
The Dynamics of β-Amyloid Proteoforms Accumulation in the Brain of a 5xFAD Mouse Model of Alzheimer’s Disease

 , , ,
, , ,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
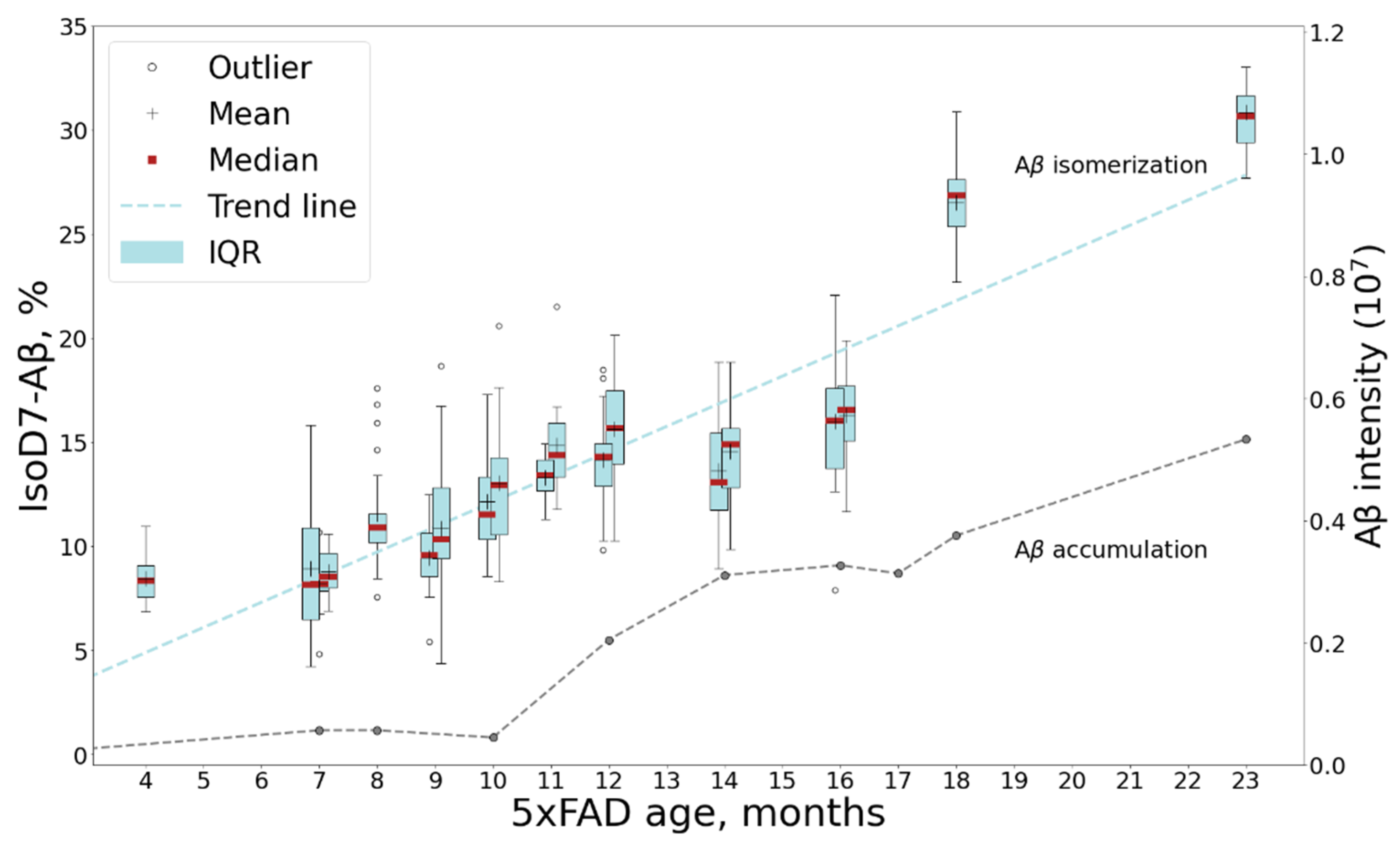
2.1. The Accumulation of Total Amyloids in the Murine Brain
2.2. Aβ Diversity
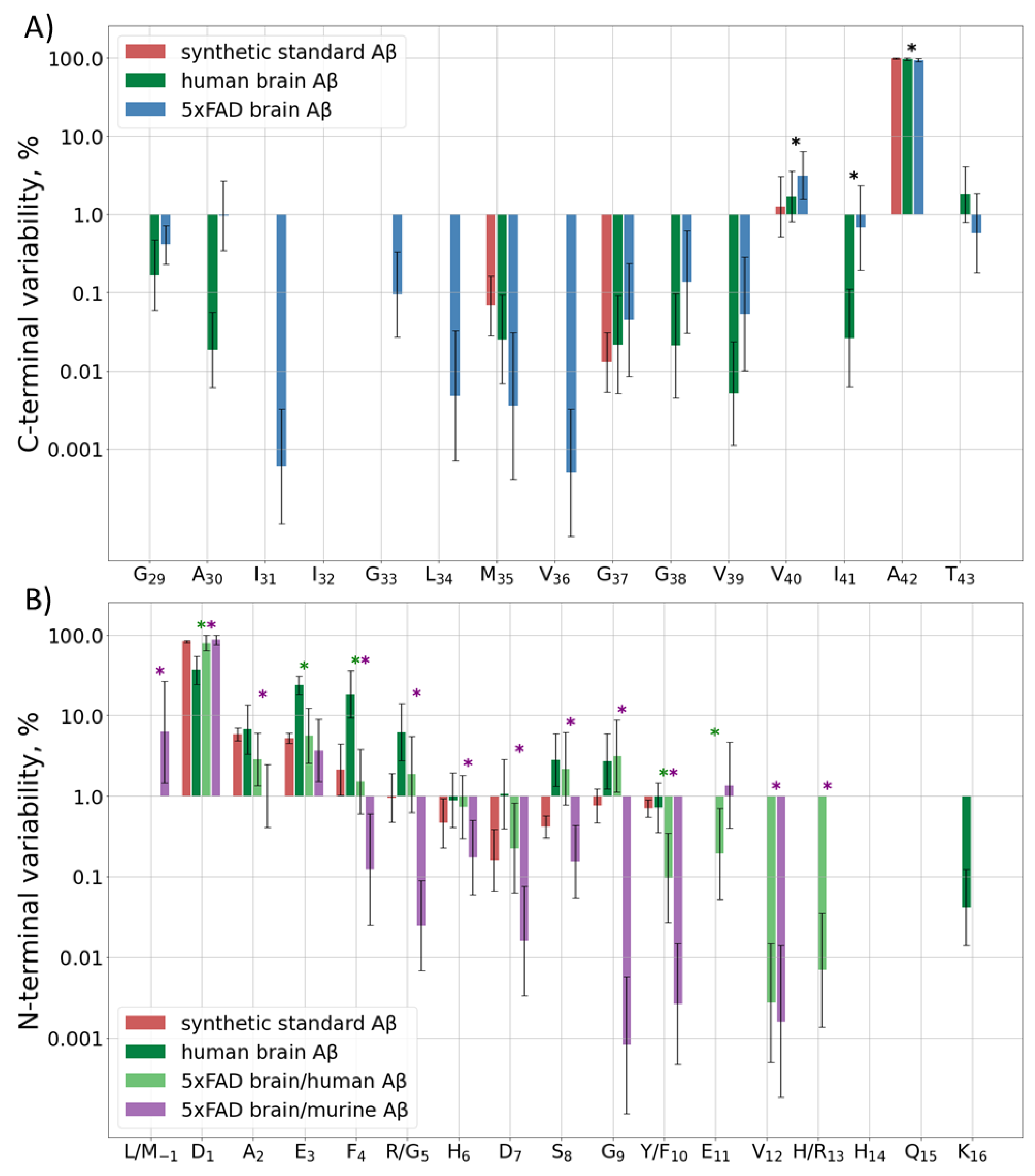
2.2.1. Diversity at the C-Terminus
2.2.2. Diversity at the N-Terminal
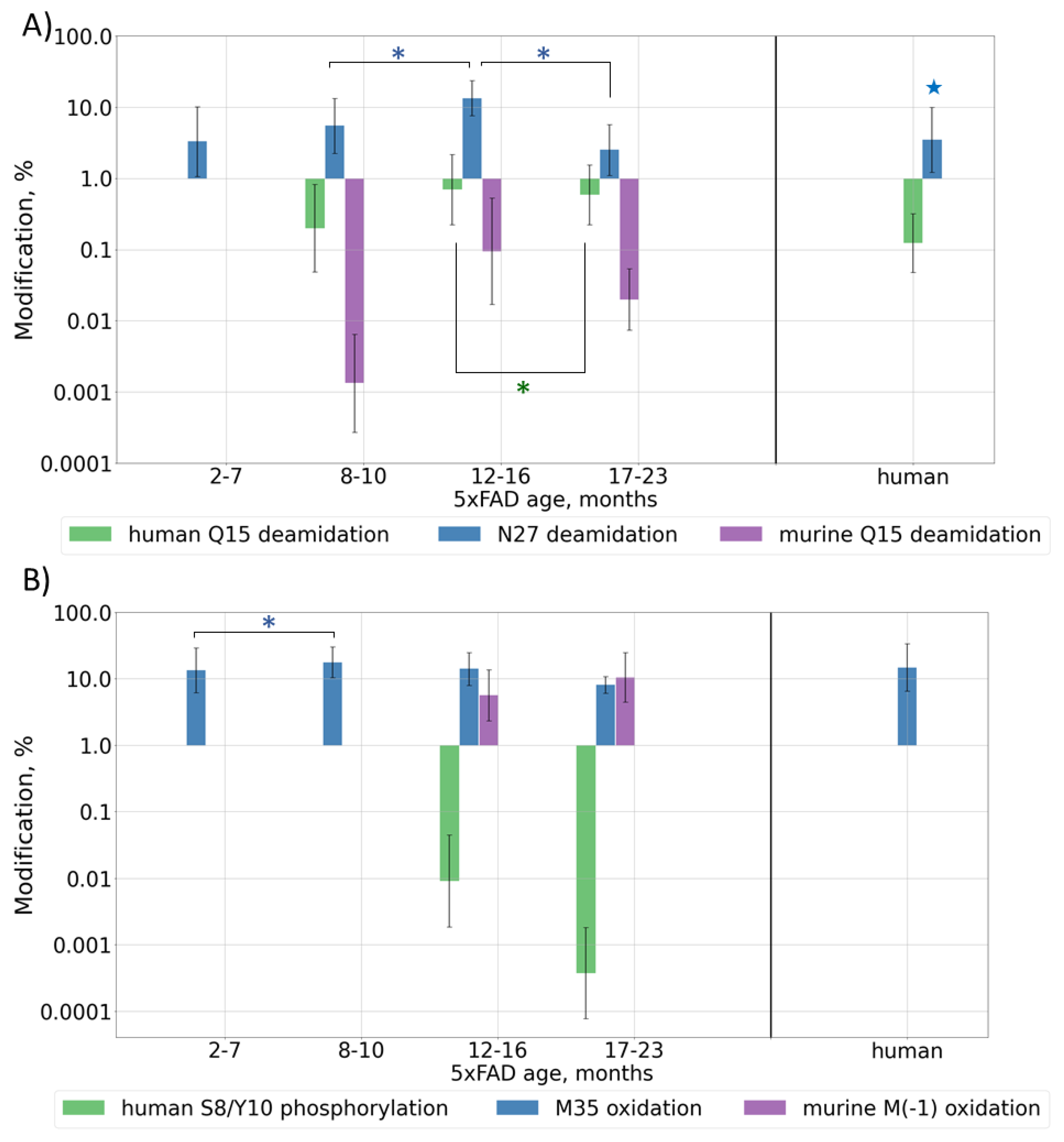
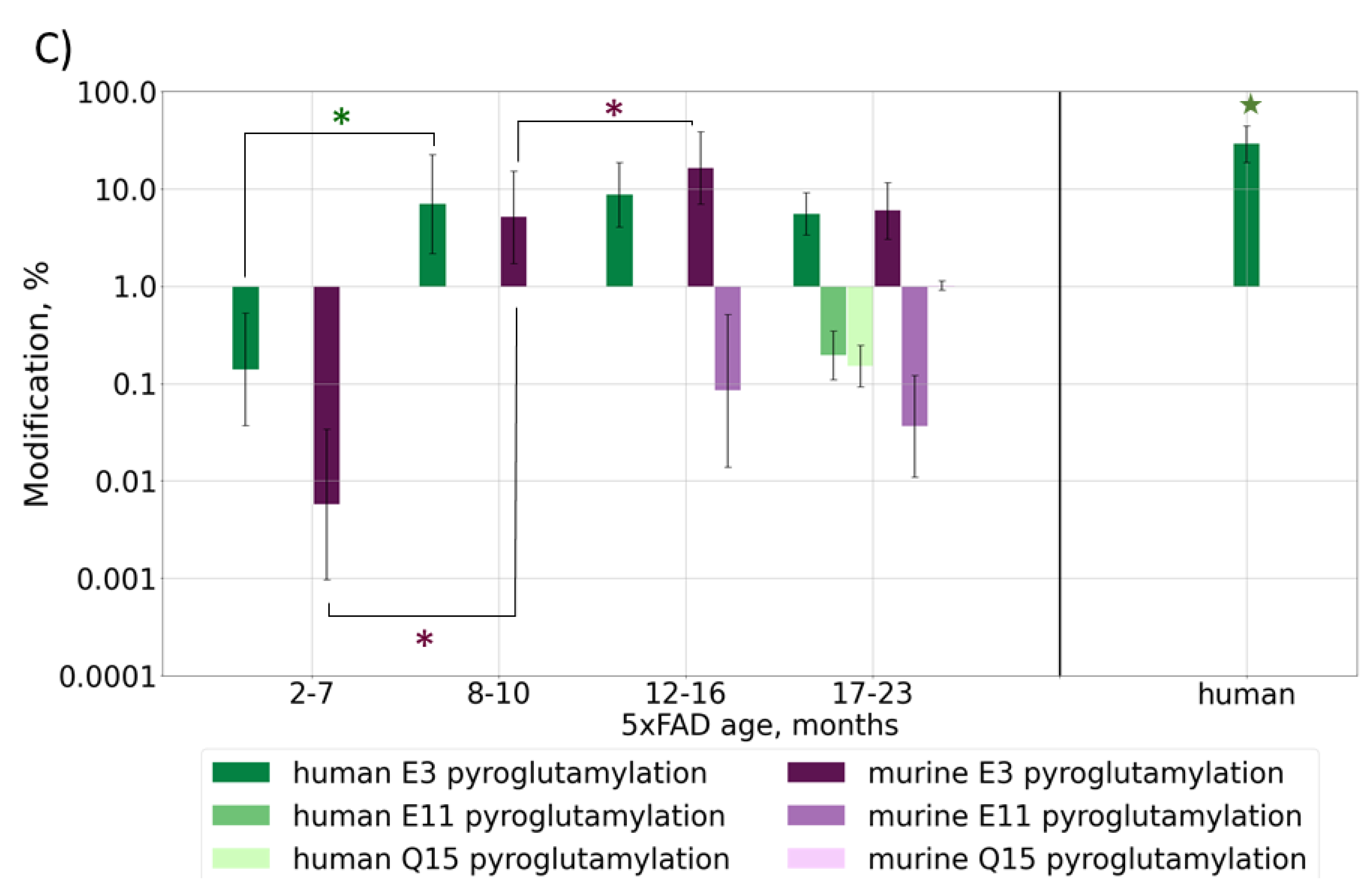
2.2.3. Post-Translational Modifications
3. Materials and Methods
3.1. Reagents and Peptides
3.2. Animals
3.3. The Extraction and Hydrolysis of Aβ Peptides
3.4. Western Blot
3.5. Human Samples
3.6. MALDI-TOF/TOF MS
3.7. The LC-MS/MS Analysis by TIMS-TOF
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gaugler, J.; James, B.; Johnson, T.; Scholz, K.; Weuve, J. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 84–185. [Google Scholar] [CrossRef]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small A β oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. β-amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.C.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-β oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Duyckaerts, C. Propagation of Aß pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 5–25. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Monteleone, D.; Manassero, G.; Vasciaveo, V.; Tabaton, M. The Unexpected Role of Aβ 1-42 Monomers in the Pathogenesis of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1241–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimers Res. Ther. 2014, 6, 28–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roher, A.E.; Kokjohn, T.A.; Clarke, S.G.; Sierks, M.R.; Maarouf, C.L.; Serrano, G.E.; Sabbagh, M.S.; Beach, T.G. APP/Aβ structural diversity and Alzheimer’s disease pathogenesis. Neurochem. Int. 2017, 110, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, Y.; Kawahara, M. Implications of metal binding and asparagine deamidation for amyloid formation. Int. J. Mol. Sci. 2018, 19, 2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, D.A.; Kanski, J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer’s amyloid β-peptide 1-42. Peptides 2002, 23, 1299–1309. [Google Scholar] [CrossRef]
- Hou, L.; Kang, I.; Marchant, R.E.; Zagorski, M.G. Methionine 35 oxidation reduces fibril assembly of the amyloid aβ-(1-42) peptide of Alzheimer’s disease. J. Biol. Chem. 2002, 277, 40173–40176. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.S.; Bergquist, J.; Volbracht, C.; Päiviö, A.; Leist, M.; Lannfelt, L.; Westlind-Danielsson, A. Attenuated amyloid-β aggregation and neurotoxicity owing to methionine oxidation. NeuroReport 2007, 18, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portelius, E.; Bogdanovic, N.; Gustavsson, M.K.; Volkmann, I.; Brinkmalm, G.; Zetterberg, H.; Winblad, B.; Blennow, K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roher, A.; Lowenson, J.; Clarke, S. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef]
