
The Energy Sensor AMPKα1 Is Critical in Rapamycin-Inhibition of mTORC1-S6K-Induced T-cell Memory

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
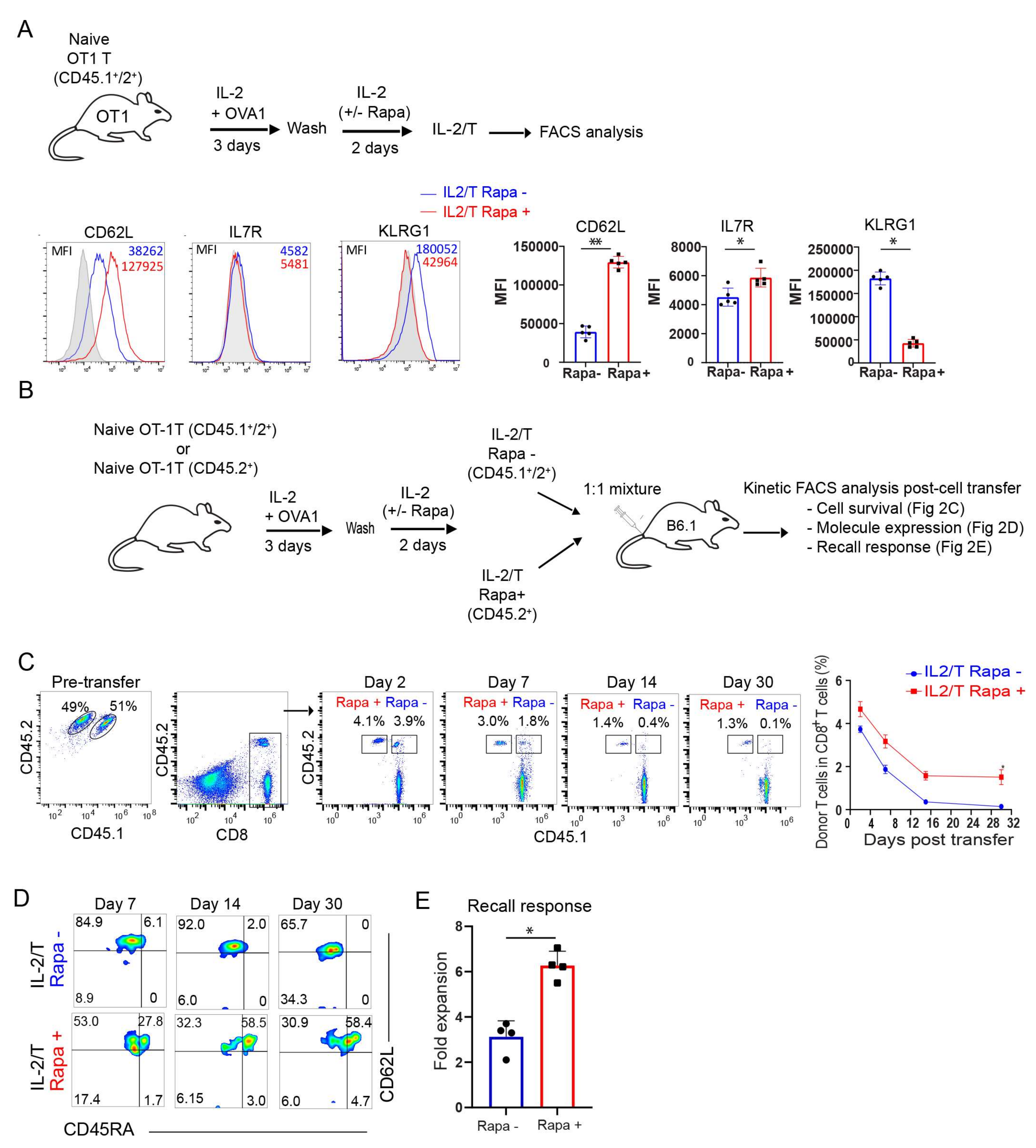
2.1. Rapamycin Promotes T-cell Survival and Memory Formation In Vivo Post Infection with Recombinant Listeria Monocytogenes rLmOVA
2.2. Rapamycin Promotes the Transition of TE into Long-Term CD45RA+ Stem Cell-Like TM Cells In Vitro
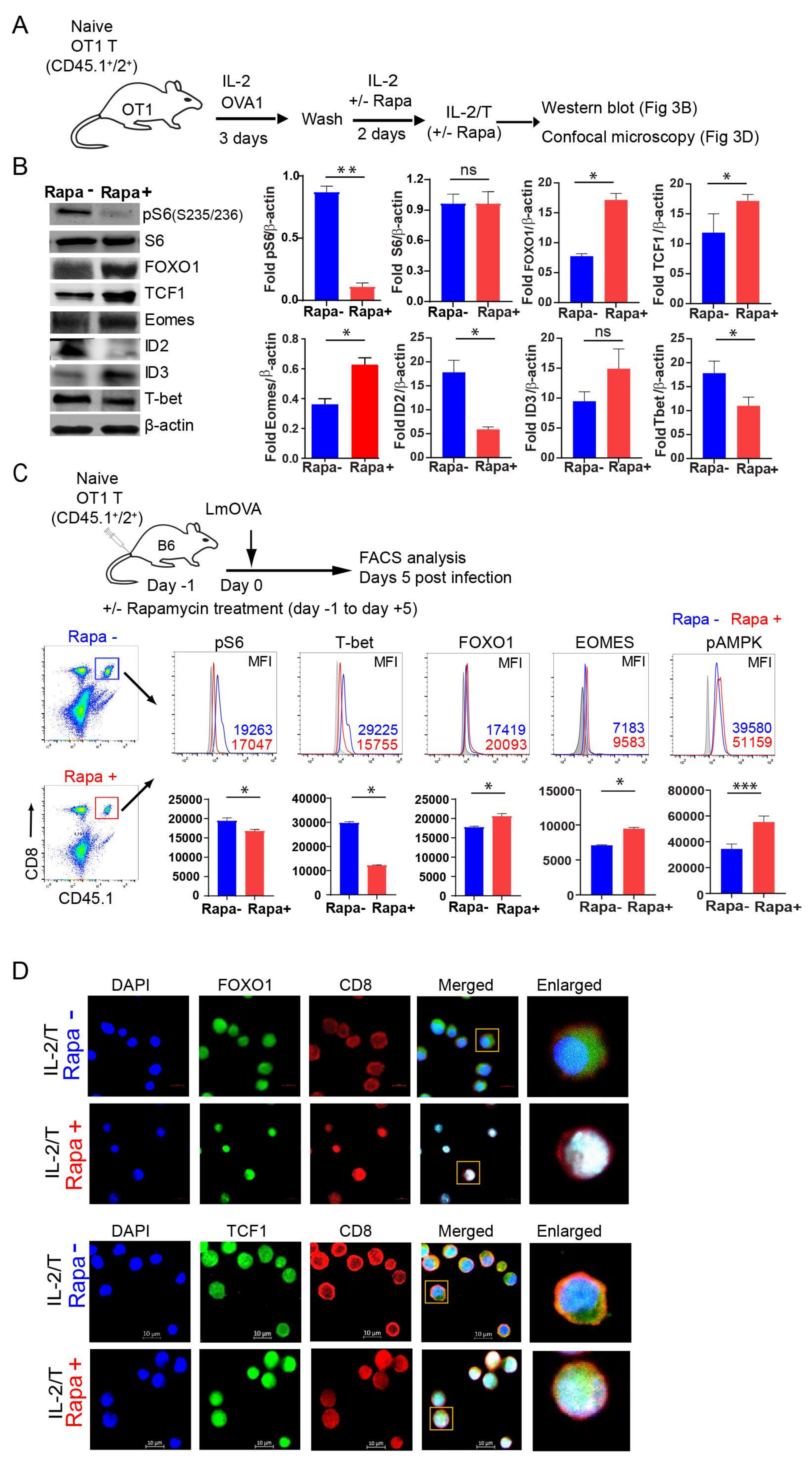
2.3. IL-2(Rapa+)/T-cells Suppress mTORC1/S6K Signaling and Activate the FOXO1-TCF1-Eomes Transcriptional Pathway
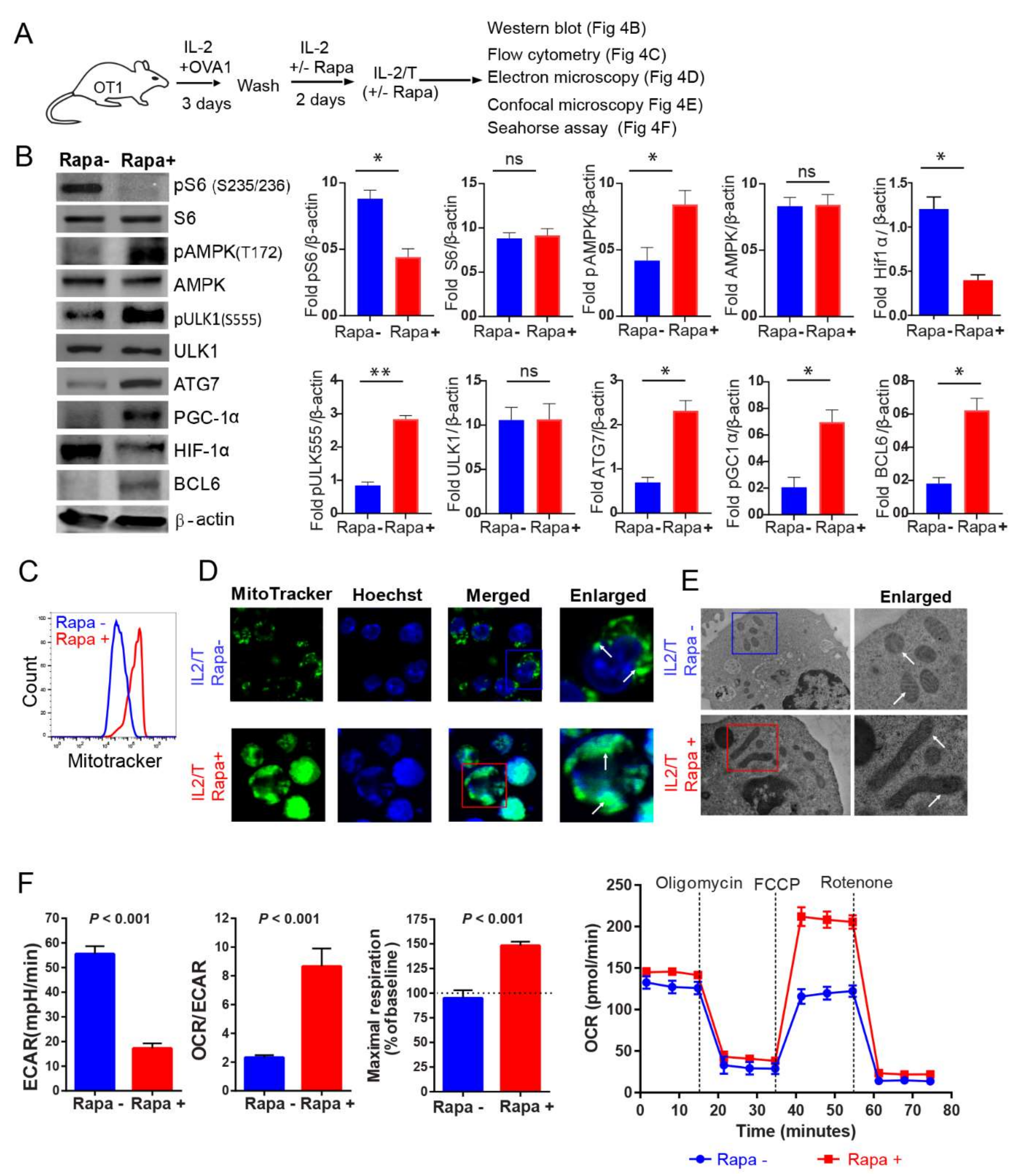
2.4. IL-2(Rapa+)/T-cells Activate the AMPKα1-ULK1-ATG7 Metabolic Axis
2.5. IL-2(Rapa+)/T-cells Promote Mitochondrial Biogenesis
2.6. IL-2(Rapa+)/T-cells Have Substantial Mitochondrial SRC and Rely on FAO
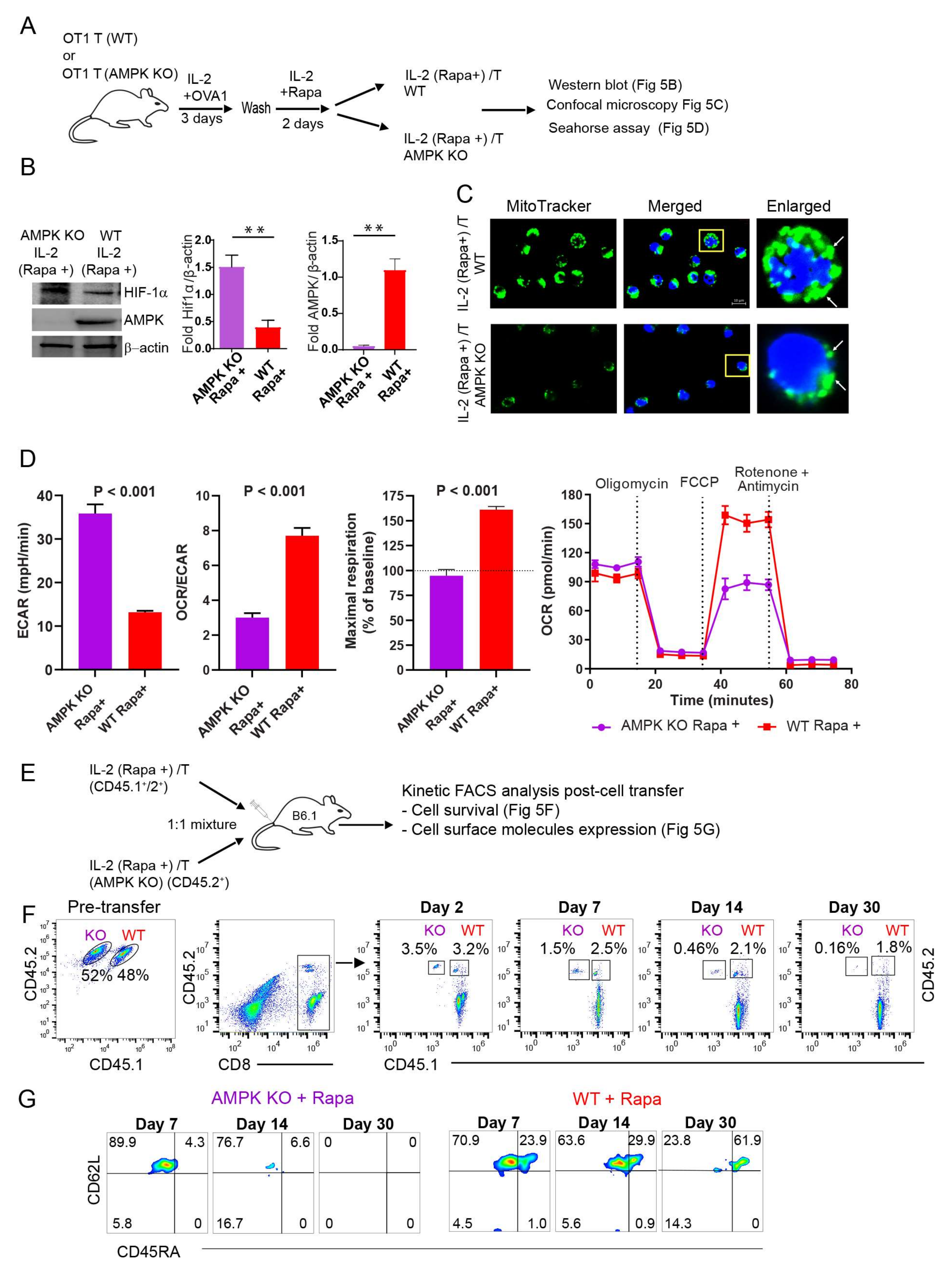
2.7. AMPKα1 Deficiency in IL-2(Rapa+)/T-cells Reduces Mitochondrial Biogenesis, but Up-Regulates HIF-1α Expression and Induces a Metabolic Switch from FAO to Glycolysis
2.8. AMPKα1 Deficiency Down-Regulates CD45RA Expression in IL-2(Rapa+)/T-cells and Abolishes Their Long-Term Survival
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Rapamycin Treatment in Mice Challenged with rLmOVA
4.3. Splenocytes and Peripheral Blood Mononuclear Cell Preparation
4.4. Preparation of In Vitro Activated T-cells
4.5. Adoptive T-cell Transfer and Kinetic Flow Cytometry Analyses
4.6. Flow Cytometry
4.7. Confocal and Electron Microscopy Imaging
4.8. Immunoblotting
4.9. Seahorse Assays
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPKα1 | adenosine monophosphate-activated protein kinase-α1 |
| ATG7 | autophagy-related gene-7 |
| ECAR | extracellular acidification rate |
| FAO | fatty acid oxidation |
| FOXO1 | forkhead box protein-O1 |
| KLRG1 | killer cell lectin-like receptor subfamily G member-1 |
| KO | knockout |
| MPEC | memory precursor effector cell |
