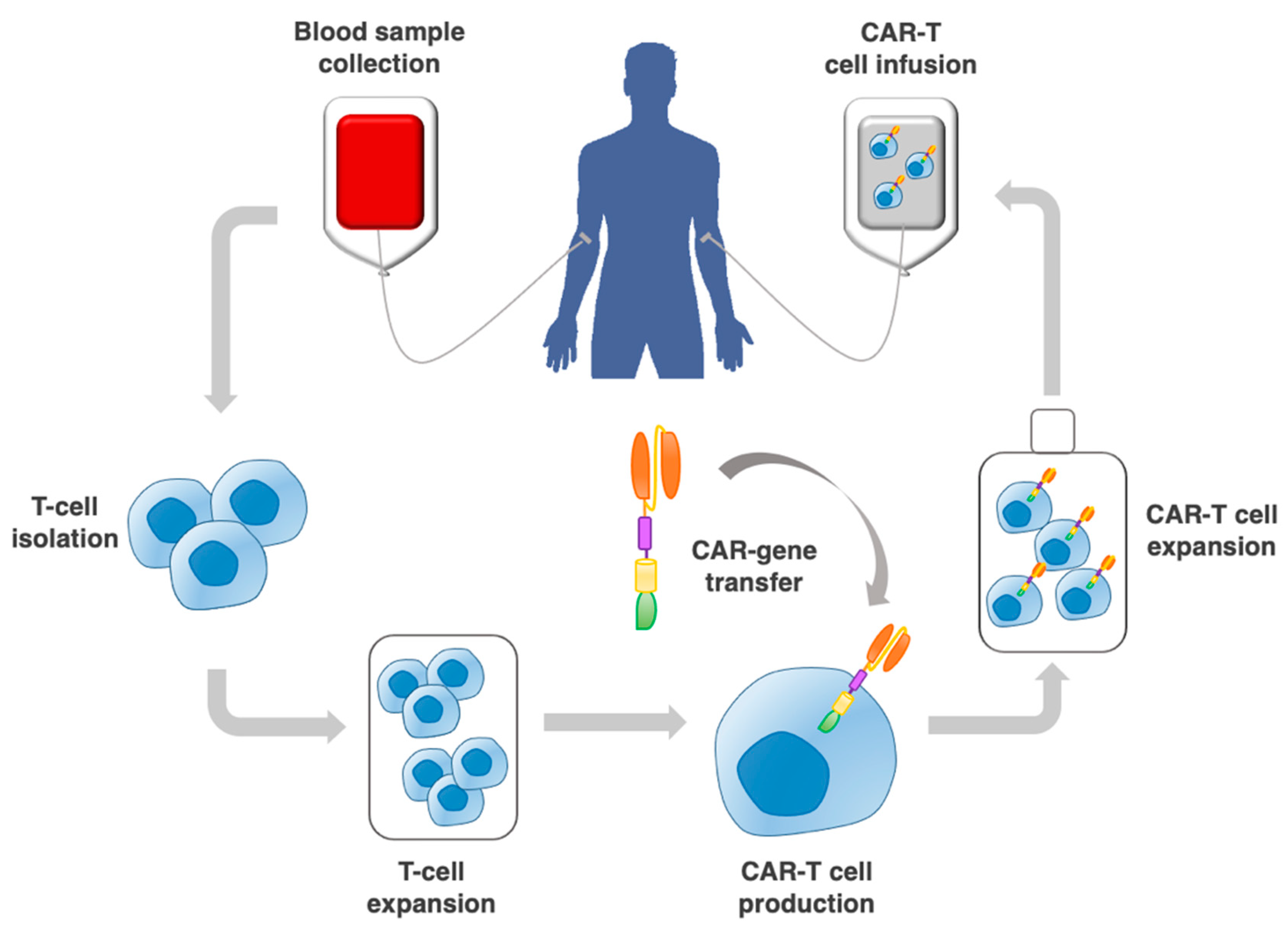
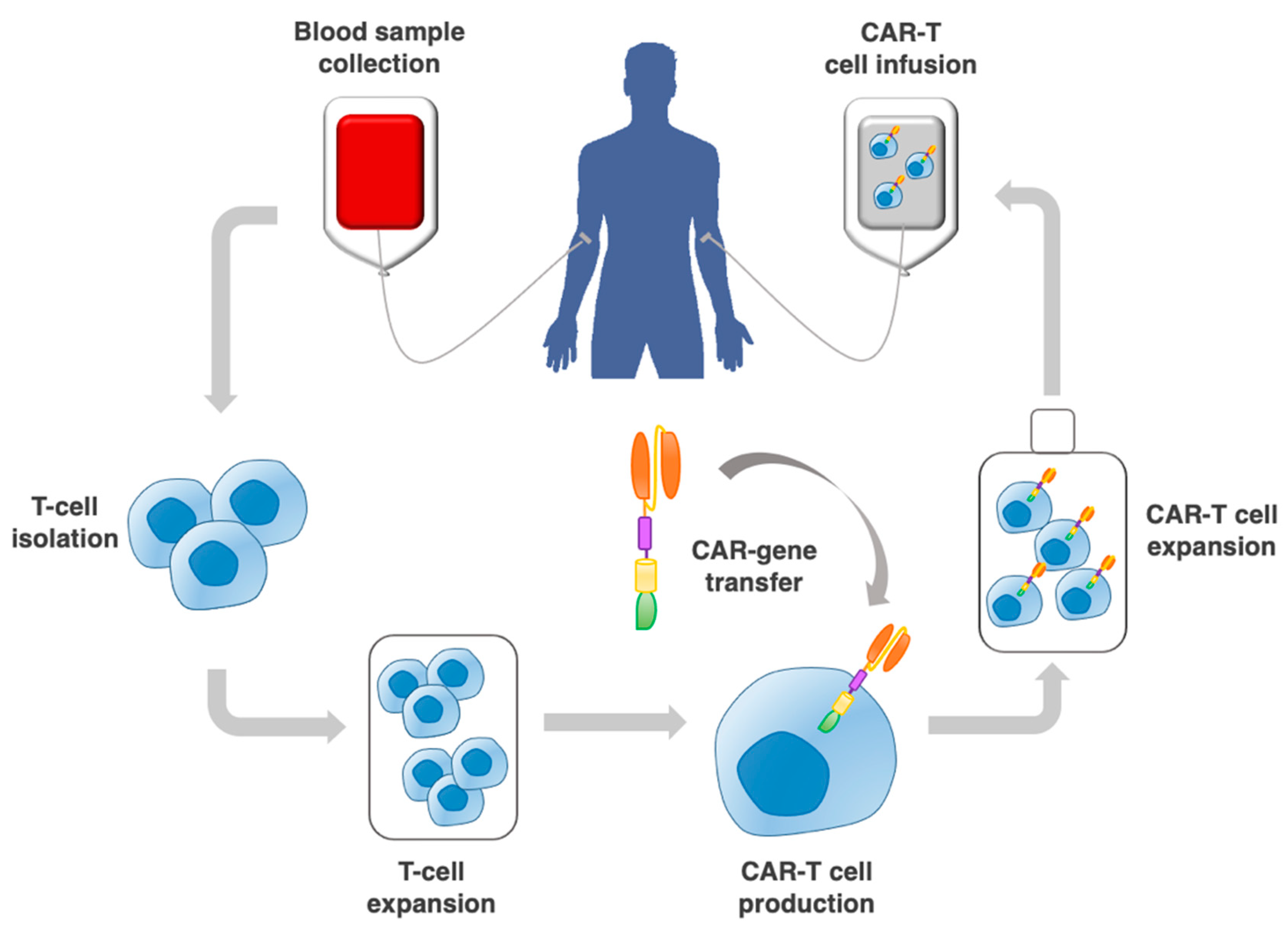
:1. Introduction
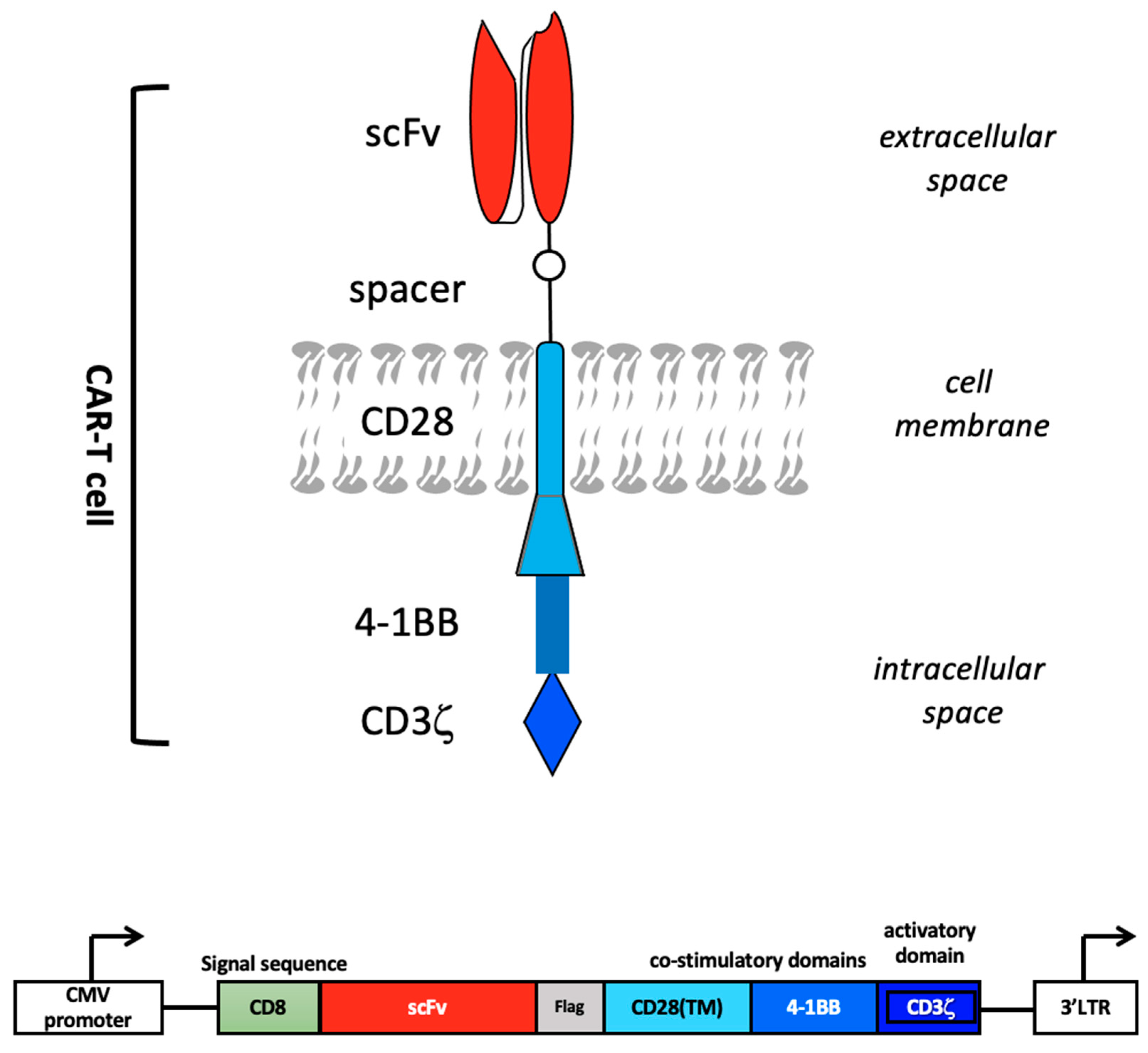
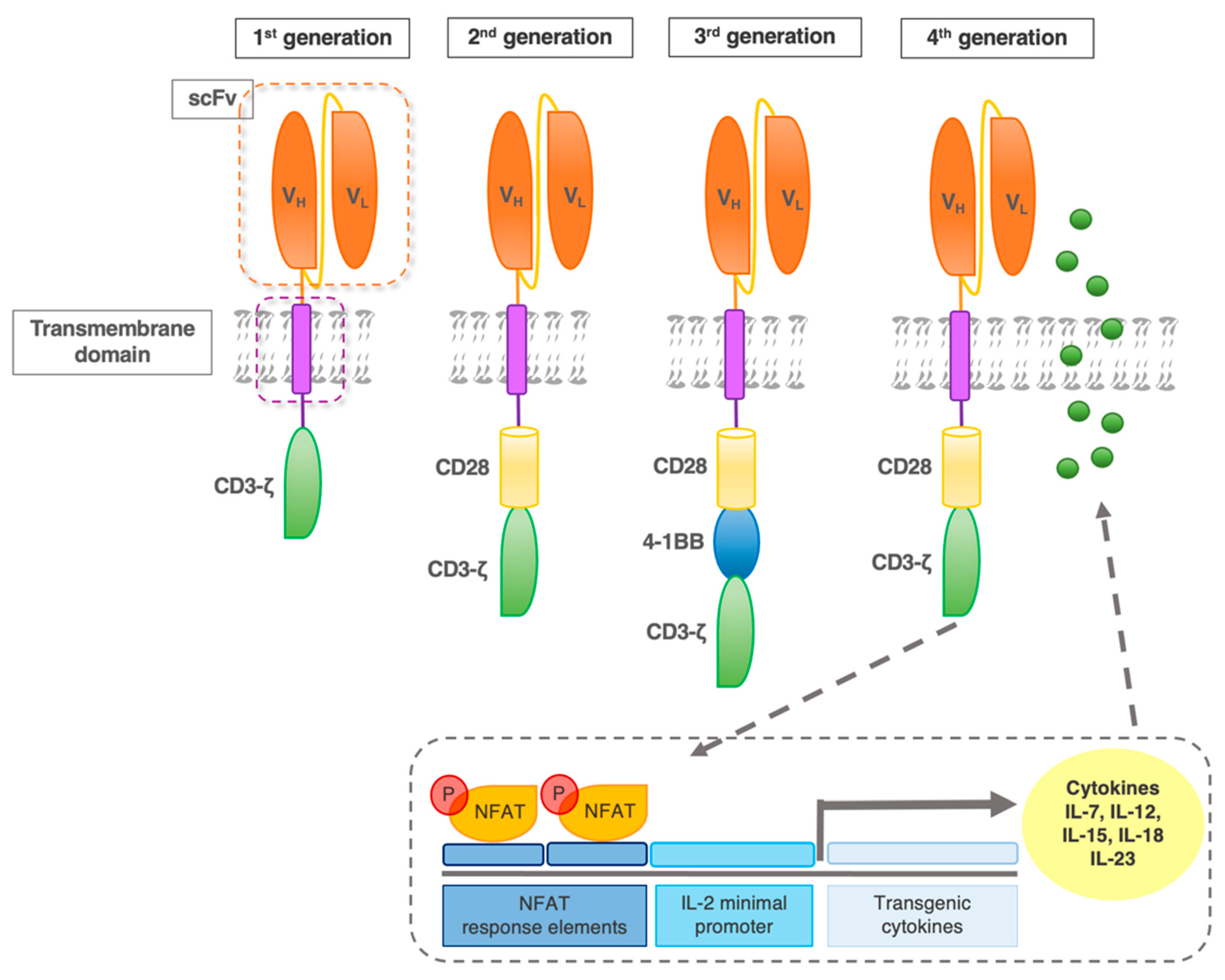
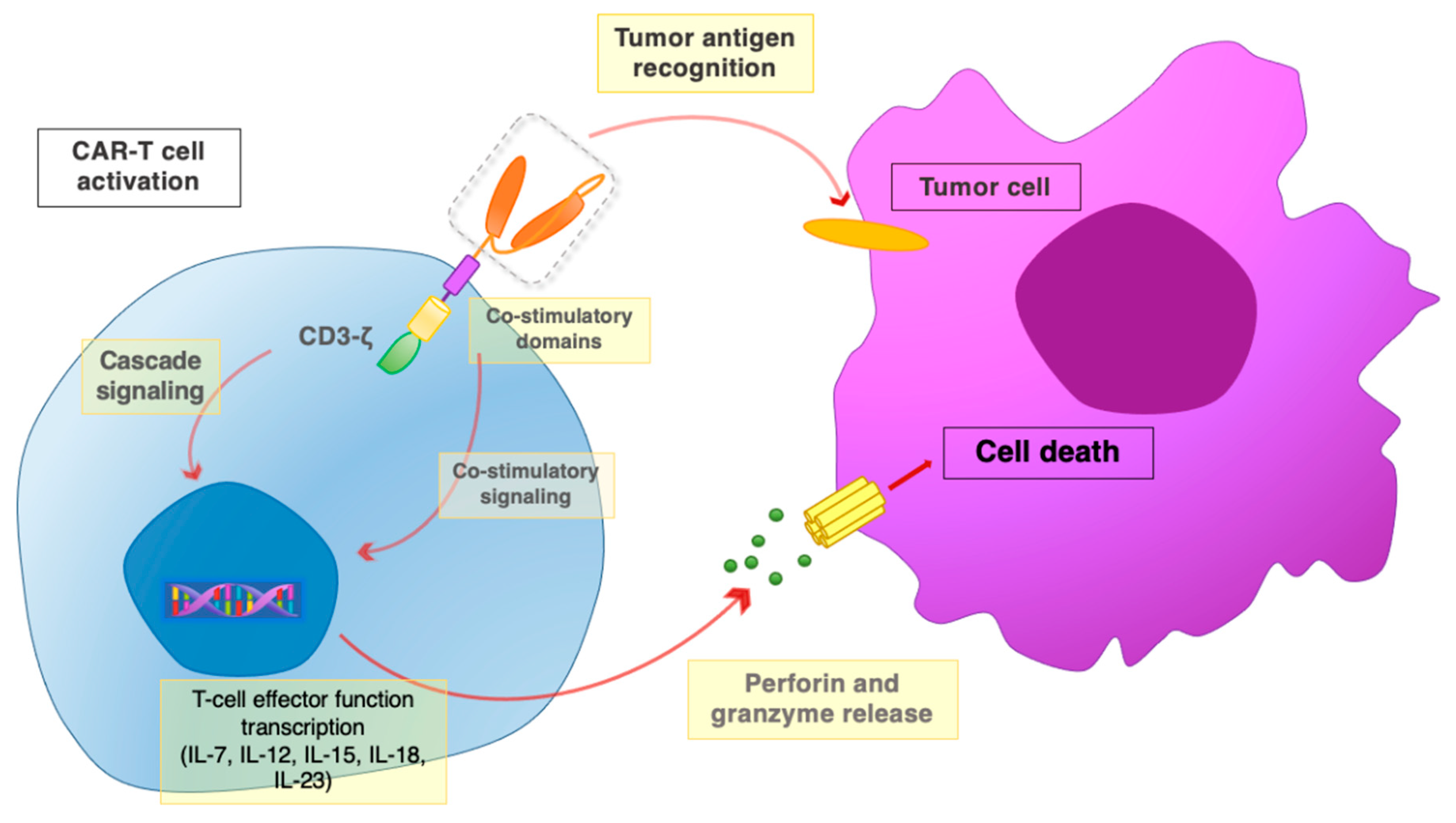
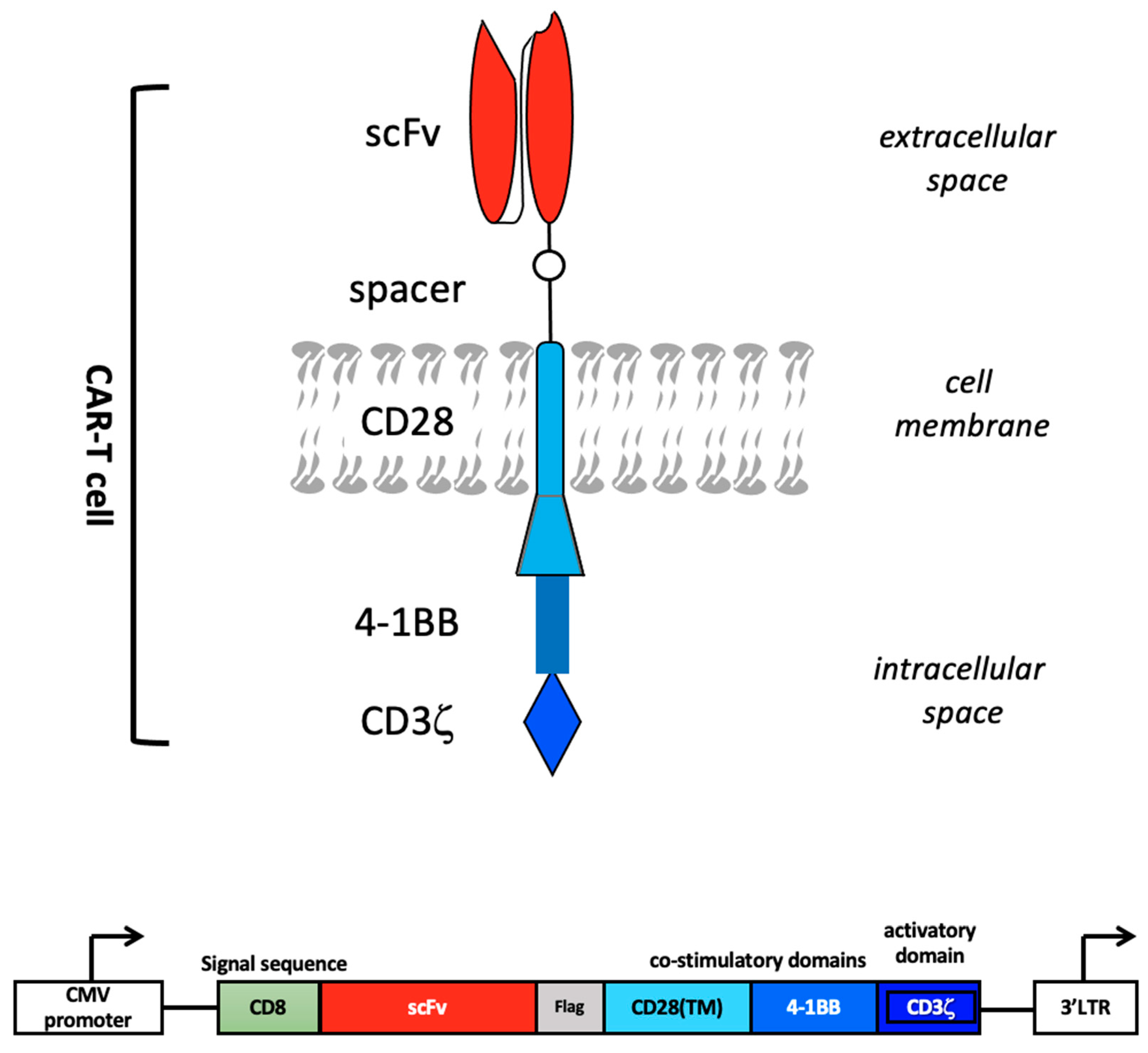
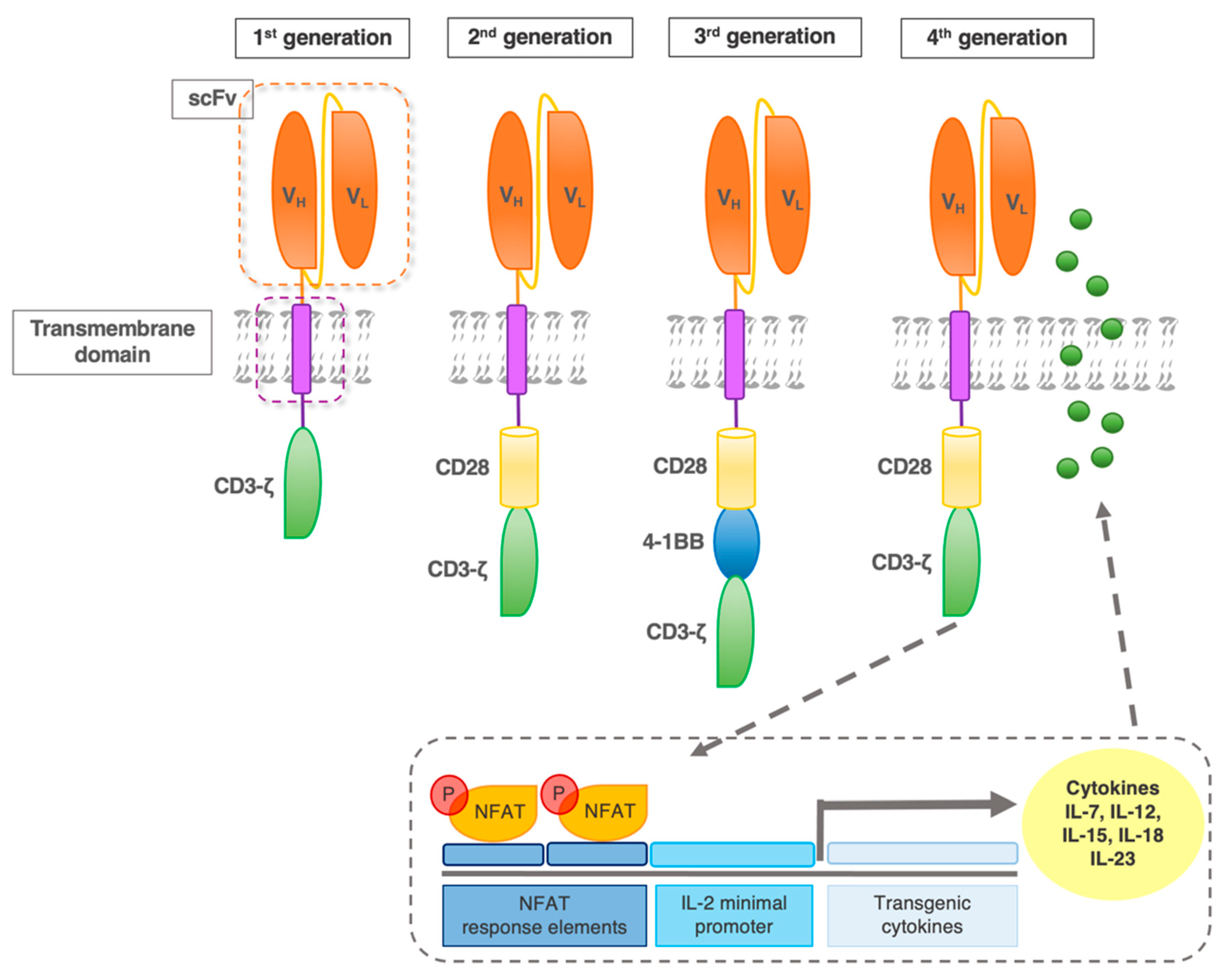
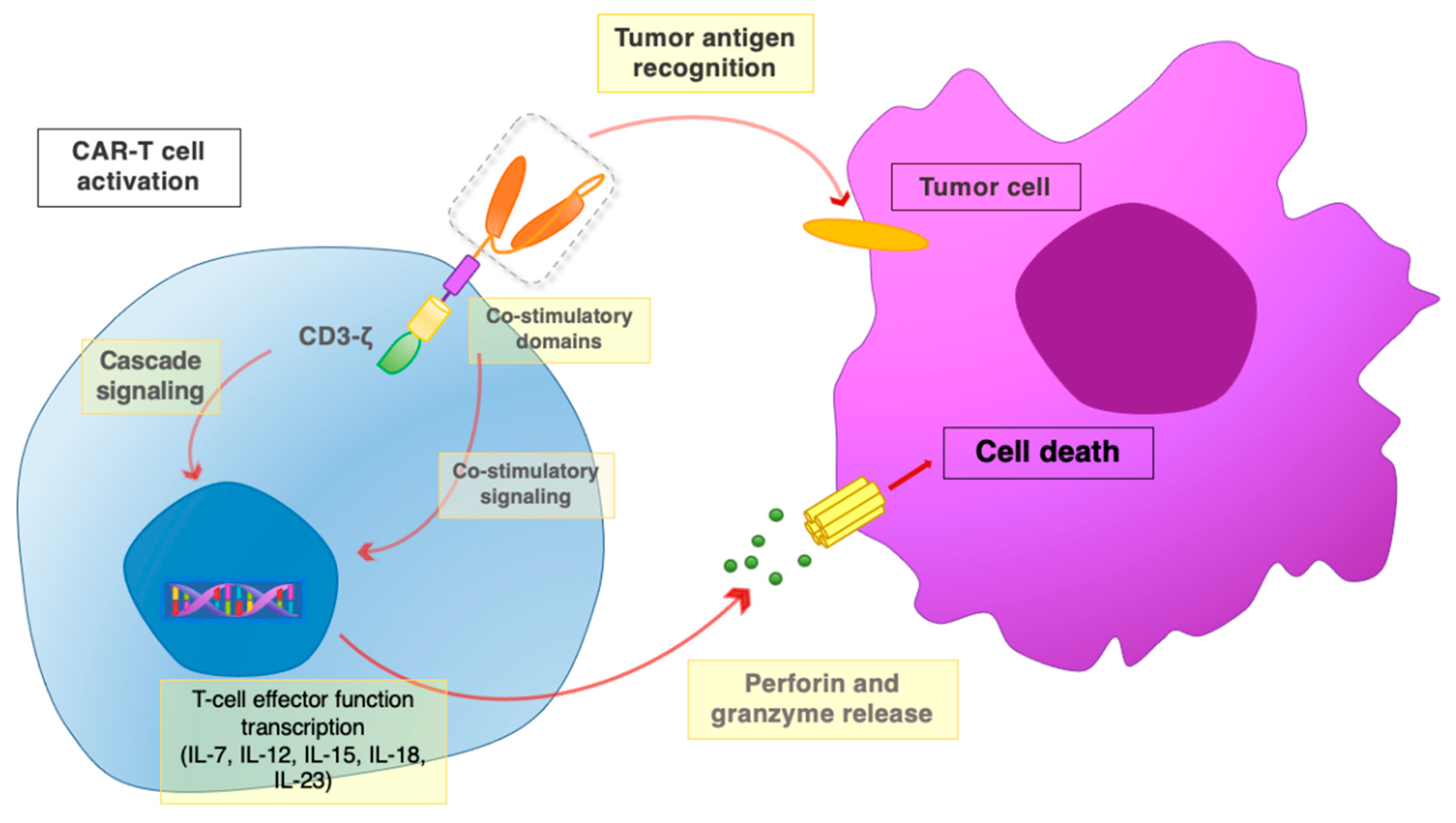
2. CAR Design
2.1. Early CAR
2.2. Fourth-Generation CAR
3. Main Challenges for CAR-T Cell Therapy against Solid Tumors
3.1. CAR Target Choice for Anticancer Therapy
3.2. Trafficking and Persistence of CAR-T Cells in the Tumor
3.3. CAR-T Treatment Toxicity
3.4. CAR-T Cell Exhaustion
3.5. Counteracting Immunosuppression against CAR-T Cells
3.6. CAR-T Cells Targeting Multiple Antigens
4. Cancer-Associated Immune Cell Populations
4.1. T Cell Subsets
4.2. Myeloid-Derived Suppressor Cells (MDSC)
4.3. Tumor-Associated Macrophages
4.4. Tumor-Reactive B Lymphocytes
4.5. NK Cells, Neutrophils, Dendritic Cells (DC)
5. Cancer-Associated Fibroblasts (CAF)
6. Immune Cell Dynamics and CAR-T Anticancer Response
6.1. CAR-T Cell Interactions with the Tumor Micro-Environment
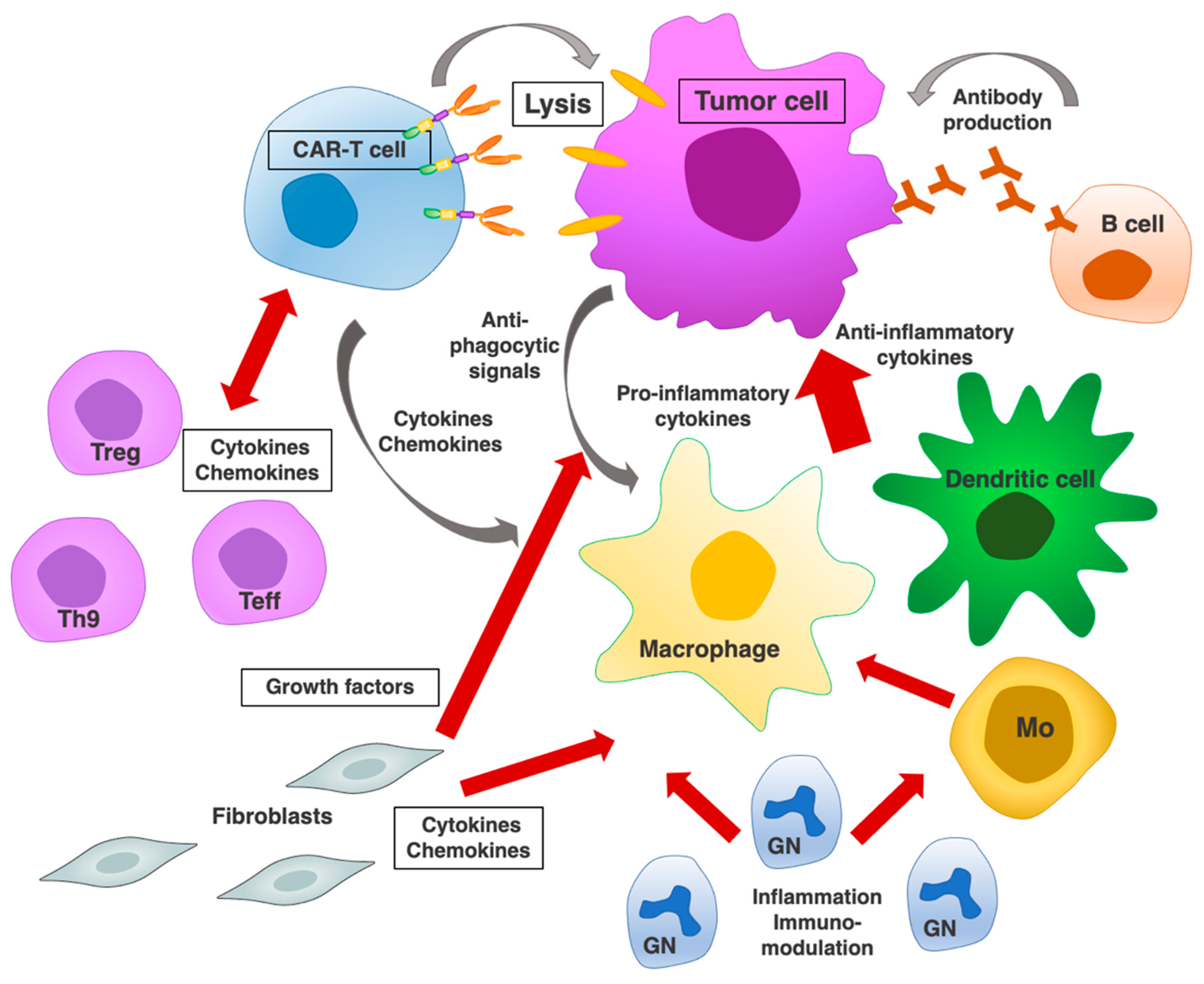
6.2. CAR-T Cell Interactions with Endogenous Immune Cell Subpopulations
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg Steven, A.; Spiess, P.; Lafreniere, R. A New Approach to the Adoptive Immunotherapy of Cancer with Tumor-Infiltrating Lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Alberti, S. A high affinity T cell receptor? Immunol. Cell Biol. 1996, 74, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.L.; Kaufman, D.S. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front. Immunol. 2015, 6, 195. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Honemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Puré, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Duong, C.P.; Yong, C.S.; Kershaw, M.H.; Slaney, C.Y.; Darcy, P.K. Cancer immunotherapy utilizing gene-modified T cells: From the bench to the clinic. Mol. Immunol. 2015, 67, 46–57. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.Y.; Sun, Y.; Zhang, A.; Hu, G.L.; Cao, W.; Wang, D.H.; Zhang, B.; Chen, H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: A non-randomised, open-label phase I trial protocol. BMJ Open 2016, 6, e013904. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Pitter, M.R.; Zou, W. Uncovering the Immunoregulatory Function and Therapeutic Potential of the PD-1/PD-L1 Axis in Cancer. Cancer Res. 2021, 81, 5141–5143. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [Green Version]
