
CRISPR/Cas9 Genome Editing vs. Over-Expression for Fluorescent Extracellular Vesicle-Labeling: A Quantitative Analysis

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
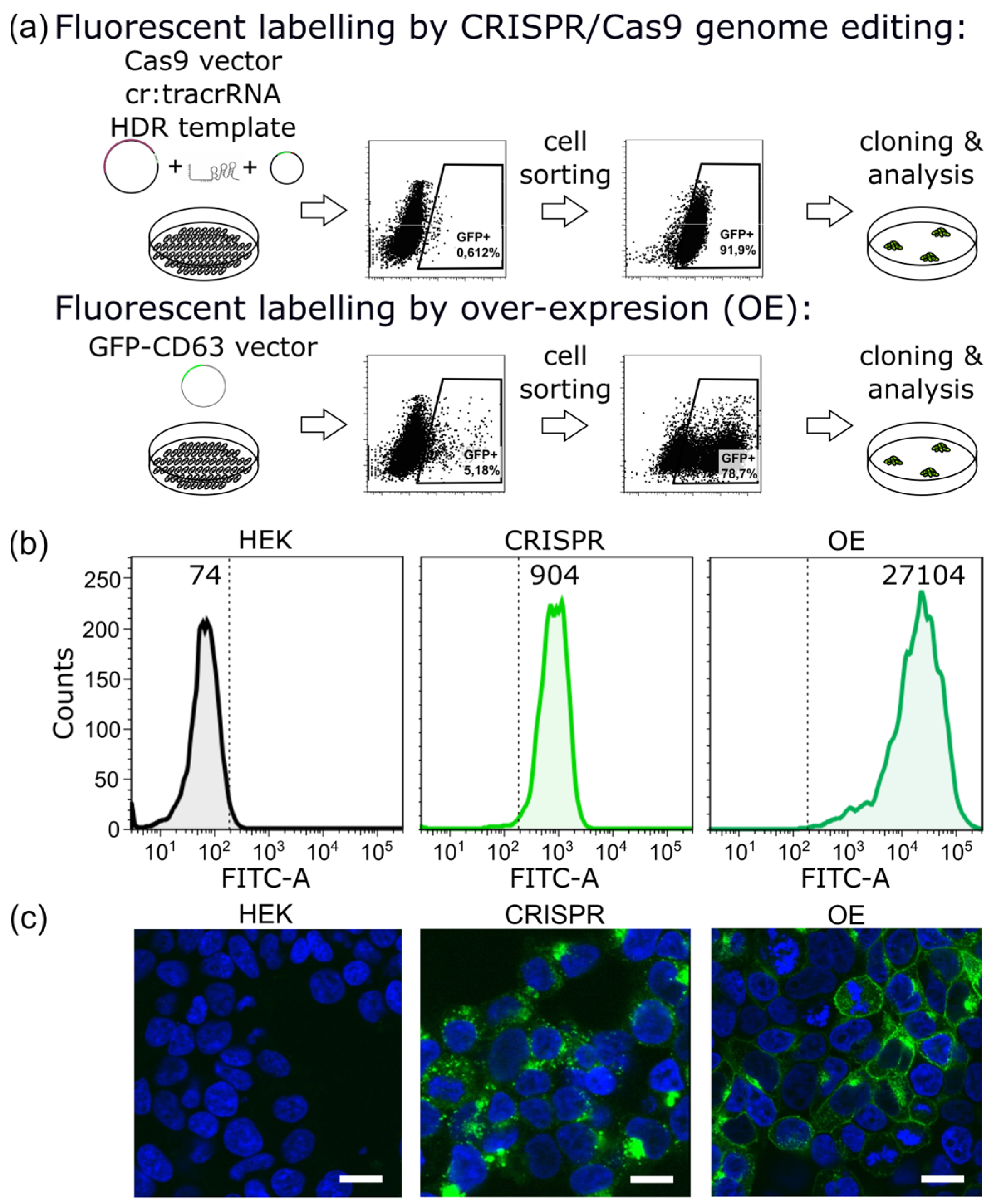
2.1. CRISPR/Cas9 Fluorescent Labeling of EVs
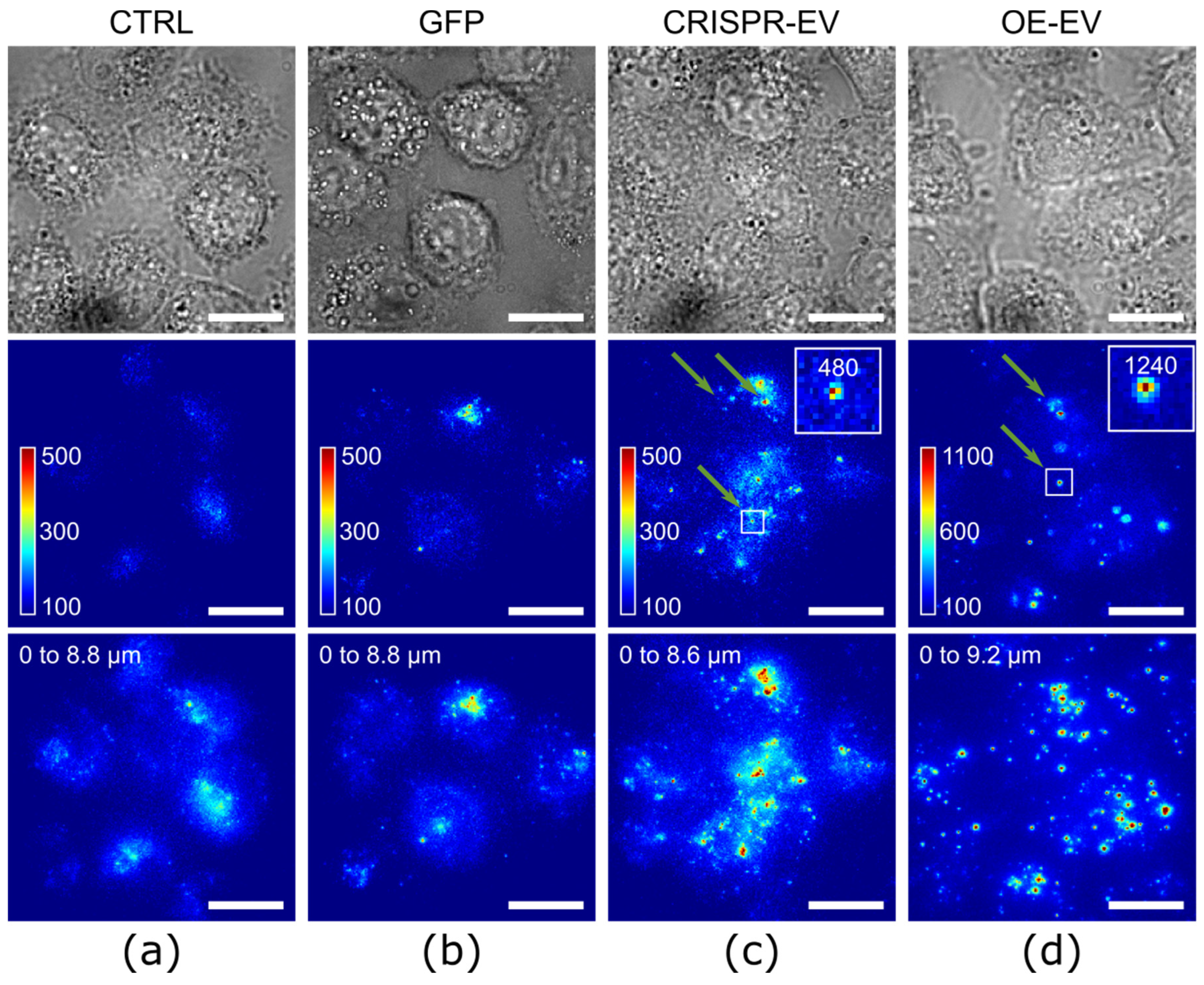
2.2. Intracellular Imaging with CRISPR/Cas9-Modified EVs
3. Discussion
4. Materials and Methods
4.1. Transfections and Cell Culture
4.2. Flow Cytometry and Cell Sorting
4.3. Integration-Specific PCR
4.4. EV Isolation
4.5. Western Blotting
4.6. High-Speed AFM Imaging
4.7. Fluorescence Microscopy
4.8. Intracellular EV Uptake
4.9. Image Analysis
4.10. Probability Density Function Convolution
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, É.; Pap, E.; Kittel, Á.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Valcz, G.; Buzás, E.I.; Sebestyén, A.; Krenács, T.; Szállási, Z.; Igaz, P.; Molnár, B. Extracellular Vesicle-Based Communication May Contribute to the Co-Evolution of Cancer Stem Cells and Cancer-Associated Fibroblasts in Anti-Cancer Therapy. Cancers 2020, 12, 2324. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]
- Abramowicz, A.; Widlak, P.; Pietrowska, M. Proteomic analysis of exosomal cargo: The challenge of high purity vesicle isolation. Mol. Biosyst. 2016, 12, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics 2013, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Lam, A.K. Liquid Biopsy for Investigation of Cancer DNA in Esophageal Adenocarcinoma: Cell-Free Plasma DNA and Exosome-Associated DNA. Methods Mol. Biol. 2018, 1756, 187–194. [Google Scholar] [CrossRef]
- Kibria, G.; Ramos, E.K.; Wan, Y.; Gius, D.R.; Liu, H. Exosomes as a Drug Delivery System in Cancer Therapy: Potential and Challenges. Mol. Pharm. 2018, 15, 3625–3633. [Google Scholar] [CrossRef]
- Urban, S.K.; Mocan, T.; Sanger, H.; Lukacs-Kornek, V.; Kornek, M. Extracellular Vesicles in Liver Diseases: Diagnostic, Prognostic, and Therapeutic Application. Semin. Liver Dis. 2019, 39, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.B.; Brennan, M.; Lötvall, J.; Breakefield, X.O.; Andaloussi, S.E.L. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, A.; Prenzler, N.; Harre, J.; Köhl, U.; Gärtner, L.; Lenarz, T.; Laner-Plamberger, S.; Wietzorrek, G.; Staecker, H.; Lassacher, T.; et al. First-in-human intracochlear application of human stromal cell-derived extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12094. [Google Scholar] [CrossRef] [PubMed]