| mTORC1 | mammalian target of rapamycin complex-1 |
| OCR | O2 consumption rate |
| OXPHOS | oxidative phosphorylation |
| Rapa | rapamycin |
| S6K1 | S6 kinase |
| SLEC | short-lived effector cell |
| SRC | spare respiratory capacity |
| TCF1 | T-cell factor-1 |
| TE cell | effector T-cell |
| TM cell | memory T-cell |
| IL-2/T-cell | IL-2-stimulated T-cell |
| IL-2(Rapa+)/T-cell | IL-2+Rapa-stimulated T-cell |
| ULK1 | Unc-51-like autophagy-activating kinase-1 |
| WT | wild-type |
References
- Buchholz, V.R.; Schumacher, T.N.; Busch, D.H. T cell fate at the single-cell level. Annu. Rev. Immunol. 2016, 34, 65–92. [Google Scholar] [CrossRef]
- Keating, R.; McGargill, M.A. mTOR regulation of lymphoid cells in immunity to pathogens. Front. Immunol. 2016, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Darrah, P.A.; Roederer, M. T-cell quality in memory and protection: Implications for vaccine design. Nat. Rev. Immunol. 2008, 8, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Doedens, A.L.; Palazon, A.; Tyrakis, P.A.; Cheung, K.P.; Johnson, R.S.; Goldrath, A.W. Constitutive glycolytic metabolism supports CD8+ T cell effector memory differentiation during viral infection. Immunity 2016, 45, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Zeng, H.; Horng, T. Metabolism as a guiding force for immunity. Nat. Cell Biol. 2019, 21, 85–93. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020, 20, 55–70. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Wolint, P.; Schwarz, K.; Oxenius, A. Recall proliferation potential of memory CD8+ T cells and antiviral protection. J. Immunol. 2005, 175, 4677–4685. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Wolint, P.; Schwarz, K.; Jager, P.; Oxenius, A. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor alpha and CD62L. J. Immunol. 2005, 175, 4686–4696. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.D.; Badovinac, V.P. Defining memory CD8 T cell. Front. Immunol. 2018, 9, 2692. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, S.; Zhao, D.M.; Harty, J.T.; Badovinac, V.P.; Xue, H.H. Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef]