- Blanco, B.; Domínguez-Alonso, C.; Alvarez-Vallina, L. Bispecific Immunomodulatory Antibodies for Cancer Immunotherapy. Clin. Cancer Res. 2021, 27, 5457. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Ding, Y.; Fang, Y.; Wang, P.; Chu, W.; Jin, Z.; Yang, X.; Wang, J.; Lou, J.; et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021, 147, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Sancho, E.; Batlle, E. Overcoming TGFβ-mediated immune evasion in cancer. Nat. Rev. Cancer 2022, 22, 25–44. [Google Scholar] [CrossRef]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.J.; Yang, Y.X.; Han, E.Q.; Cao, N.; Wang, Y.F.; Wang, Y.; Zhao, Y.Y.; Zhao, L.M.; Cui, J.; Gupta, P.; et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J. Hematol. Oncol. 2013, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.L.; Sheard, V.E.; Kalaitsidou, M.; Blount, D.; Lad, Y.; Cheadle, E.J.; Edmondson, R.J.; Kooner, G.; Gilham, D.E.; Harrop, R. Preclinical Assessment of CAR T-Cell Therapy Targeting the Tumor Antigen 5T4 in Ovarian Cancer. J. Immunother. 2018, 41, 130–140. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, G.; Liu, J.; Yang, M.; Fu, J.; Liu, G.; Li, D.; Gu, Z.; Zhang, L.; Pan, Y.; et al. The antitumor capacity of mesothelin-CAR-T cells in targeting solid tumors in mice. Mol. Ther.-Oncolytics 2021, 20, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef]
- Hong, H.; Brown, C.E.; Ostberg, J.R.; Priceman, S.J.; Chang, W.C.; Weng, L.; Lin, P.; Wakabayashi, M.T.; Jensen, M.C.; Forman, S.J. L1 Cell Adhesion Molecule-Specific Chimeric Antigen Receptor-Redirected Human T Cells Exhibit Specific and Efficient Antitumor Activity against Human Ovarian Cancer in Mice. PLoS ONE 2016, 11, e0146885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, P.B.; Giralt, S.A.; Saltz, L.B. FDA Approval of Tisagenlecleucel: Promise and Complexities of a $475000 Cancer Drug. JAMA 2017, 318, 1861–1862. [Google Scholar] [CrossRef]
- Fala, L. Yescarta (Axicabtagene Ciloleucel) Second CAR T-Cell Therapy Approved for Patients with Certain Types of Large B-Cell Lymphoma. Am. Health Drug Benefits 2018, 11, 109–111. [Google Scholar]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Cazaux, M.; Grandjean, C.L.; Lemaitre, F.; Garcia, Z.; Beck, R.J.; Milo, I.; Postat, J.; Beltman, J.B.; Cheadle, E.J.; Bousso, P. Single-cell imaging of CAR T cell activity in vivo reveals extensive functional and anatomical heterogeneity. J. Exp. Med. 2019, 216, 1038–1049. [Google Scholar] [CrossRef]