- Gimona, M.; Brizzi, M.F.; Choo, A.B.H.; Dominici, M.; Davidson, S.M.; Grillari, J.; Hermann, D.M.; Hill, A.F.; de Kleijn, D.; Lai, R.C.; et al. Critical considerations for the development of potency tests for therapeutic applications of mesenchymal stromal cell-derived small extracellular vesicles. Cytotherapy 2021, 23, 373–380. [Google Scholar] [CrossRef]
- Saint-Pol, J.; Gosselet, F.; Duban-Deweer, S.; Pottiez, G.; Karamanos, Y. Targeting and Crossing the Blood-Brain Barrier with Extracellular Vesicles. Cells 2020, 9, 851. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- György, B.; Módos, K.; Pállinger, É.; Pálóczi, K.; Pásztói, M.; Misják, P.; Deli, M.A.; Sipos, Á.; Szalai, A.; Voszka, I.; et al. Detection and isolation of cell-derived microparticles are compromised by protein complexes resulting from shared biophysical parameters. Blood 2011, 117, 39–48. [Google Scholar] [CrossRef]
- Sharma, S.; LeClaire, M.; Gimzewski, J.K. Ascent of atomic force microscopy as a nanoanalytical tool for exosomes and other extracellular vesicles. Nanotechnology 2018, 29, 132001. [Google Scholar] [CrossRef]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Han, C.; Kang, H.; Yi, J.; Kang, M.; Lee, H.; Kwon, Y.; Jung, J.; Lee, J.; Park, J. Single-vesicle imaging and co-localization analysis for tetraspanin profiling of individual extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12047. [Google Scholar] [CrossRef]
- Yi, Y.W.; Lee, J.H.; Kim, S.Y.; Pack, C.G.; Ha, D.H.; Park, S.R.; Youn, J.; Cho, B.S. Advances in analysis of biodistribution of exosomes by molecular imaging. Int. J. Mol. Sci. 2020, 21, 665. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Heusermann, W.; Trojer, D.; Görgens, A.; Steib, E.; Voshol, J.; Graff, A.; Genoud, C.; Hean, J.; Nordin, J.Z.; et al. Systematic characterization of extracellular vesicle sorting domains and quantification at the single molecule—Single vesicle level by fluorescence correlation spectroscopy and single particle imaging. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Verweij, F.J.; Balaj, L.; Boulanger, C.M.; Carter, D.R.F.; Compeer, E.B.; D’Angelo, G.; El Andaloussi, S.; Goetz, J.G.; Gross, J.C.; Hyenne, V.; et al. The power of imaging to understand extracellular vesicle biology in vivo. Nat. Methods 2021, 18, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Parisse, P.; Rago, I.; Ulloa Severino, L.; Perissinotto, F.; Ambrosetti, E.; Paoletti, P.; Ricci, M.; Beltrami, A.P.; Cesselli, D.; Casalis, L. Atomic force microscopy analysis of extracellular vesicles. Eur. Biophys. J. 2017, 46, 813–820. [Google Scholar] [CrossRef]