- Rao, R.R.; Li, Q.; Gubbels Bupp, M.R.; Shrikant, P.A. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8+ T cell differentiation. Immunity 2012, 36, 374–387. [Google Scholar] [CrossRef]
- Hess Michelini, R.; Doedens, A.L.; Goldrath, A.W.; Hedrick, S.M. Differentiation of CD8 memory T cells depends on Foxo1. J. Exp. Med. 2013, 210, 1189–1200. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Takemoto, N.; Wherry, E.J.; Longworth, S.A.; Northrup, J.T.; Palanivel, V.R.; Mullen, A.C.; Gasink, C.R.; Kaech, S.M.; Miller, J.D.; et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 2005, 6, 1236–1244. [Google Scholar] [CrossRef]
- Zhang, L.; Tschumi, B.O.; Lopez-Mejia, I.C.; Oberle, S.G.; Meyer, M.; Samson, G.; Ruegg, M.A.; Hall, M.N.; Fajas, L.; Zehn, D.; et al. Mammalian target of rapamycin complex 2 controls CD8 T cell memory differentiation in a Foxo1-dependent manner. Cell Rep. 2016, 14, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Chapman, N.M.; Chi, H. mTOR links environmental signals to T cell fate decisions. Front. Immunol. 2014, 5, 686. [Google Scholar] [CrossRef]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, A.; Castro, I.; Levay, A.; Malek, T.R.; Gilboa, E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J. Clin. Investig. 2014, 124, 188–197. [Google Scholar] [CrossRef]
- Li, Q.; Rao, R.; Vazzana, J.; Goedegebuure, P.; Odunsi, K.; Gillanders, W.; Shrikant, P.A. Regulating mammalian target of rapamycin to tune vaccination-induced CD8+ T cell responses for tumor immunity. J. Immunol. 2012, 188, 3080–3087. [Google Scholar] [CrossRef]