- Van De Vyver, A.J.; Marrer-Berger, E.; Wang, K.; Lehr, T.; Walz, A.-C. Cytokine Release Syndrome By T-cell–Redirecting Therapies: Can We Predict and Modulate Patient Risk? Clin. Cancer Res. 2021, 27, 6083. [Google Scholar] [CrossRef]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Sheth, V.S.; Gauthier, J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. 2021, 56, 552–566. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Nastoupil, L.J.; Adkins, S.; Lacchetti, C.; Schneider, B.J.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated With Chimeric Antigen Receptor T-Cell Therapy: ASCO Guideline. J. Clin. Oncol. 2021, 39, 3978–3992. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Lopez de Rodas, M.; Schalper, K.A. Tumour antigen-induced T cell exhaustion—The archenemy of immune-hot malignancies. Nat. Rev. Clin. Oncol. 2021, 18, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Miggelbrink, A.M.; Jackson, J.D.; Lorrey, S.J.; Srinivasan, E.S.; Waibl-Polania, J.; Wilkinson, D.S.; Fecci, P.E. CD4 T-Cell Exhaustion: Does It Exist and What Are Its Roles in Cancer? Clin. Cancer Res. 2021, 27, 5742. [Google Scholar] [CrossRef]
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59. [Google Scholar] [CrossRef]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef]
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494. [Google Scholar] [CrossRef] [Green Version]
- Good, C.R.; Aznar, M.A.; Kuramitsu, S.; Samareh, P.; Agarwal, S.; Donahue, G.; Ishiyama, K.; Wellhausen, N.; Rennels, A.K.; Ma, Y.; et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 2021, 184, 6081–6100.e26. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef]
- Boyerinas, B.; Miller, S.M.; Murray, R.C.; Evans, J.W.; Parsons, G.B.; Seidl, K.J.; Friedman, K.M.; Morgan, R.A. A Novel TGF-β2/Interleukin Receptor Signal Conversion Platform That Protects CAR/TCR T Cells from TGF-β2-Mediated Immune Suppression and Induces T Cell Supportive Signaling Networks. Blood 2017, 130, 1911. [Google Scholar]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [Green Version]
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The Bioenergetic Signature of Cancer: A Marker of Tumor Progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar]
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Rössig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Puré, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol. Res. 2016, 4, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, H.; Chen, G.; Wei, X.; Wang, C.; Zhou, S.; Huang, A.; Zhang, Z.; Zhan, C.; Wu, Y.; et al. Arming Anti-EGFRvIII CAR-T With TGFβ Trap Improves Antitumor Efficacy in Glioma Mouse Models. Front. Oncol. 2020, 10, 1117. [Google Scholar] [CrossRef]
