
Genetic Background of Fetal Growth Restriction

 ,
,  , , and
, , and
Abstract
:1. Introduction
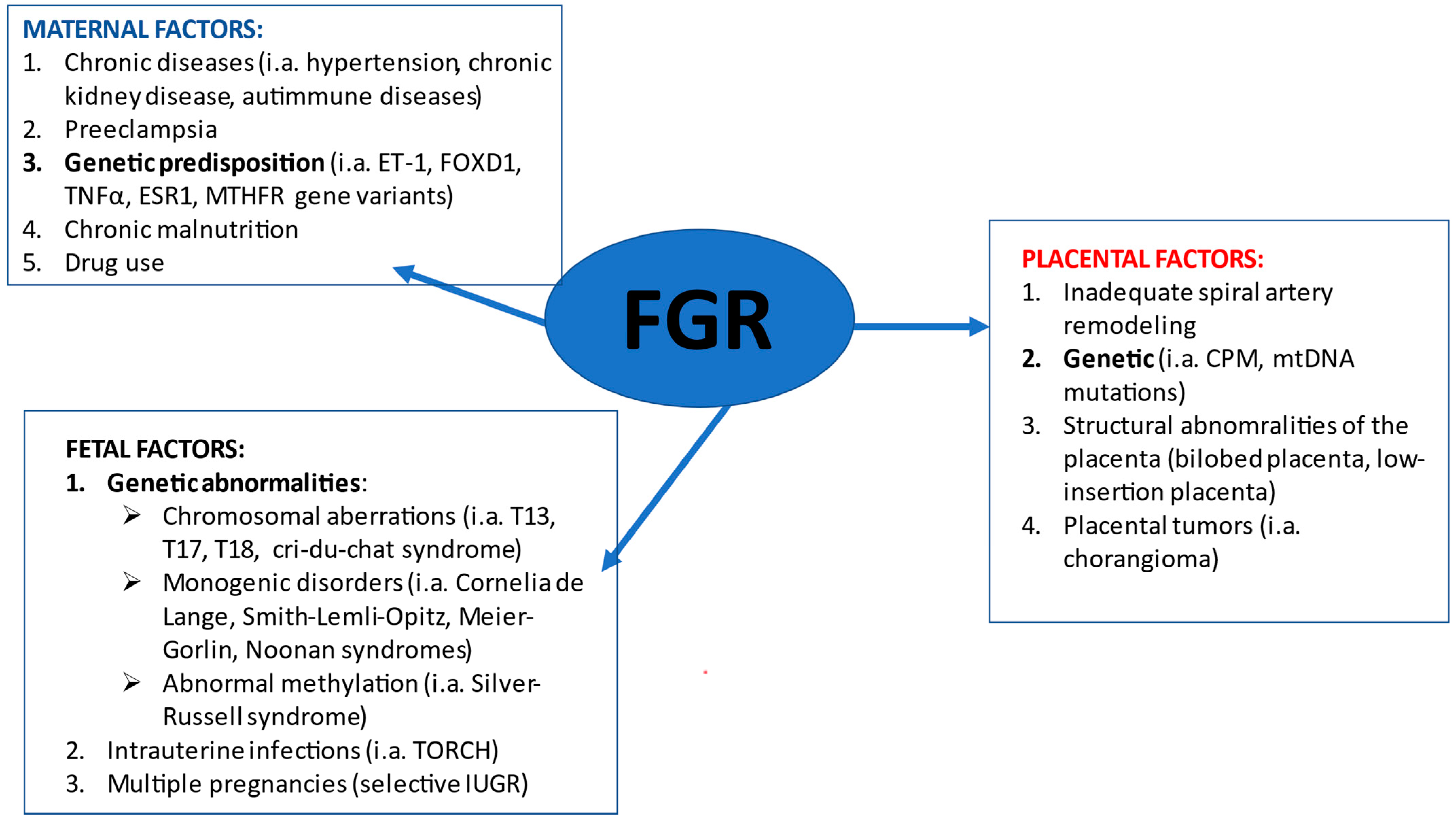
2. Fetal FGR Causes
3. Placental FGR Causes
4. Maternal FGR Causes
5. Fetal Growth Restriction and Preeclampsia as a Maternal Factor
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hammami, A.; Mazer Zumaeta, A.; Syngelaki, A.; Akolekar, R.; Nicolaides, K.H. Ultrasonographic Estimation of Fetal Weight: Development of New Model and Assessment of Performance of Previous Models. Ultrasound Obstet. Gynecol. 2018, 52, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Lees, C.C.; Stampalija, T.; Baschat, A.; da Silva Costa, F.; Ferrazzi, E.; Figueras, F.; Hecher, K.; Poon, L.C.; Salomon, L.J.; Unterscheider, J. ISUOG Practice Guidelines: Diagnosis and Management of Small-for-Gestational-Age Fetus and Fetal Growth Restriction. Ultrasound Obstet. Gynecol. 2020, 56, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Gordijn, S.J.; Beune, I.M.; Thilaganathan, B.; Papageorghiou, A.; Baschat, A.A.; Baker, P.N.; Silver, R.M.; Wynia, K.; Ganzevoort, W. Consensus Definition of Fetal Growth Restriction: A Delphi Procedure. Ultrasound Obstet. Gynecol. 2016, 48, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Nowacka, U.; Papastefanou, I.; Bouariu, A.; Syngelaki, A.; Akolekar, R.; Nicolaides, K.H. Second Trimester Contingent Screening for Small for Gestational Age Neonates. Ultrasound Obstet. Gynecol. 2021. [Google Scholar] [CrossRef]
- Nowacka, U.; Kosińska-Kaczyńska, K.; Krajewski, P.; Saletra-Bielińska, A.; Walasik, I.; Szymusik, I. Predictive Accuracy of Singleton Versus Customized Twin Growth Chart for Adverse Perinatal Outcome: A Cohort Study. Int. J. Environ. Res. Public Health 2021, 18, 2016. [Google Scholar] [CrossRef] [PubMed]
- Nohuz, E.; Rivière, O.; Coste, K.; Vendittelli, F. Prenatal Identification of Small-for-Gestational Age and Risk of Neonatal Morbidity and Stillbirth. Ultrasound Obstet. Gynecol. 2020, 55, 621–628. [Google Scholar] [CrossRef]
- Barker, D.J.P.; Osmond, C.; Forsén, T.J.; Kajantie, E.; Eriksson, J.G. Trajectories of Growth among Children Who Have Coronary Events as Adults. N. Engl. J. Med. 2005, 353, 1802–1809. [Google Scholar] [CrossRef]
- Meler, E.; Sisterna, S.; Borrell, A. Genetic Syndromes Associated with Isolated Fetal Growth Restriction. Prenat. Diagn. 2020, 40, 432–446. [Google Scholar] [CrossRef]
- Nardozza, L.M.M.; Caetano, A.C.R.; Zamarian, A.C.P.; Mazzola, J.B.; Silva, C.P.; Marçal, V.M.G.; Lobo, T.F.; Peixoto, A.B.; Araujo Júnior, E. Fetal Growth Restriction: Current Knowledge. Arch. Gynecol. Obstet. 2017, 295, 1061–1077. [Google Scholar] [CrossRef]
- Merriel, A.; Alberry, M.; Abdel-Fattah, S. Implications of Non-Invasive Prenatal Testing for Identifying and Managing High-Risk Pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 256, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Snijders, R.J.; Sherrod, C.; Gosden, C.M.; Nicolaides, K.H. Fetal Growth Retardation: Associated Malformations and Chromosomal Abnormalities. Am. J. Obstet. Gynecol. 1993, 168, 547–555. [Google Scholar] [CrossRef]
- Anandakumar, C.; Chew, S.; Wong, Y.C.; Malarvishy, G.; Po, L.U.; Ratnam, S.S. Early Asymmetric IUGR and Aneuploidy. J. Obstet. Gynaecol. Res. 1996, 22, 365–370. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Shi, Q.; Li, L.; Jiang, Y.; Liu, R.; Zhang, H. Ultrasonographic Findings and Prenatal Diagnosis of Complete Trisomy 17p Syndrome: A Case Report and Review of the Literature. J. Clin. Lab. Anal. 2021, 35, e23582. [Google Scholar] [CrossRef]
- Peng, Y.; Pang, J.; Hu, J.; Jia, Z.; Xi, H.; Ma, N.; Yang, S.; Liu, J.; Huang, X.; Tang, C.; et al. Clinical and Molecular Characterization of 12 Prenatal Cases of Cri-Du-Chat Syndrome. Mol. Genet. Genom. Med. 2020, 8, e1312. [Google Scholar] [CrossRef] [PubMed]