- Gammon, J.M.; Gosselin, E.A.; Tostanoski, L.H.; Chiu, Y.C.; Zeng, X.; Zeng, Q.; Jewell, C.M. Low-dose controlled release of mTOR inhibitors maintains T cell plasticity and promotes central memory T cells. J. Control. Release 2017, 263, 151–161. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Xu, X.; Araki, K.; Li, S.; Han, J.H.; Ye, L.; Tan, W.G.; Konieczny, B.T.; Bruinsma, M.W.; Martinez, J.; Pearce, E.L.; et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat. Immunol. 2014, 15, 1152–1161. [Google Scholar] [CrossRef]
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPKalpha1: A glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vazquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Luo, G.; Li, Y. The roles of stem cell memory T cells in hematological malignancies. J. Hematol. Oncol. 2015, 8, 113. [Google Scholar] [CrossRef]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Wherry, E.J.; Goldrath, A.W. Molecular regulation of effector and memory T cell differentiation. Nat. Immunol. 2014, 15, 1104–1115. [Google Scholar] [CrossRef]
- Carrio, R.; Bathe, O.F.; Malek, T.R. Initial antigen encounter programs CD8+ T cells competent to develop into memory cells that are activated in an antigen-free, IL-7- and IL-15-rich environment. J. Immunol. 2004, 172, 7315–7323. [Google Scholar] [CrossRef] [PubMed]
- Van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef]
- Chen, Y.; Zander, R.; Khatun, A.; Schauder, D.M.; Cui, W. Transcriptional and epigenetic regulation of effector and memory CD8 T cell differentiation. Front. Immunol. 2018, 9, 2826. [Google Scholar] [CrossRef]
- Gullicksrud, J.A.; Li, F.; Xing, S.; Zeng, Z.; Peng, W.; Badovinac, V.P.; Harty, J.T.; Xue, H.H. Differential requirements for Tcf1 long isoforms in CD8+ and CD4+ T cell responses to acute viral infection. J. Immunol. 