- Durgin, J.S.; Henderson, F., Jr.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front. Oncol. 2021, 11, 669071. [Google Scholar] [CrossRef]
- Duong, C.P.; Westwood, J.A.; Berry, L.J.; Darcy, P.K.; Kershaw, M.H. Enhancing the specificity of T-cell cultures for adoptive immunotherapy of cancer. Immunotherapy 2011, 3, 33–48. [Google Scholar] [CrossRef]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Muliaditan, T.; Halim, L.; Whilding, L.M.; Draper, B.; Achkova, D.Y.; Kausar, F.; Glover, M.; Bechman, N.; Arulappu, A.; Sanchez, J.; et al. Synergistic T cell signaling by 41BB and CD28 is optimally achieved by membrane proximal positioning within parallel chimeric antigen receptors. Cell Rep. Med. 2021, 2, 100457. [Google Scholar] [CrossRef]
- Castellanos-Rueda, R.; Di Roberto, R.B.; Schlatter, F.S.; Reddy, S.T. Leveraging Single-Cell Sequencing for Chimeric Antigen Receptor T Cell Therapies. Trends Biotechnol. 2021, 39, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Coles, M.; Thomas, T.; Kollias, G.; Ludewig, B.; Turley, S.; Brenner, M.; Buckley, C.D. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol. 2021, 21, 704–717. [Google Scholar] [CrossRef]
- Luca, B.A.; Steen, C.B.; Matusiak, M.; Azizi, A.; Varma, S.; Zhu, C.; Przybyl, J.; Espín-Pérez, A.; Diehn, M.; Alizadeh, A.A.; et al. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell 2021, 184, 5482–5496.e28. [Google Scholar] [CrossRef]
- Oh, D.Y.; Fong, L.; Newell, E.W.; Turk, M.J.; Chi, H.; Chang, H.Y.; Satpathy, A.T.; Fairfax, B.; Silva-Santos, B.; Lantz, O. Toward a better understanding of T cells in cancer. Cancer Cell 2021, 39, 1549–1552. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Duell, J.; Dittrich, M.; Bedke, T.; Mueller, T.; Eisele, F.; Rosenwald, A.; Rasche, L.; Hartmann, E.; Dandekar, T.; Einsele, H.; et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 2017, 31, 2181–2190. [Google Scholar] [CrossRef]
- Oh, D.Y.; Fong, L. Cytotoxic CD4+ T cells in cancer: Expanding the immune effector toolbox. Immunity 2021, 54, 2701–2711. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med. 2017, 23, 540–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelletti, L.; Yeo, D.; van Zandwijk, N.; Rasko, J.E.J. Anti-Mesothelin CAR T cell therapy for malignant mesothelioma. Biomark. Res. 2021, 9, 11. [Google Scholar] [CrossRef]