- Priglinger, E.; Strasser, J.; Buchroithner, B.; Weber, F.; Wolbank, S.; Auer, D.; Grasmann, E.; Arzt, C.; Sivun, D.; Grillari, J.; et al. Label-free characterization of an extracellular vesicle-based therapeutic. J. Extracell. Vesicles 2021, 10, e12156. [Google Scholar] [CrossRef]
- Colombo, F.; Norton, E.G.; Cocucci, E. Microscopy approaches to study extracellular vesicles. Biochim. Biophys. Acta Gen. Subj. 2020, 1865, 129752. [Google Scholar] [CrossRef]
- Lai, C.P.; Kim, E.Y.; Badr, C.E.; Weissleder, R.; Mempel, T.R.; Tannous, B.A.; Breakefield, X.O. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef]
- Stickney, Z.; Losacco, J.; McDevitt, S.; Zhang, Z.; Lu, B. Development of exosome surface display technology in living human cells. Biochem. Biophys. Res. Commun. 2016, 472, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Dong, B.; Zeng, E.; Wang, F.; Jiang, Y.; Li, D.; Liu, D. In Vivo Tracking of Multiple Tumor Exosomes Labeled by Phospholipid-Based Bioorthogonal Conjugation. Anal. Chem. 2018, 90, 11273–11279. [Google Scholar] [CrossRef]
- Chen, C.; Zong, S.; Wang, Z.; Lu, J.; Zhu, D.; Zhang, Y.; Cui, Y. Imaging and Intracellular Tracking of Cancer-Derived Exosomes Using Single-Molecule Localization-Based Super-Resolution Microscope. ACS Appl. Mater. Interfaces 2016, 8, 25825–25833. [Google Scholar] [CrossRef] [PubMed]
- Chuo, S.T.Y.; Chien, J.C.Y.; Lai, C.P.K. Imaging extracellular vesicles: Current and emerging methods. J. Biomed. Sci. 2018, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.A.; Davidson, M.W.; Piston, D.W. Fluorescent protein tracking and detection: Applications using fluorescent proteins in living cells. Cold Spring Harb. Protoc. 2009, 2009, pdb.top64. [Google Scholar] [CrossRef] [PubMed]
- Birchler, J.A.; Veitia, R.A. Gene balance hypothesis: Connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. USA 2012, 109, 14746–14753. [Google Scholar] [CrossRef]