- Honjo, R.S.; Mello, C.B.; Pimenta, L.S.E.; Nuñes-Vaca, E.C.; Benedetto, L.M.; Khoury, R.B.F.; Befi-Lopes, D.M.; Kim, C.A. Cri Du Chat Syndrome: Characteristics of 73 Brazilian Patients. J. Intellect. Disabil. Res. 2018, 62, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Espirito Santo, L.D.; Moreira, L.M.A.; Riegel, M. Cri-Du-Chat Syndrome: Clinical Profile and Chromosomal Microarray Analysis in Six Patients. BioMed Res. Int. 2016, 2016, 5467083. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Deng, L.; Yang, Y.; Sun, L. Intrauterine Phenotype Features of Fetuses with Williams-Beuren Syndrome and Literature Review. Ann. Hum. Genet. 2020, 84, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Tzadikevitch Geffen, K.; Singer, A.; Maya, I.; Ben-Shachar, S.; Sagi-Dain, L.; Daum, H.; Michaelson-Cohen, R.; Greenbaum, L.; Feingold-Zadok, M.; Sukenik Halevy, R. The Yield of Chromosomal Microarray in Pregnancies Complicated with Fetal Growth Restriction Can Be Predicted According to Clinical Parameters. Fetal Diagn. Ther. 2021, 48, 140–148. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Lin, Y.; Xu, L.P.; Huang, H.L.; Liu, S.P.; Yu, Y.H.; Yang, F. Application of Chromosomal Microarray to Investigate Genetic Causes of Isolated Fetal Growth Restriction. Mol. Cytogenet. 2018, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- de Wit, M.C.; Srebniak, M.I.; Joosten, M.; Govaerts, L.C.P.; Kornelisse, R.F.; Papatsonis, D.N.M.; de Graaff, K.; Knapen, M.F.C.M.; Bruggenwirth, H.T.; de Vries, F.a.T.; et al. Prenatal and Postnatal Findings in Small-for-Gestational-Age Fetuses without Structural Ultrasound Anomalies at 18-24 Weeks. Ultrasound Obstet. Gynecol. 2017, 49, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Sagi-Dain, L.; Peleg, A.; Sagi, S. Risk for Chromosomal Aberrations in Apparently Isolated Intrauterine Growth Restriction: A Systematic Review. Prenat. Diagn. 2017, 37, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-P.; Chen, M.; Wang, L.-K.; Chern, S.-R.; Wu, P.-S.; Ma, G.-C.; Chang, S.-P.; Chen, S.-W.; Wu, F.-T.; Lee, C.-C.; et al. Low-Level Mosaicism for Trisomy 16 at Amniocentesis in a Pregnancy Associated with Intrauterine Growth Restriction and a Favorable Outcome. Taiwan J. Obstet. Gynecol. 2021, 60, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Naert, M.; Lam-Rachlin, J.; Monteagudo, A.; Rebarber, A.; Saltzman, D.; Fox, N.S. Outcomes in Patients with Early-Onset Fetal Growth Restriction without Fetal or Genetic Anomalies. J. Matern. Fetal Neonatal Med. 2019, 32, 2662–2666. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.G. Review and Hypothesis: Syndromes with Severe Intrauterine Growth Restriction and Very Short Stature—Are They Related to the Epigenetic Mechanism(s) of Fetal Survival Involved in the Developmental Origins of Adult Health and Disease? Am. J. Med. Genet. Part A 2010, 152, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Shettigar, S.K.G.; Kumar, S.; Kantharia, S.; Kurva, J.; Cherian, S. Novel Mutation in Cul7 Gene in a Family Diagnosed with 3M Syndrome. J. Genet. 2019, 98, 21. [Google Scholar] [CrossRef] [PubMed]
- de Munnik, S.A.; Hoefsloot, E.H.; Roukema, J.; Schoots, J.; Knoers, N.V.A.M.; Brunner, H.G.; Jackson, A.P.; Bongers, E.M.H.F. Meier-Gorlin Syndrome. Orphanet J. Rare Dis. 2015, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, M.I.; Jespersgaard, C.; Brøndum-Nielsen, K.; Bisgaard, A.-M.; Tümer, Z. Cornelia de Lange Syndrome. Clin. Genet. 2015, 88, 1–12. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Legeai-Mallet, L. Achondroplasia: Development, Pathogenesis, and Therapy. Dev. Dyn. 2017, 246, 291–309. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan Syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Simeoni, U.; Armengaud, J.-B.; Siddeek, B.; Tolsa, J.-F. Perinatal Origins of Adult Disease. Neonatology 2018, 113, 393–399. [Google Scholar] [CrossRef]
- Barbieri, M.R.; Fontes, A.M.; Barbieri, M.A.; Saraiva, M.C.P.; Simões, V.M.F.; da Silva, A.A.M.; Abraham, K.J.; Bettiol, H. Effects of FTO and PPARγ Variants on Intrauterine Growth Restriction in a Brazilian Birth Cohort. Braz. J. Med. Biol. Res. 2021, 54, e10465. [Google Scholar] [CrossRef]