2017, 199, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Tanaka, K.; Fukamizu, A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. USA 2003, 100, 11285–11290. [Google Scholar] [CrossRef]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 68 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Read, K.A.; Gilbertson, S.E.; Hough, K.P.; McDonald, P.W.; Krishnamoorthy, V.; Weinmann, A.S. Bcl-6 directly represses the gene program of the glycolysis pathway. Nat. Immunol. 2014, 15, 957–964. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Bhanumathy, K.K.; Wu, J.; Ye, Z.; Freywald, A.; Leary, S.C.; Li, R.; Xiang, J. IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci. 2016, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Klein Geltink, R.I.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial priming by CD28. Cell 2017, 171, 385–397.e11. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Prlic, M.; Bevan, M.J. Immunology: A metabolic switch to memory. Nature 2009, 460, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Mehta, G.U.; Patel, S.J.; Roychoudhuri, R.; Crompton, J.G.; Klebanoff, C.A.; Ji, Y.; Li, P.; Yu, Z.; et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016, 23, 63–76. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Friias, M.A.; Chatterjee, A.; Yellen, P.; Foster, D.A. The enigma of rapamycin dosage. Mol. Cancer Ther. 2016, 15, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Hermiston, M.L.; Xu, Z.; Weiss, A. CD45: A critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 2003, 21, 107–137. [Google Scholar] [CrossRef]
- Booth, N.J.; McQuaid, A.J.; Sobande, T.; Kissane, S.; Agius, E.; Jackson, S.E.; Salmon, M.; Falciani, F.; Yong, K.; Rustin, M.H.; et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J. Immunol. 2010, 184, 4317–4326. [Google Scholar] [CrossRef]