- Jensen, E.C. Use of fluorescent probes: Their effect on cell biology and limitations. Anat. Rec. 2012, 295, 2031–2036. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Mori, T.; Kurihara, T.; Kawase, S.; Koike, R.; Satoga, M.; Cao, X.; Li, X.; Yanagawa, T.; Sakurai, T.; et al. Fluorescent protein tagging of endogenous protein in brain neurons using CRISPR/Cas9-mediated knock-in and in utero electroporation techniques. Sci. Rep. 2016, 6, 35861. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Roberts, B.; Haupt, A.; Tucker, A.; Grancharova, T.; Arakaki, J.; Fuqua, M.A.; Nelson, A.; Hookway, C.; Ludmann, S.A.; Mueller, I.A.; et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol. Biol. Cell 2017, 28, 2854–2874. [Google Scholar] [CrossRef] [PubMed]
- White, C.W.; Vanyai, H.K.; See, H.B.; Johnstone, E.K.M.; Pfleger, K.D.G. Using nanoBRET and CRISPR/Cas9 to monitor proximity to a genome-edited protein in real-time. Sci. Rep. 2017, 7, 3187. [Google Scholar] [CrossRef] [PubMed]
- Ratz, M.; Testa, I.; Hell, S.W.; Jakobs, S. CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci. Rep. 2015, 5, 9592. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Simms, V.A.; Pike, J.A.; Thomas, S.G.; Morgan, N.V. CRISPR-Cas9 Mediated Labelling Allows for Single Molecule Imaging and Resolution. Sci. Rep. 2017, 7, 8450. [Google Scholar] [CrossRef] [PubMed]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- England, C.G.; Luo, H.; Cai, W. HaloTag Technology: A Versatile Platform for Biomedical Applications. Bioconjug. Chem. 2015, 26, 975–986. [Google Scholar] [CrossRef]
- Bottanelli, F.; Kromann, E.B.; Allgeyer, E.S.; Erdmann, R.S.; Baguley, S.W.; Sirinakis, G.; Schepartz, A.; Baddeley, D.; Toomre, D.K.; Rothman, J.E.; et al. Two-colour live-cell nanoscale imaging of intracellular targets. Nat. Commun. 2016, 7, 10778. [Google Scholar] [CrossRef] [PubMed]
- Duc, H.; Ren, X. Labelling HaloTag Fusion Proteins with HaloTag Ligand in Living Cells. Bio-Protocol 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.Á.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [PubMed]
- Gildea, J.J.; Seaton, J.E.; Victor, K.G.; Reyes, C.M.; Bigler Wang, D.; Pettigrew, A.C.; Courtner, C.E.; Shah, N.; Tran, H.T.; Van Sciver, R.E.; et al. Exosomal transfer from human renal proximal tubule cells to distal tubule and collecting duct cells. Clin. Biochem. 2014, 47, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Koumangoye, R.B.; Sakwe, A.M.; Goodwin, J.S.; Patel, T.; Ochieng, J. Detachment of breast tumor cells induces rapid secretion of exosomes which subsequently mediate cellular adhesion and spreading. PLoS ONE 2011, 6, e20592. [Google Scholar] [CrossRef]