- Karimi-Zarchi, M.; Zanbagh, L.; Javaheri, A.; Tabatabaei, R.S.; Abbasi, H.; Meibodi, B.; Hadadan, A.; Bahrami, R.; Mirjalili, S.R.; Neamatzadeh, H. Association of Insulin-like Growth Factor-II Apa1 and MspI Polymorphisms with Intrauterine Growth Restriction Risk. Fetal Pediatr. Pathol. 2020, 40, 605–611. [Google Scholar] [CrossRef]
- Krishna, R.G.; Vishnu Bhat, B.; Bobby, Z.; Papa, D.; Badhe, B.; Kalidoss, V.K.; Karli, S. Identification of Differentially Methylated Candidate Genes and Their Biological Significance in IUGR Neonates by Methylation EPIC Array. J. Matern.-Fetal Neonatal Med. 2020, 24, 1–9. [Google Scholar] [CrossRef]
- Cammarata-Scalisi, F.; Callea, M.; Stock, F.; Zambito, V.; Sparago, Á.; Riccio, A. Silver-Russell Syndrome. Clinical and Etiopathological Aspects of a Model Genomic Imprinting Entity. Arch. Argent. Pediatr. 2020, 118, e258–e264. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, B.S.; Stafrace, Y.; Calleja-Agius, J. Silver-Russell Syndrome: A Review. Neonatal Netw. 2017, 36, 206–212. [Google Scholar] [CrossRef]
- Gardner, R.; McKinlay, J.; Amor, D.J. Chromosome Abnormalities and Genetic Counseling, 5th ed.; Gardner and Sutherland’s: London, UK, 2018. [Google Scholar]
- Kalousek, D.K.; Dill, F.J.; Pantzar, T.; McGillivray, B.C.; Yong, S.L.; Wilson, R.D. Confined Chorionic Mosaicism in Prenatal Diagnosis. Hum. Genet. 1987, 77, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Toutain, J.; Goutte-Gattat, D.; Horovitz, J.; Saura, R. Confined Placental Mosaicism Revisited: Impact on Pregnancy Characteristics and Outcome. PLoS ONE 2018, 13, e0195905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Yoshiura, K.-I.; Miura, S.; Kondoh, T.; Harada, N.; Yamasaki, K.; Fujimoto, Y.; Yamasaki, Y.; Tanigawa, T.; Kitajima, Y.; et al. Clinical Outcome of Infants with Confined Placental Mosaicism and Intrauterine Growth Restriction of Unknown Cause. Am. J. Med. Genet. Part A 2006, 140, 1827–1833. [Google Scholar] [CrossRef] [PubMed]
- Grati, F.R.; Malvestiti, F.; Branca, L.; Agrati, C.; Maggi, F.; Simoni, G. Chromosomal Mosaicism in the Fetoplacental Unit. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 42, 39–52. [Google Scholar] [CrossRef]
- Johnson, A.; Wapner, R.J.; Davis, G.H.; Jackson, L.G. Mosaicism in Chorionic Villus Sampling: An Association with Poor Perinatal Outcome. Obstet. Gynecol. 1990, 75, 573–577. [Google Scholar]
- Lazier, J.; Martin, N.; Stavropoulos, J.D.; Chitayat, D. Maternal Uniparental Disomy for Chromosome 6 in a Patient with IUGR, Ambiguous Genitalia, and Persistent Mullerian Structures. Am. J. Med. Genet. Part A 2016, 170, 3227–3230. [Google Scholar] [CrossRef] [PubMed]
- Grau Madsen, S.; Uldbjerg, N.; Sunde, L.; Becher, N. Danish Fetal Medicine Study Group; Danish Clinical Genetics Study Group Prognosis for Pregnancies with Trisomy 16 Confined to the Placenta: A Danish Cohort Study. Prenat. Diagn. 2018, 38, 1103–1110. [Google Scholar] [CrossRef]
- Neiswanger, K.; Hohler, P.M.; Hively-Thomas, L.B.; McPherson, E.W.; Hogge, W.A.; Surti, U. Variable Outcomes in Mosaic Trisomy 16: Five Case Reports and Literature Analysis. Prenat. Diagn. 2006, 26, 454–461. [Google Scholar] [CrossRef]
- Post, J.G.; Nijhuis, J.G. Trisomy 16 Confined to the Placenta. Prenat. Diagn. 1992, 12, 1001–1007. [Google Scholar] [CrossRef]
- Del Gobbo, G.F.; Yuan, V.; Robinson, W.P. Confined Placental Mosaicism Involving Multiple de Novo Copy Number Variants Associated with Fetal Growth Restriction: A Case Report. Am. J. Med. Genet. Part A 2021, 185, 1908–1912. [Google Scholar] [CrossRef] [PubMed]