- Shimizu, T.; Watanabe, A.; Ogawara, M.; Mori, H.; Shirasawa, T. Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Arch. Biochem. Biophys. 2000, 381, 225–234. [Google Scholar] [CrossRef]
- Shimizu, T.; Matsuoka, Y.; Shirasawa, T. Biological significance of isoaspartate and its repair system. Biol. Pharm. Bull. 2005, 28, 1590–1596. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Perez, K.A.; Lago, L.C.; Klatt, S.; McLean, C.A.; Birchall, I.E.; Barnham, K.J.; Masters, C.L.; Roberts, B.R. Quantification of N-terminal amyloid-β isoforms reveals isomers are the most abundant form of the amyloid-β peptide in sporadic Alzheimer’s disease. Brain Commun. 2021, 3, fcab028. [Google Scholar] [CrossRef]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Hosaka, D.; Mochizuki, N.; Akatsu, H.; Tsutsumiuchi, K.; Hashizume, Y.; Matsukawa, N.; Yamamoto, T.; Toyo’oka, T. Simultaneous determination of post-translational racemization and isomerization of N -terminal amyloid-β in alzheimer’s brain tissues by covalent chiral derivatized ultraperformance liquid chromatography tandem mass spectrometry. Anal. Chem. 2014, 86, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Indeykina, M.I.; Popov, I.A.; Kozin, S.A.; Kononikhin, A.S.; Kharybin, O.N.; Tsvetkov, P.O.; Makarov, A.A.; Nikolaev, E.N. Capabilities of MS for analytical quantitative determination of the ratio of α- And βasp7 isoforms of the amyloid-β peptide in binary mixtures. Anal. Chem. 2011, 83, 3205–3210. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, P.B.; Cournoyer, J.J.; Pitteri, S.J.; Chrisman, P.A.; McLuckey, S.A. Differentiation of aspartic and isoaspartic acids using electron transfer dissociation. J. Am. Soc. Mass Spectrom. 