- Bobrie, A.; Colombo, M.; Krumeich, S.; Raposo, G.; Thery, C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J. Extracell. Vesicles 2012, 1, 18397. [Google Scholar] [CrossRef]
- Mayr, S.; Hauser, F.; Peterbauer, A.; Tauscher, A.; Naderer, C.; Axmann, M.; Plochberger, B.; Jacak, J. Localization microscopy of actin cytoskeleton in human platelets. Int. J. Mol. Sci. 2018, 19, 1150. [Google Scholar] [CrossRef] [PubMed]
- Borgmann, D.M.; Mayr, S.; Polin, H.; Schaller, S.; Dorfer, V.; Obritzberger, L.; Endmayr, T.; Gabriel, C.; Winkler, S.M.; Jacak, J. Single Molecule Fluorescence Microscopy and Machine Learning for Rhesus D Antigen Classification. Sci. Rep. 2016, 6, 32317. [Google Scholar] [CrossRef] [PubMed]
- Buchroithner, B.; Mayr, S.; Hauser, F.; Priglinger, E.; Stangl, H.; Santa-Maria, A.R.; Deli, M.A.; Der, A.; Klar, T.A.; Axmann, M.; et al. Dual Channel Microfluidics for Mimicking the Blood−Brain Barrier. ACS Nano 2021, 15, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- Hauser, F.; Jacak, J. Real-time 3D single-molecule localization microscopy analysis using lookup tables. Biomed. Opt. Express 2021, 12, 4955. [Google Scholar] [CrossRef]
- Schmidt, T.; Schütz, G.J.; Gruber, H.J.; Schindler, H. Local Stoichiometries Determined by Counting Individual Molecules. Anal. Chem. 1996, 68, 4397–4401. [Google Scholar] [CrossRef]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef]
- Mayr, S.; Hauser, F.; Puthukodan, S.; Axmann, M.; Göhring, J.; Jacak, J. Statistical analysis of 3D localisation microscopy images for quantification of membrane protein distributions in a platelet clot model. PLoS Comput. Biol. 2020, 16, e1007902. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, D.; Santos, M.F.; Karbanová, J.; Kurth, T.; Rappa, G.; Lorico, A. Uptake and Fate of Extracellular Membrane Vesicles: Nucleoplasmic Reticulum-Associated Late Endosomes as a New Gate to Intercellular Communication. Cells 2020, 9, 1931. [Google Scholar] [CrossRef] [PubMed]