- Van Opstal, D.; van Maarle, M.C.; Lichtenbelt, K.; Weiss, M.M.; Schuring-Blom, H.; Bhola, S.L.; Hoffer, M.J.V.; Huijsdens-van Amsterdam, K.; Macville, M.V.; Kooper, A.J.A.; et al. Origin and Clinical Relevance of Chromosomal Aberrations Other than the Common Trisomies Detected by Genome-Wide NIPS: Results of the TRIDENT Study. Genet. Med. 2018, 20, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Grati, F.R.; Ferreira, J.; Benn, P.; Izzi, C.; Verdi, F.; Vercellotti, E.; Dalpiaz, C.; D’Ajello, P.; Filippi, E.; Volpe, N.; et al. Outcomes in Pregnancies with a Confined Placental Mosaicism and Implications for Prenatal Screening Using Cell-Free DNA. Genet. Med. 2020, 22, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Scotchman, E.; Chandler, N.; Chitty, L.S. PREIMPLANTATION GENETIC TESTING: Non-Invasive Prenatal Testing for Aneuploidy, Copy-Number Variants and Single-Gene Disorders. Reproduction 2020, 160, A1–A11. [Google Scholar] [CrossRef] [PubMed]
- Cremer, M.; Treiss, I.; Cremer, T.; Hager, D.; Franke, W.W. Characterization of Cells of Amniotic Fluids by Immunological Identification of Intermediate-Sized Filaments: Presence of Cells of Different Tissue Origin. Hum. Genet. 1981, 59, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grati, F.R.; Bajaj, K.; Zanatta, V.; Malvestiti, F.; Malvestiti, B.; Marcato, L.; Grimi, B.; Maggi, F.; Simoni, G.; Gross, S.J.; et al. Implications of Fetoplacental Mosaicism on Cell-Free DNA Testing for Sex Chromosome Aneuploidies. Prenat. Diagn. 2017, 37, 1017–1027. [Google Scholar] [CrossRef]
- Hayata, K.; Hiramatsu, Y.; Masuyama, H.; Eto, E.; Mitsui, T.; Tamada, S. Discrepancy between Non-Invasive Prenatal Genetic Testing (NIPT) and Amniotic Chromosomal Test Due to Placental Mosaicism: A Case Report and Literature Review. Acta Med. Okayama 2017, 71, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Wapner, R.J.; Simpson, J.L.; Golbus, M.S.; Zachary, J.M.; Ledbetter, D.H.; Desnick, R.J.; Fowler, S.E.; Jackson, L.G.; Lubs, H.; Mahony, R.J. Chorionic Mosaicism: Association with Fetal Loss but Not with Adverse Perinatal Outcome. Prenat. Diagn. 1992, 12, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, J. Confined Placental Mosaicism for Trisomies 2, 3, 7, 8, 9, 16, and 22: Their Incidence, Likely Origins, and Mechanisms for Cell Lineage Compartmentalization. Prenat. Diagn. 1996, 16, 511–524. [Google Scholar] [CrossRef]
- Wilkins-Haug, L.; Quade, B.; Morton, C.C. Confined Placental Mosaicism as a Risk Factor among Newborns with Fetal Growth Restriction. Prenat. Diagn. 2006, 26, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Sharma, P.; Shastri, S. Genetic, Metabolic and Endocrine Aspect of Intrauterine Growth Restriction: An Update. J. Matern. Fetal Neonatal Med. 2017, 30, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.; Lipka, A.; Paukszto, L.; Jastrzebski, J.P.; Szeszko, K.; Gowkielewicz, M.; Lepiarczyk, E.; Jozwik, M.; Majewski, M.K. Placenta Transcriptome Profiling in Intrauterine Growth Restriction (IUGR). Int. J. Mol. Sci. 2019, 20, 1510. [Google Scholar] [CrossRef] [Green Version]
- Murthi, P. Review: Placental Homeobox Genes and Their Role in Regulating Human Fetal Growth. Placenta 2014, 35, S46–S50. [Google Scholar] [CrossRef]
- Chiu, Y.-H.; Yang, M.-R.; Wang, L.-J.; Chen, M.-H.; Chang, G.-D.; Chen, H. New Insights into the Regulation of Placental Growth Factor Gene Expression by the Transcription Factors GCM1 and DLX3 in Human Placenta. J. Biol. Chem. 2018, 293, 9801–9811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelbi, S.T.; Wilson, M.L.; Veillard, A.-C.; Ingles, S.A.; Zhang, J.; Mondon, F.; Gascoin-Lachambre, G.; Doridot, L.; Mignot, T.-M.; Rebourcet, R.; et al. Genetic and Epigenetic Mechanisms Collaborate to Control SERPINA3 Expression and Its Association with Placental Diseases. Hum. Mol. Genet. 2012, 21, 1968–1978. [Google Scholar] [CrossRef] [Green Version]
- Gascoin-Lachambre, G.; Buffat, C.; Rebourcet, R.; Chelbi, S.T.; Rigourd, V.; Mondon, F.; Mignot, T.-M.; Legras, E.; Simeoni, U.; Vaiman, D.; et al. Cullins in Human Intra-Uterine Growth Restriction: Expressional and Epigenetic Alterations. Placenta 2010, 31, 151–157. [Google Scholar] [CrossRef]