2006, 17, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zubarev, R.A. Mass spectrometric analysis of asparagine deamidation and aspartate isomerization in polypeptides. Electrophoresis 2010, 31, 1764–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekov, S.I.; Ivanov, D.G.; Bugrova, A.E.; Indeykina, M.I.; Zakharova, N.V.; Popov, I.A.; Kononikhin, A.S.; Kozin, S.A.; Makarov, A.A.; Nikolaev, E.N. Evaluation of MALDI-TOF/TOF Mass Spectrometry Approach for Quantitative Determination of Aspartate Residue Isomerization in the Amyloid-β Peptide. J. Am. Soc. Mass Spectrom. 2019, 30, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.G.; Indeykina, M.I.; Pekov, S.I.; Iusupov, A.E.; Bugrova, A.E.; Kononikhin, A.S.; Nikolaev, E.N.; Popov, I.A. Probabilistic model applied to ion abundances in product-ion spectra: Quantitative analysis of aspartic acid isomerization in peptides. Anal. Bioanal. Chem. 2019, 411, 7783–7789. [Google Scholar] [CrossRef]
- Ivanov, D.G.; Indeykina, M.I.; Pekov, S.I.; Bugrova, A.E.; Kechko, O.I.; Iusupov, A.E.; Kononikhin, A.S.; Makarov, A.A.; Nikolaev, E.N.; Popov, I.A. Relative Quantitation of Beta-Amyloid Peptide Isomers with Simultaneous Isomerization of Multiple Aspartic Acid Residues by Matrix Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 411, 7783–7789. [Google Scholar] [CrossRef]