- Kim, M.V.; Ouyang, W.; Liao, W.; Zhang, M.Q.; Li, M.O. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity 2013, 39, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Verma, K.; Croft, W.; White, A.; Zuo, J.; Nagy, Z.; Kissane, S.; Anderson, G.; Moss, P.; Chen, F.E. Homeostatic Cytokines Drive Epigenetic Reprogramming of Activated T Cells into a "Naive-Memory" Phenotype. iScience 2020, 23, 100989. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.; Cao, X. Metabolic control of T-cell immunity via epigenetic mechanisms. Cell Mol. Immunol. 2018, 15, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.X.Y.; Kaczmarek, A.; Hoque, A.; Davie, E.; Ngoei, K.R.W.; Morrison, K.R.; Smiles, W.J.; Forte, G.M.; Wang, T.; Lie, S.; et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2020, 2, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.V.; Melo, A.C.L.; Low, J.S.; de Castro, I.A.; Braga, T.T.; Almeida, D.C.; de Lima, A.G.U.; Hiyane, M.I.; Correa-Costa, M.; Origassa, C.; et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018, 9, 25808–25825. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ashford, M.L. AMPK: Regulating energy balance at the cellular and whole body levels. Physiology 2014, 29, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Allison, J.P.; Krummel, M.F. The yin and yang of T cell costimulation. Science 1995, 270, 932–933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Yin and yang interplay of IFN-γ in inflammation and autoimmune disease. J. Clin. Investig. 2007, 117, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Immunology in China: The past, present and future. Nat. Immunol. 2008, 9, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The yin and yang of cellular nutrient sensing and growth control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Foreman, D.; O’Brien, S.A.; Jin, Y.; Zhang, W. Phospholipase D in TCR-Mediated Signaling and T Cell Activation. J. Immunol. 2018, 200, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.M.; Suarez-Alvarez, B.; Lavin, J.L.; Mosen-Ansorena, D.; Baragano Raneros, A.; Marquez-Kisinousky, L.; Aransay, A.M.; Lopez-Larrea, C. Epigenetic Networks Regulate the Transcriptional Program in Memory and Terminally Differentiated CD8+ T Cells. J. Immunol. 