- Mamsen, L.S.; Zafeiri, A.; Bøtkjær, J.A.; Hardlei, J.R.; Ernst, E.; Oxvig, C.; Fowler, P.A.; Andersen, C.Y. Expression of the Insulin-like Growth Factor System in First- and Second-Trimester Human Embryonic and Fetal Gonads. J. Clin. Endocrinol. Metab. 2020, 105, dgaa470. [Google Scholar] [CrossRef]
- Hellström, A.; Ley, D.; Hansen-Pupp, I.; Hallberg, B.; Löfqvist, C.; van Marter, L.; van Weissenbruch, M.; Ramenghi, L.A.; Beardsall, K.; Dunger, D.; et al. Insulin-like Growth Factor 1 Has Multisystem Effects on Foetal and Preterm Infant Development. Acta Paediatr. 2016, 105, 576–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Börzsönyi, B.; Demendi, C.; Nagy, Z.; Tóth, K.; Csanád, M.; Pajor, A.; Rig, J.; Joó, J.G. Gene Expression Patterns of Insulin-like Growth Factor 1, Insulin-like Growth Factor 2 and Insulin-like Growth Factor Binding Protein 3 in Human Placenta from Pregnancies with Intrauterine Growth Restriction. J. Perinat. Med. 2011, 39, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Jeon, Y.J.; Lee, S.M.; Park, M.H.; Jung, S.-C.; Kim, Y.J. Placental Gene Expression Is Related to Glucose Metabolism and Fetal Cord Blood Levels of Insulin and Insulin-like Growth Factors in Intrauterine Growth Restriction. Early Hum Dev. 2010, 86, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Giabicani, E.; Willems, M.; Steunou, V.; Chantot-Bastaraud, S.; Thibaud, N.; Abi Habib, W.; Azzi, S.; Lam, B.; Bérard, L.; Bony-Trifunovic, H.; et al. Increasing Knowledge in IGF1R Defects: Lessons from 35 New Patients. J. Med. Genet. 2020, 57, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Naha, R.; Anees, A.; Chakrabarty, S.; Naik, P.S.; Pandove, M.; Pandey, D.; Satyamoorthy, K. Placental Mitochondrial DNA Mutations and Copy Numbers in Intrauterine Growth Restricted (IUGR) Pregnancy. Mitochondrion 2020, 55, 85–94. [Google Scholar] [CrossRef]
- Margarit, L.; Griffiths, A.N.; Tsapanos, V.; Tsakas, S.; Gumenos, D.; Decavalas, G. Second Trimester Amniotic Fluid Endothelin-1 Concentrations and Subsequent Development of Intrauterine Growth Restriction. Eur. J. Obstet. Gynecol. Reprod. Biol. 2007, 134, 192–195. [Google Scholar] [CrossRef]
- Thaete, L.G.; Dewey, E.R.; Neerhof, M.G. Endothelin and the Regulation of Uterine and Placental Perfusion in Hypoxia-Induced Fetal Growth Restriction. J. Soc. Gynecol. Investig. 2004, 11, 16–21. [Google Scholar] [CrossRef]
- Aydin, H.I.; Eser, A.; Kaygusuz, I.; Yildirim, S.; Celik, T.; Gunduz, S.; Kalman, S. Adipokine, Adropin and Endothelin-1 Levels in Intrauterine Growth Restricted Neonates and Their Mothers. J. Perinat. Med. 2016, 44, 669–676. [Google Scholar] [CrossRef]
- Arslan, M.; Yazici, G.; Erdem, A.; Erdem, M.; Arslan, E.O.; Himmetoglu, O. Endothelin 1 and Leptin in the Pathophysiology of Intrauterine Growth Restriction. Int. J. Gynaecol. 2004, 84, 120–126. [Google Scholar] [CrossRef]
- Quintero-Ronderos, P.; Jiménez, K.M.; Esteban-Pérez, C.; Ojeda, D.A.; Bello, S.; Fonseca, D.J.; Coronel, M.A.; Moreno-Ortiz, H.; Sierra-Díaz, D.C.; Lucena, E.; et al. FOXD1 Mutations Are Related to Repeated Implantation Failure, Intra-Uterine Growth Restriction and Preeclampsia. Mol. Med. 2019, 25, 37. [Google Scholar] [CrossRef] [PubMed]
- Kaluba-Skotarczak, A.; Magiełda, J.; Romała, A.; Kurzawińska, G.; Barlik, M.; Drews, K.; Ożarowski, M.; Łoziński, T.; Seremak-Mrozikiewicz, A. Importance of Polymorphic Variants of Tumour Necrosis Factor-α Gene in the Etiology of Intrauterine Growth Restriction. Ginekol. Pol. 2018, 89, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golovchenko, O.; Abramova, M.; Ponomarenko, I.; Reshetnikov, E.; Aristova, I.; Polonikov, A.; Dvornyk, V.; Churnosov, M. Functionally Significant Polymorphisms of ESR1and PGR and Risk of Intrauterine Growth Restriction in Population of Central Russia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 253, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Banjac, L.; Kotur-Stevuljević, J.; Gojković, T.; Bokan-Mirković, V.; Banjac, G.; Banjac, G. Relationship between Insulin-Like Growth Factor Type 1 and Intrauterine Growth. Acta Clin. Croat. 