- Zhen, A.; Carrillo, M.A.; Mu, W.; Rezek, V.; Martin, H.; Hamid, P.; Chen, I.S.Y.; Yang, O.O.; Zack, J.A.; Kitchen, S.G. Robust CAR-T memory formation and function via hematopoietic stem cell delivery. PLoS Pathog. 2021, 17, e1009404. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, T.C.; Alice, A.F.; Zebertavage, L.; Crittenden, M.R.; Gough, M.J. The Dynamic Entropy of Tumor Immune Infiltrates: The Impact of Recirculation, Antigen-Specific Interactions, and Retention on T Cells in Tumors. Front. Oncol. 2021, 11, 1350. [Google Scholar] [CrossRef]
- Dong, Y.; Wan, Z.; Gao, X.; Yang, G.; Liu, L. Reprogramming Immune Cells for Enhanced Cancer Immunotherapy: Targets and Strategies. Front. Immunol. 2021, 12, 609762. [Google Scholar] [CrossRef]
- Liu, M.; Wei, F.; Wang, J.; Yu, W.; Shen, M.; Liu, T.; Zhang, D.; Wang, Y.; Ren, X.; Sun, Q. Myeloid-derived suppressor cells regulate the immunosuppressive functions of PD-1−PD-L1+ Bregs through PD-L1/PI3K/AKT/NF-κB axis in breast cancer. Cell Death Dis. 2021, 12, 465. [Google Scholar] [CrossRef]
- Lightman, S.M.; Peresie, J.L.; Carlson, L.M.; Holling, G.A.; Honikel, M.M.; Chavel, C.A.; Nemeth, M.J.; Olejniczak, S.H.; Lee, K.P. Indoleamine 2,3-dioxygenase 1 is essential for sustaining durable antibody responses. Immunity 2021, 54, 2772–2783.e5. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Pan, P.Y.; Wang, G.X.; Yin, B.; Ozao, J.; Ku, T.; Divino, C.M.; Chen, S.H. Reversion of immune tolerance in advanced malignancy: Modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood 2008, 111, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Sumida, K.; Wakita, D.; Narita, Y.; Masuko, K.; Terada, S.; Watanabe, K.; Satoh, T.; Kitamura, H.; Nishimura, T. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur. J. Immunol. 2012, 42, 2060–2072. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Lecker, L.S.M.; Berlato, C.; Maniati, E.; Delaine-Smith, R.; Pearce, O.M.T.; Heath, O.; Nichols, S.J.; Trevisan, C.; Novak, M.; McDermott, J.; et al. TGFBI Production by Macrophages Contributes to an Immunosuppressive Microenvironment in Ovarian Cancer. Cancer Res. 2021, 81, 5706. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Lynn, R.; Poussin, M.; Eiva, M.; Shaw, L.; O’Connor, R.; Minutolo, N.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Möckl, A.; Salamero-Boix, A.; Alekseeva, T.; Schäffer, A.; Schulz, M.; Niesel, K.; Maas, R.R.; Groth, M.; Elie, B.T.; et al. Compensatory CSF2-driven macrophage activation promotes adaptive resistance to CSF1R inhibition in breast-to-brain metastasis. Nat. Cancer 2021, 2, 1086–1101. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.L.