2017, 198, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.A.; Xiang, J. mTORC1 regulates mannose-6-phosphate receptor transport and T-cell vulnerability to regulatory T cells by controlling kinesin KIF13A. Cell Discov. 2017, 3, 17011. [Google Scholar] [CrossRef][Green Version]
- Xu, A.; Zhang, L.; Yuan, J.; Babikr, F.; Freywald, A.; Chibbar, R.; Moser, M.; Zhang, W.; Zhang, B.; Fu, Z.; et al. TLR9 agonist enhances radiofrequency ablation-induced CTL responses, leading to the potent inhibition of primary tumor growth and lung metastasis. Cell. Mol. Immunol. 2019, 16, 820–832. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ara, A.; Xu, A.; Ahmed, K.A.; Leary, S.C.; Islam, M.F.; Wu, Z.; Chibbar, R.; Xiang, J. The Energy Sensor AMPKα1 Is Critical in Rapamycin-Inhibition of mTORC1-S6K-Induced T-cell Memory. Int. J. Mol. Sci. 2022, 23, 37. https://doi.org/10.3390/ijms23010037
Ara A, Xu A, Ahmed KA, Leary SC, Islam MF, Wu Z, Chibbar R, Xiang J. The Energy Sensor AMPKα1 Is Critical in Rapamycin-Inhibition of mTORC1-S6K-Induced T-cell Memory. International Journal of Molecular Sciences. 2022; 23(1):37. https://doi.org/10.3390/ijms23010037
Chicago/Turabian StyleAra, Anjuman, Aizhang Xu, Khawaja Ashfaque Ahmed, Scot C. Leary, Md. Fahmid Islam, Zhaojia Wu, Rajni Chibbar, and Jim Xiang. 2022. "The Energy Sensor AMPKα1 Is Critical in Rapamycin-Inhibition of mTORC1-S6K-Induced T-cell Memory" International Journal of Molecular Sciences 23, no. 1: 37. https://doi.org/10.3390/ijms23010037
APA StyleAra, A., Xu, A., Ahmed, K. A., Leary, S. C., Islam, M. F., Wu, Z., Chibbar, R., & Xiang, J. (2022). The Energy Sensor AMPKα1 Is Critical in Rapamycin-Inhibition of mTORC1-S6K-Induced T-cell Memory. International Journal of Molecular Sciences, 23(1), 37. https://doi.org/10.3390/ijms23010037