2020, 59, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, L.; Zhu, L.-H.; Zhang, S.-T.; Wu, Y.-L. Association of Methylenetetrahydrofolate Reductase (MTHFR) C677T Polymorphism with Preterm Delivery and Placental Abruption: A Systematic Review and Meta-Analysis. Acta Obstet. Gynecol. Scand. 2016, 95, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, X.; Xiong, X.; Chuan, J.; Zhong, L.; Chen, G.; Yu, D. The Association between Maternal Methylenetetrahydrofolate Reductase C677T and A1298C Polymorphism and Birth Defects and Adverse Pregnancy Outcomes. Prenat. Diagn. 2019, 39, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Hogeveen, M.; Blom, H.J.; den Heijer, M. Maternal Homocysteine and Small-for-Gestational-Age Offspring: Systematic Review and Meta-Analysis. Am. J. Clin. Nutr. 2012, 95, 130–136. [Google Scholar] [CrossRef]
- Bahrami, R.; Schwartz, D.A.; Asadian, F.; Karimi-Zarchi, M.; Dastgheib, S.A.; Tabatabaie, R.S.; Meibodi, B.; Neamatzadeh, H. Association of MTHFR 677C>T Polymorphism with IUGR and Placental Abruption Risk: A Systematic Review and Meta-Analysis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 256, 130–139. [Google Scholar] [CrossRef]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. The Hypertensive Disorders of Pregnancy: ISSHP Classification, Diagnosis & Management Recommendations for International Practice. Pregnancy Hypertens. 2018, 13, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.-H.; Hsieh, T.-T.; Chen, S.-F. Risk of Abnormal Fetal Growth in Women with Early- and Late-Onset Preeclampsia. Pregnancy Hypertens. 2018, 12, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, K.; Fijałkowska, A.; Issat, T.; Maciejewski, T.M. Insight into the Key Points of Preeclampsia Pathophysiology: Uterine Artery Remodeling and the Role of MicroRNAs. Int. J. Mol. Sci. 2021, 22, 3132. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Dołegowska, B.; Kwiatkowska, E.; Rzepka, R.; Marczuk, N.; Loj, B.; Torbè, A. Maternal Endothelial Damage as a Disorder Shared by Early Preeclampsia, Late Preeclampsia and Intrauterine Growth Restriction. J. Perinat. Med. 2017, 45, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Benny, P.A.; Alakwaa, F.M.; Schlueter, R.J.; Lassiter, C.B.; Garmire, L.X. A Review of Omics Approaches to Study Preeclampsia. Placenta 2020, 92, 17–27. [Google Scholar] [CrossRef]
- McGinnis, R.; Steinthorsdottir, V.; Williams, N.O.; Thorleifsson, G.; Shooter, S.; Hjartardottir, S.; Bumpstead, S.; Stefansdottir, L.; Hildyard, L.; Sigurdsson, J.K.; et al. Variants in the Fetal Genome near FLT1 Are Associated with Risk of Preeclampsia. Nat. Genet. 2017, 49, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Bracken, M.B.; DeWan, A.T. Genome-Wide Association Study of Pre-Eclampsia Detects Novel Maternal Single Nucleotide Polymorphisms and Copy-Number Variants in Subsets of the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) Study Cohort. Ann. Hum. Genet. 2013, 77, 277–287. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, M.; van Bezu, J.; van Abel, D.; Dunk, C.; Blankenstein, M.A.; Oudejans, C.B.M.; Lye, S.J. The STOX1 Genotype Associated with Pre-Eclampsia Leads to a Reduction of Trophoblast Invasion by Alpha-T-Catenin Upregulation. Hum. Mol. Genet. 2010, 19, 2658–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakou, K.; Evangelou, E.; Papatheodorou, S.I. Genetic and Non-Genetic Risk Factors for Pre-Eclampsia: Umbrella Review of Systematic Reviews and Meta-Analyses of Observational Studies. Ultrasound Obstet. Gynecol. 2018, 51, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Bracken, M.B.; Dewan, A.T.; Chen, S. Association between the SERPINE1 (PAI-1) 4G/5G Insertion/Deletion Promoter Polymorphism (Rs1799889) and Pre-Eclampsia: A Systematic Review and Meta-Analysis. Mol. Hum. Reprod. 