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007, 67, 9417–9424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonapace, L.; Coissieux, M.M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Khong, H.; Dai, Z.; Huang, X.F.; Wargo, J.A.; Cooper, Z.A.; Vasilakos, J.P.; Hwu, P.; Overwijk, W.W. Effective innate and adaptive antimelanoma immunity through localized TLR7/8 activation. J. Immunol. 2014, 193, 4722–4731. [Google Scholar] [CrossRef] [Green Version]
- Baer, C.; Squadrito, M.L.; Laoui, D.; Thompson, D.; Hansen, S.K.; Kiialainen, A.; Hoves, S.; Ries, C.H.; Ooi, C.H.; De Palma, M. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 2016, 18, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Karp, J.E.; Emadi, A. Elevated lactate dehydrogenase (LDH) can be a marker of immune suppression in cancer: Interplay between hematologic and solid neoplastic clones and their microenvironments. Cancer Biomark. 2017, 19, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kumar, A.; Vilgelm, A.E.; Chen, S.C.; Ayers, G.D.; Novitskiy, S.V.; Joyce, S.; Richmond, A. Loss of CXCR4 in Myeloid Cells Enhances Antitumor Immunity and Reduces Melanoma Growth through NK Cell and FASL Mechanisms. Cancer Immunol. Res. 2018, 6, 1186–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.; Suh, Y.; Jung, K. Context Drives Diversification of Monocytes and Neutrophils in Orchestrating the Tumor Microenvironment. Front. Immunol. 2019, 10, 1817. [Google Scholar] [CrossRef]
- Kochetkova, M.; Samuel, M.S. Differentiation of the tumor microenvironment: Are CAFs the Organizer? Trends Cell Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef]
- Cao, H.; Eppinga, R.D.; Razidlo, G.L.; Krueger, E.W.; Chen, J.; Qiang, L.; McNiven, M.A. Stromal fibroblasts facilitate cancer cell invasion by a novel invadopodia-independent matrix degradation process. Oncogene 2016, 35, 1099–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polette, M.; Nawrocki-Raby, B.; Gilles, C.; Clavel, C.; Birembaut, P. Tumour invasion and matrix metalloproteinases. Crit. Rev. Oncol. Hematol. 2004, 49, 179–186. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.; Levi-Galibov, O.; David, E.; Bornstein, C.; Giladi, A.; Dadiani, M.; Mayo, A.; Halperin, C.; Pevsner-Fischer, M.; Lavon, H.; et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat. Cancer 2020, 1, 692–708. [Google Scholar] [CrossRef]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Hanna, B.S.; Llaó-Cid, L.; Iskar, M.; Roessner, P.M.; Klett, L.C.; Wong, J.K.L.; Paul, Y.; Ioannou, N.; Öztürk, S.; Mack, N.; et al. Interleukin-10 receptor signaling promotes the maintenance of a PD-1/TCF-1/CD8+/T cell population that sustains anti-tumor immunity. Immunity 2021, 54, 2825–2841.e10. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2020, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T.; et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 2016, 213, 2315–2331. [Google Scholar] [CrossRef]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latouche, J.B.; Sadelain, M. Induction of human cytotoxic T lymphocytes by artificial antigen- presenting cells. Nat. Biotechnol. 2000, 18, 405–409. [Google Scholar] [CrossRef]
- Mangino, G.; Capri, M.G.; Barnaba, V.; Alberti, S. Presentation of native Trop-2 tumor antigens to human cytotoxic T lymphocytes by engineered antigen-presenting cells. Int. J. Cancer 2002, 101, 353–359. [Google Scholar] [CrossRef]
- Gangaev, A.; Rozeman, E.A.; Rohaan, M.W.; Isaeva, O.I.; Philips, D.; Patiwael, S.; van den Berg, J.H.; Ribas, A.; Schadendorf, D.; Schilling, B.; et al. Differential effects of PD-1 and CTLA-4 blockade on the melanoma-reactive CD8 T cell response. Proc. Natl. Acad. Sci. USA 2021, 118, e2102849118. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Scharping, N.E.; Goldrath, A.W. CD8+ T cell metabolism in infection and cancer. Nat. Rev. Immunol. 2021, 21, 718–738. [Google Scholar] [CrossRef]
- Payne, R.P.; Longet, S.; Austin, J.A.; Skelly, D.T.; Dejnirattisai, W.; Adele, S.; Meardon, N.; Faustini, S.; Al-Taei, S.; Moore, S.C.; et al. Immunogenicity of standard and extended dosing intervals of BNT162b2 mRNA vaccine. Cell 2021, 184, 5699–5714. [Google Scholar] [CrossRef]
- Xue, G.; Zheng, N.; Fang, J.; Jin, G.; Li, X.; Dotti, G.; Yi, Q.; Lu, Y. Adoptive cell therapy with tumor-specific Th9 cells induces viral mimicry to eliminate antigen-loss-variant tumor cells. Cancer Cell 2021, 39, 1610–1622. [Google Scholar] [CrossRef]
- Sek, K.; Chan, C.W.; Beavis, P.A.; Darcy, P.K. Adoptive transfer of tumor-specific Th9 cells eradicates heterogeneous antigen-expressing tumor cells. Cancer Cell 2021, 39, 1564–1566. [Google Scholar] [CrossRef] [PubMed]
- Beatson, R.E.; Parente-Pereira, A.C.; Halim, L.; Cozzetto, D.; Hull, C.; Whilding, L.M.; Martinez, O.; Taylor, C.A.; Obajdin, J.; Luu Hoang, K.N.; et al. TGF-b1 potentiates Vg9Vd2 T cell adoptive immunotherapy of cancer. Cell Rep. Med. 2021, 2, 100473. [Google Scholar] [CrossRef]
- Hou, A.J.; Chang, Z.L.; Lorenzini, M.H.; Zah, E.; Chen, Y.Y. TGF-β-responsive CAR-T cells promote anti-tumor immune function. Bioeng. Transl. Med. 2018, 3, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Sanseviero, E.; Timosenko, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells: A Propitious Road to Clinic. Cancer Discov. 2021, 11, 2693–2706. [Google Scholar] [CrossRef] [PubMed]