- French, K.C.; Antonyak, M.A.; Cerione, R.A. Extracellular vesicle docking at the cellular port: Extracellular vesicle binding and uptake. Semin. Cell Dev. Biol. 2017, 67, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, A.; Carmi, I.; Oszvald, Á.; Lőrincz, P.; Petővári, G.; Tölgyes, T.; Dede, K.; Bursics, A.; Buzás, E.I.; Wiener, Z. IFITM1 expression determines extracellular vesicle uptake in colorectal cancer. Cell. Mol. Life Sci. 2021, 78, 7009–7024. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Kodera, N.; Takai, E.; Maruyama, D.; Saito, K.; Toda, A. A high-speed atomic force microscope for studying biological macromolecules. Proc. Natl. Acad. Sci. USA 2001, 98, 12468–12472. [Google Scholar] [CrossRef] [PubMed]
- Preiner, J.; Horner, A.; Karner, A.; Ollinger, N.; Siligan, C.; Pohl, P.; Hinterdorfer, P. High-speed AFM images of thermal motion provide stiffness map of interfacial membrane protein moieties. Nano Lett. 2015, 15, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Preiner, J.; Kodera, N.; Tang, J.; Ebner, A.; Brameshuber, M.; Blaas, D.; Gelbmann, N.; Gruber, H.J.; Ando, T.; Hinterdorfer, P. IgGs are made for walking on bacterial and viral surfaces. Nat. Commun. 2014, 5, 4394. [Google Scholar] [CrossRef]
- Karner, A.; Nimmervoll, B.; Plochberger, B.; Klotzsch, E.; Horner, A.; Knyazev, D.G.; Kuttner, R.; Winkler, K.; Winter, L.; Siligan, C.; et al. Tuning membrane protein mobility by confinement into nanodomains. Nat. Nanotechnol. 2017, 12, 260–266. [Google Scholar] [CrossRef]
- Sams, M.; Silye, R.; Göhring, J.; Muresan, L.; Schilcher, K.; Jacak, J. Spatial cluster analysis of nanoscopically mapped serotonin receptors for classification of fixed brain tissue. J. Biomed. Opt. 2013, 19, 011021. [Google Scholar] [CrossRef][Green Version]
- Wolter, S.; Loschberger, A.; Holm, T.; Aufmkolk, S.; Dabauvalle, M.C.; van de Linde, S.; Sauer, M. rapidSTORM: Accurate, fast open-source software for localization microscopy. Nat. Methods 2012, 9, 1040–1041. [Google Scholar] [CrossRef]
- Gohring, J.; Fulcher, N.; Schilcher, K.; Barta, A.; Jacak, J. Suitable transfection methods for single particle tracing in plant suspension cells. Plant Methods 2014, 10, 15. [Google Scholar] [CrossRef]