2013, 19, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Gannoun, M.B.A.; Raguema, N.; Zitouni, H.; Mehdi, M.; Seda, O.; Mahjoub, T.; Lavoie, J.L. MMP-2 and MMP-9 Polymorphisms and Preeclampsia Risk in Tunisian Arabs: A Case-Control Study. J. Clin. Med. 2021, 10, 2647. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Song, G.; Zhao, G.; Meng, T. Gene Polymorphism Associated with TGF-Β1 and Susceptibility to Preeclampsia: A Meta-Analysis and Trial Sequential Analysis. J. Obstet. Gynaecol. Res. 2021, 47, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Benian, A.; Madazli, R.; Aksu, F.; Uzun, H.; Aydin, S. Plasma and Placental Levels of Interleukin-10, Transforming Growth Factor-Beta1, and Epithelial-Cadherin in Preeclampsia. Obstet. Gynecol. 2002, 100, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Nasri, F.; Zare, M.; Hesampour, F.; Ahmadi, M.; Ali-Hassanzadeh, M.; Mostafaei, S.; Gharesi-Fard, B. Are Genetic Variations in IL-1β and IL-6 Cytokines Associated with the Risk of Pre-Eclampsia? Evidence from a Systematic Review and Meta-Analysis. J. Matern. Neonatal Med. 2021, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mazlum, F.; Gharesi-Fard, B.; Hadinedoushan, H.; Bakhshizadeh Ghashti, Y. Association between Interleukin-32 Gene Polymorphism and Susceptibility to Preeclampsia. Hypertens. Pregnancy 2021, 40, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, K.; Szczerba, E.; Maciejewski, T.; Fijałkowska, A. Non-Obstetric Complications in Preeclampsia. Menopause Rev. 2019, 18, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Khaliq, O.P.; Konoshita, T.; Moodley, J.; Naicker, T. The Association of NPHS1 and ACNT4 Gene Polymorphisms with Pre-Eclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 266, 9–14. [Google Scholar] [CrossRef]
- Ma, C.; Zheng, Y.; Liu, X.; Zhang, W. Association Study of Polymorphisms of Endoplasmic Reticulum Aminopeptidase 1 Gene with Preeclampsia in Chinese Populations. Clin. Exp. Hypertens. 2021, 43, 550–554. [Google Scholar] [CrossRef]
- Burton, G.J.; Yung, H.-W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental Endoplasmic Reticulum Stress and Oxidative Stress in the Pathophysiology of Unexplained Intrauterine Growth Restriction and Early Onset Preeclampsia. Placenta 2009, 30 (Suppl. A), S43–S48. [Google Scholar] [CrossRef] [Green Version]
- Vatish, M.; Randeva, H.S.; Grammatopoulos, D.K. Hormonal Regulation of Placental Nitric Oxide and Pathogenesis of Pre-Eclampsia. Trends Mol. Med. 2006, 12, 223–233. [Google Scholar] [CrossRef]
- Kulandavelu, S.; Whiteley, K.J.; Qu, D.; Mu, J.; Bainbridge, S.A.; Adamson, S.L. Endothelial Nitric Oxide Synthase Deficiency Reduces Uterine Blood Flow, Spiral Artery Elongation, and Placental Oxygenation in Pregnant Mice. Hypertension 2012, 60, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Oe, Y.; Ko, M.; Fushima, T.; Sato, E.; Karumanchi, S.A.; Sato, H.; Sugawara, J.; Ito, S.; Takahashi, N. Hepatic Dysfunction and Thrombocytopenia Induced by Excess SFlt1 in Mice Lacking Endothelial Nitric Oxide Synthase. Sci. Rep. 2018, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Sljivancanin Jakovljevic, T.; Kontic-Vucinic, O.; Nikolic, N.; Carkic, J.; Stamenkovic, J.; Soldatovic, I.; Milasin, J. Association Between Endothelial Nitric Oxide Synthase (ENOS)-786 T/C and 27-Bp VNTR 4b/a Polymorphisms and Preeclampsia Development. Reprod. Sci. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, G.; Jahan, S.; Bibi, N.; Ullah, A.; Faryal, R.; Almajwal, A.; Afsar, T.; Al-Disi, D.; Abulmeaty, M.; Al Khuraif, A.A.; et al. Association of Endothelial Nitric Oxide Synthase Gene Variants with Preeclampsia. Reprod. Health 2021, 18, 163. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Guo, Z.; Zhang, J.; Lu, Q.; Tian, Q.; Liu, S.; Li, K.; Wang, K.; Tao, Z.; Li, C.; et al. Non-Invasive Prediction of Fetal Growth Restriction by Whole-Genome Promoter Profiling of Maternal Plasma DNA: A Nested Case–Control Study. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 458–466. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Origin | Type of Genetic Abnormality | Examples | Referenes |