- Hanes, M.R.; Giacomantonio, C.A.; Marshall, J.S. Mast Cells and Skin and Breast Cancers: A Complicated and Microenvironment-Dependent Role. Cells 2021, 10, 986. [Google Scholar] [CrossRef]
- Simonetta, F.; Lohmeyer, J.K.; Hirai, T.; Maas-Bauer, K.; Alvarez, M.; Wenokur, A.S.; Baker, J.; Aalipour, A.; Ji, X.; Haile, S.; et al. Allogeneic CAR Invariant Natural Killer T Cells Exert Potent Antitumor Effects through Host CD8 T-Cell Cross-Priming. Clin. Cancer Res. 2021, 27, 6054. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Curran, K.J.; Seinstra, B.A.; Nikhamin, Y.; Yeh, R.; Usachenko, Y.; van Leeuwen, D.G.; Purdon, T.; Pegram, H.J.; Brentjens, R.J. Enhancing Antitumor Efficacy of Chimeric Antigen Receptor T Cells Through Constitutive CD40L Expression. Mol. Ther. 2015, 23, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488.e6. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Chan, J.D.; et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 914–926. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, X.; Shen, L.; Wang, X.; Xu, C. Exploiting T cell signaling to optimize engineered T cell therapies. Trends Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Lipschitz, M.; Weirather, J.L.; Jacobson, C.; Armand, P.; Wright, K.; Hodi, F.S.; Roberts, Z.J.; Sievers, S.A.; Rossi, J.; et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight 2020, 5, e134612. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, E.; Di Pietro, R.; Basile, M.; Trerotola, M.; Alberti, S. Cancer-Homing CAR-T Cells and Endogenous Immune Population Dynamics. Int. J. Mol. Sci. 2022, 23, 405. https://doi.org/10.3390/ijms23010405
Guerra E, Di Pietro R, Basile M, Trerotola M, Alberti S. Cancer-Homing CAR-T Cells and Endogenous Immune Population Dynamics. International Journal of Molecular Sciences. 2022; 23(1):405. https://doi.org/10.3390/ijms23010405
Chicago/Turabian StyleGuerra, Emanuela, Roberta Di Pietro, Mariangela Basile, Marco Trerotola, and Saverio Alberti. 2022. "Cancer-Homing CAR-T Cells and Endogenous Immune Population Dynamics" International Journal of Molecular Sciences 23, no. 1: 405. https://doi.org/10.3390/ijms23010405
APA StyleGuerra, E., Di Pietro, R., Basile, M., Trerotola, M., & Alberti, S. (2022). Cancer-Homing CAR-T Cells and Endogenous Immune Population Dynamics. International Journal of Molecular Sciences, 23(1), 405. https://doi.org/10.3390/ijms23010405