- Tsvetkov, P.O.; Popov, I.A.; Nikolaev, E.N.; Archakov, A.I.; Makarov, A.A.; Kozin, S.A. Isomerization of the Asp7 residue results in zinc-induced oligomerization of Alzheimer’s disease amyloid β (1-16) peptide. ChemBioChem 2008, 9, 164–168. [Google Scholar] [CrossRef]
- Mitkevich, V.A.; Petrushanko, I.Y.; Yegorov, Y.E.; Simonenko, O.V.; Vishnyakova, K.S.; Kulikova, A.A.; Tsvetkov, P.O.; Makarov, A.A.; Kozin, S.A. Isomerization of Asp7 leads to increased toxic effect of amyloid-b42 on human neuronal cells. Cell Death Dis. 2013, 4, e939-1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barykin, E.P.; Garifulina, A.I.; Kruykova, E.V.; Spirova, E.N.; Anashkina, A.A.; Adzhubei, A.A.; Shelukhina, I.V.; Kasheverov, I.E.; Mitkevich, V.A.; Kozin, S.A.; et al. Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the α7 Nicotinic Receptor and Promotes Neurotoxicity. Cells 2019, 8, 771. [Google Scholar] [CrossRef] [Green Version]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally applied synthetic peptide isoAsp7-Aβ(1-42) triggers cerebral β-amyloidosis. Neurotox. Res. 2013, 24, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Gnoth, K.; Piechotta, A.; Kleinschmidt, M.; Konrath, S.; Schenk, M.; Taudte, N.; Ramsbeck, D.; Rieckmann, V.; Geissler, S.; Eichentopf, R.; et al. Targeting isoaspartate-modified Aβ rescues behavioral deficits in transgenic mice with Alzheimer’s disease-like pathology. Alzheimers Res. Ther. 2020, 12, 149. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease (review). Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawhar, S.; Wirths, O.; Bayer, T.A. Pyroglutamate amyloid-β (Aβ): A hatchet man in alzheimer disease. J. Biol. Chem. 2011, 286, 38825–38832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Wirths, O.; Theil, S.; Gerth, J.; Bayer, T.A.; Walter, J. Early intraneuronal accumulation and increased aggregation of phosphorylated Abeta in a mouse model of Alzheimer’s disease. Acta Neuropathol. 2013, 125, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Baldauf, K.; Wetzel, W.; Reymann, K.G. Effects of methylphenidate on the behavior of male 5xFAD mice. Pharmacol. Biochem. Behav. 2015, 128, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Chernyuk, D.; Zhang, H.; Postnikova, T.Y.; Pats, K.; Fedorova, E.; Poroikov, V.; Zaitsev, A.V.; Bezprozvanny, I. Derivatives of piperazines as potential therapeutic agents for Alzheimer’s disease. Mol. Pharmacol. 2019, 95, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lame, M.E.; Chambers, E.E.; Blatnik, M. Quantitation of amyloid beta peptides Aβ 1-38, Aβ 1-40, and Aβ 1-42 in human cerebrospinal fluid by ultra-performance liquid chromatography-tandem mass spectrometry. Anal. Biochem. 2011, 419, 133–139. [Google Scholar] [CrossRef]
- Haider, S.R.; Reid, H.J.; Sharp, B.L. Tricine-SDS-PAGE. Methods Mol. Biol. 2012, 869, 81–91. [Google Scholar] [CrossRef]
- Pekov, S.; Indeykina, M.; Popov, I.; Kononikhin, A.; Bocharov, K.; Kozin, S.A.; Makarov, A.A.; Nikolaev, E. Application of MALDI-TOF/TOF-MS for relative quantitation of α- and β-Asp7 isoforms of amyloid-β peptide. Eur. J. Mass Spectrom. 2018, 24, 141–144. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bugrova, A.E.; Strelnikova, P.A.; Indeykina, M.I.; Kononikhin, A.S.; Zakharova, N.V.; Brzhozovskiy, A.G.; Barykin, E.P.; Pekov, S.I.; Gavrish, M.S.; Babaev, A.A.; et al. The Dynamics of β-Amyloid Proteoforms Accumulation in the Brain of a 5xFAD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 27. https://doi.org/10.3390/ijms23010027
Bugrova AE, Strelnikova PA, Indeykina MI, Kononikhin AS, Zakharova NV, Brzhozovskiy AG, Barykin EP, Pekov SI, Gavrish MS, Babaev AA, et al. The Dynamics of β-Amyloid Proteoforms Accumulation in the Brain of a 5xFAD Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(1):27. https://doi.org/10.3390/ijms23010027
Chicago/Turabian StyleBugrova, Anna E., Polina A. Strelnikova, Maria I. Indeykina, Alexey S. Kononikhin, Natalia V. Zakharova, Alexander G. Brzhozovskiy, Evgeny P. Barykin, Stanislav I. Pekov, Maria S. Gavrish, Alexey A. Babaev, and et al. 2022. "The Dynamics of β-Amyloid Proteoforms Accumulation in the Brain of a 5xFAD Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 1: 27. https://doi.org/10.3390/ijms23010027
APA StyleBugrova, A. E., Strelnikova, P. A., Indeykina, M. I., Kononikhin, A. S., Zakharova, N. V., Brzhozovskiy, A. G., Barykin, E. P., Pekov, S. I., Gavrish, M. S., Babaev, A. A., Kosyreva, A. M., Morozova, A. Y., Degterev, D. A., Mitkevich, V. A., Popov, I. A., Makarov, A. A., & Nikolaev, E. N. (2022). The Dynamics of β-Amyloid Proteoforms Accumulation in the Brain of a 5xFAD Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 23(1), 27. https://doi.org/10.3390/ijms23010027