|---|---|---|---|
| Fetal | Chromosomal aberrations | Trisomy 13, 17 and 18 | [11,12,13] |
| Cri-du-chat syndrome (5p15.2 or 5p15.3 deletion) | [14,15,16] | ||
| Williams–Beuren syndrome (7q11.23 deletion) | [17] | ||
| Monogenic syndromes | Cornelia de Lange syndrome (NIPBL mutation) | [10,25,26,27] | |
| Smith–Lemli–Opitz syndrome (DHCR7 mutation) | |||
| Meier–Gorlin syndrome (ORC1, ORC4, ORC6, CDT1 or CDC6 mutation) | |||
| 3 M syndrome (CUL7 mutation) | |||
| Noonan syndrome (PTP11 mutation) | |||
| Achondroplasia or Hypochondroplasia syndrome (FGFR3 mutation) | [10,28,29] | ||
| Abnormal methylation | Silver–Russel syndrome (11p15.5 epigenetic changes) | [34,35] | |
| Placental | Confined placental mosaicism | Most common chromosomes: 2, 6, 7–10, 13–18, 21, 22 | [36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55] |
| Alterations in gene expression | Upregulation | ||
| DLX3 and 4 | [56,57,58] | ||
| TGIF-1 | |||
| HLX1 | |||
| CUL1 | [61] | ||
| CUL4B, 4a | |||
| CUL7 | |||
| IGF-2 and IGFBP-3 | [62,63,64,65] | ||
| Downregulation | |||
| ESX1L | [56,57,58] | ||
| HLX1 | |||
| IGF-1 | [65] | ||
| Single nucleotide variants | IGF1R | [66] | |
| Mitochondrial increased expression | Sirtuin-3 | [67] | |
| Maternal | Gene mutations causing its abnormal function | ET-1 FOXD1 TNFα | [68,69,70,71] [72] [73] |
| Single nucleotide polymorphisms | rs2234693-rs9340799 ESR1 variants MTHFR 677C > T variant | [74,75] [76,77,78,79] | |
| Genetic causes of preeclampsia | |||
| Gene | Variant/Genotype | Effect Size | References |
|---|---|---|---|
| FGR | |||
| TNFα | -308GA | OR 1.65 (95% CI 0.93–2.93) p = 0.06 OR 2.8 p = 0.01 with uterine arteries flow abnormatlities | [72] |
| ESR1 | rs2234693/rs9340799 TG genotype | OR 1.94 p = 0.001 | [73] |
| MTHFR | 677C > T TT + TC vs. CC overall Caucasian ethnicity African ethnicity | OR 0.14 (95% CI 0.049–0.045) p < 0.001 OR 0.15 (95% CI 0.04–0.561) p = 0.005 OR 0.162 (95% CI 0.06–0.436) p < 0.001 | [78] |
| PE | |||
| FLT1 | rs4769613 C allele (fetal genome) | OR 1.22 (95% CI 1.14–1.31) | [84] |
| PAI-1 | 4G/5G | OR 1.36 (95% CI 1.13–1.64) | [88] |
| MMP-9 | rs3918242 (-562C > T) | OR 1.62 (95% CI 1.03–2.56) | [89] |
| TGFβ1 | rs1800469 | OR 1.17 (95% CI 1.02–1.35) OR 1.35 (95% CI 1.06–1.72) OR 1.48 (95% CI 1.07–2.05) Allele, recessive and homozygous model respectively | [90] |
| IL-1β | rs1143634 T allele | OR 1.28 (95% CI 1.04–1.58) | [92] |
| NPHS1 | rs437168 C allele C vs. T CC vs. CT/TT | OR 1.287 (95% CI 1.021–1.622) OR 1.398 (95% CI 1.005–1.945) | [95] |
| ERAP-1 | 96121524 TC genotype | OR 2.002 (95% CI 0.687–5.831) p = 0.02 | [96] |
| eNOS | -86T/C (CC variant) | p = 0.006 PE p = 0.01 9 early onset PE p = 0.012 late onset PE | [101] |
| VNTR 4a4a homozygote | OR 7.68 (95% CI 0.89–65.98) p = 0.04 | [101] | |
| IL-32 | rs4786370 T allele CT/GT TT/TT TT/GT | OR 2.75 (95% CI 1.34–5.642) OR 3.266 (95% CI 1.279–8.339) OR 3.438 (95% CI 1.354–8.73) | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowakowska, B.A.; Pankiewicz, K.; Nowacka, U.; Niemiec, M.; Kozłowski, S.; Issat, T. Genetic Background of Fetal Growth Restriction. Int. J. Mol. Sci. 2022, 23, 36. https://doi.org/10.3390/ijms23010036
Nowakowska BA, Pankiewicz K, Nowacka U, Niemiec M, Kozłowski S, Issat T. Genetic Background of Fetal Growth Restriction. International Journal of Molecular Sciences. 2022; 23(1):36. https://doi.org/10.3390/ijms23010036
Chicago/Turabian StyleNowakowska, Beata Anna, Katarzyna Pankiewicz, Urszula Nowacka, Magdalena Niemiec, Szymon Kozłowski, and Tadeusz Issat. 2022. "Genetic Background of Fetal Growth Restriction" International Journal of Molecular Sciences 23, no. 1: 36. https://doi.org/10.3390/ijms23010036
APA StyleNowakowska, B. A., Pankiewicz, K., Nowacka, U., Niemiec, M., Kozłowski, S., & Issat, T. (2022). Genetic Background of Fetal Growth Restriction. International Journal of Molecular Sciences, 23(1), 36. https://doi.org/10.3390/ijms23010036