- Jacak, J.; Schaller, S.; Borgmann, D.; Winkler, S.M. Characterization of the Distance Relationship Between Localized Serotonin Receptors and Glia Cells on Fluorescence Microscopy Images of Brain Tissue. Microsc. Microanal. 2015, 21, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Schwartz, S.L.; Byars, J.M.; Lidke, K.A. Simultaneous multiple-emitter fitting for single molecule super-resolution imaging. Biomed. Opt. Express 2011, 2, 1377–1393. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K.I.; Churchman, L.S.; Spudich, J.A.; Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 2010, 7, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Wiesbauer, M.; Wollhofen, R.; Vasic, B.; Schilcher, K.; Jacak, J.; Klar, T.A. Nano-Anchors with Single Protein Capacity Produced with STED Lithography. Nano Lett. 2013, 13, 5672–5678. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gRNA | Sequence [5′–3′] | PAM [5′–3′] |
| gRNA#3 | GAGGGCGGGGGGATTAAAAC | TGG |
| gRNA#4 | CCGGCAGCCATGGCGGTGGA | AGG |
| Primer | Sequence [5′–3′] | |
| GFP 18 F | ATGGTGAGCAAGGGCGAG | |
| CD63 gDNA F | CCCTCTCCTGCGGGTAAAGA | |
| Halo integration_F | CCGCTGACTGAAGTCGAGATGG | |
| gDNA Exb 3_R | CCCACTGCACAGGCCTAAGAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strohmeier, K.; Hofmann, M.; Hauser, F.; Sivun, D.; Puthukodan, S.; Karner, A.; Sandner, G.; Le Renard, P.-E.; Jacak, J.; Mairhofer, M. CRISPR/Cas9 Genome Editing vs. Over-Expression for Fluorescent Extracellular Vesicle-Labeling: A Quantitative Analysis. Int. J. Mol. Sci. 2022, 23, 282. https://doi.org/10.3390/ijms23010282
Strohmeier K, Hofmann M, Hauser F, Sivun D, Puthukodan S, Karner A, Sandner G, Le Renard P-E, Jacak J, Mairhofer M. CRISPR/Cas9 Genome Editing vs. Over-Expression for Fluorescent Extracellular Vesicle-Labeling: A Quantitative Analysis. International Journal of Molecular Sciences. 2022; 23(1):282. https://doi.org/10.3390/ijms23010282
Chicago/Turabian StyleStrohmeier, Karin, Martina Hofmann, Fabian Hauser, Dmitry Sivun, Sujitha Puthukodan, Andreas Karner, Georg Sandner, Pol-Edern Le Renard, Jaroslaw Jacak, and Mario Mairhofer. 2022. "CRISPR/Cas9 Genome Editing vs. Over-Expression for Fluorescent Extracellular Vesicle-Labeling: A Quantitative Analysis" International Journal of Molecular Sciences 23, no. 1: 282. https://doi.org/10.3390/ijms23010282
APA StyleStrohmeier, K., Hofmann, M., Hauser, F., Sivun, D., Puthukodan, S., Karner, A., Sandner, G., Le Renard, P.-E., Jacak, J., & Mairhofer, M. (2022). CRISPR/Cas9 Genome Editing vs. Over-Expression for Fluorescent Extracellular Vesicle-Labeling: A Quantitative Analysis. International Journal of Molecular Sciences, 23(1), 282. https://doi.org/10.3390/ijms23010282