
Pros and Cons of Pharmacological Manipulation of cGMP-PDEs in the Prevention and Treatment of Breast Cancer

 , ,
, ,  ,
,
Abstract
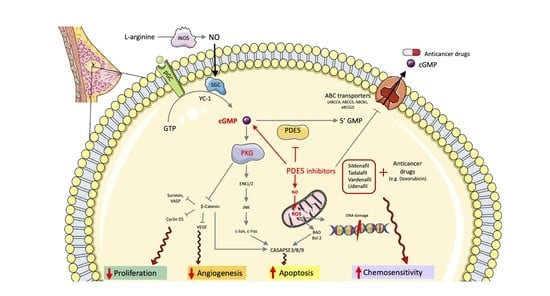
1. Introduction
2. Double-Edged Role of NO/cGMP Signaling in Breast Cancer
3. Challenges of Targeting cGMP Signaling in Breast Cancer
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wilcock, P.; Webster, R.M. The breast cancer drug market. Nat. Rev. Drug Discov. 2021, 20, 339–340. [Google Scholar] [CrossRef]
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2013, 64, 52–62. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Maughan, K.L.; Lutterbie, M.A.; Ham, P.S. Treatment of breast cancer. Am. Fam. Phys. 2010, 81, 1339–1346. [Google Scholar]
- Azimi, I.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention. Br. J. Pharmacol. 2014, 171, 945–960. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Gileadi, O. Structures of soluble guanylate cyclase: Implications for regulatory mechanisms and drug development. Biochem. Soc. Trans. 2014, 42, 108–113. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and Physiology of Cyclic Nucleotide Phosphodiesterases: Essential Components in Cyclic Nucleotide Signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef]
- Omori, K.; Kotera, J. Overview of PDEs and Their Regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef]
- Rehmann, H.; Wittinghofer, A.; Bos, J.L. Capturing cyclic nucleotides in action: Snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 2007, 8, 63–73. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D. cGMP-Dependent Protein Kinases and cGMP Phosphodiesterases in Nitric Oxide and cGMP Action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Bhargava, P.; Janda, J.; Schnellmann, R.G. Elucidation of cGMP-dependent induction of mitochondrial biogenesis through PKG and p38 MAPK in the kidney. Am. J. Physiol. Physiol. 2020, 318, F322–F328. [Google Scholar] [CrossRef]
- Chiche, J.-D.; Schlutsmeyer, S.M.; Bloch, D.B.; de la Monte, S.M.; Roberts, J.D.; Filippov, G.; Janssens, S.P.; Rosenzweig, A.; Bloch, K.D. Adenovirus-mediated Gene Transfer of cGMP-dependent Protein Kinase Increases the Sensitivity of Cultured Vascular Smooth Muscle Cells to the Antiproliferative and Pro-apoptotic Effects of Nitric Oxide/cGMP. J. Biol. Chem. 1998, 273, 34263–34271. [Google Scholar] [CrossRef]
- Feil, R.; Lohmann, S.M.; de Jonge, H.; Walter, U.; Hofmann, F. Cyclic GMP-Dependent Protein Kinases and the Cardiovascular System. Circ. Res. 2003, 93, 907–916. [Google Scholar] [CrossRef]
- Blanton, R.M. cGMP Signaling and Modulation in Heart Failure. J. Cardiovasc. Pharmacol. 2020, 75, 385–398. [Google Scholar] [CrossRef]
- Browning, D.D.; Kwon, I.-K.; Wang, R. cGMP-dependent protein kinases as potential targets for colon cancer prevention and treatment. Futur. Med. Chem. 2010, 2, 65–80. [Google Scholar] [CrossRef]
- Di Iorio, P.; Beggiato, S.; Ronci, M.; Nedel, C.B.; Tasca, C.I.; Zuccarini, M. Unfolding New Roles for Guanine-Based Purines and Their Metabolizing Enzymes in Cancer and Aging Disorders. Front. Pharmacol. 2021, 12, 653549. [Google Scholar] [CrossRef]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The Role of Cyclic Nucleotide Signaling Pathways in Cancer: Targets for Prevention and Treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef]
- Friebe, A.; Koesling, D. Regulation of Nitric Oxide-Sensitive Guanylyl Cyclase. Circ. Res. 2003, 93, 96–105. [Google Scholar] [CrossRef]
- Kim, P.K.; Zamora, R.; Petrosko, P.; Billiar, T.R. The regulatory role of nitric oxide in apoptosis. Int. Immunopharmacol. 2001, 1, 1421–1441. [Google Scholar] [CrossRef]
- Änggård, E. Nitric oxide: Mediator, murderer, and medicine. Lancet 1994, 343, 1199–1206. [Google Scholar] [CrossRef]
- Jenkins, D.C.; Charles, I.G.; Thomsen, L.L.; Moss, D.W.; Holmes, L.S.; Baylis, S.A.; Rhodes, P.; Westmore, K.; Emson, P.C.; Moncada, S. Roles of nitric oxide in tumor growth. Proc. Natl. Acad. Sci. USA 1995, 92, 4392–4396. [Google Scholar] [CrossRef]
- Muntané, J. Nitric oxide and cancer. World J. Hepatol. 2010, 2, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.N.; Lu, J.; Xue, E.; Ruan, J.; Liu, Y.; Zhang, L.; Dhar, R.; Li, Y.; Hu, Z.; Zhou, J.; et al. PDE9 Inhibitor PF-04447943 Attenuates DSS-Induced Colitis by Suppressing Oxidative Stress, Inflammation, and Regulating T-Cell Polarization. Front. Pharmacol. 2021, 12, 643215. [Google Scholar] [CrossRef] [PubMed]
- Senga, S.S.; Grose, R.P. Hallmarks of cancer—the new testament. Open Biol. 2021, 11, 200358. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Turner, M.; Belaid, A.; Lam, H.C.; Miller, S.K.; McNamara, M.C.; Baglini, C.; Housden, B.E.; Perrimon, N.; Kwiatkowski, D.J.; et al. mTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer Cell 2017, 32, 624–638.e5. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A.; Saha, A.; Chan, A.; Casteel, D.E.; Pilz, R.B.; Boss, G.R. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem. J. 2013, 454, 91–99. [Google Scholar] [CrossRef]
- Cunningham, J.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and Nucleotide Biosynthesis Are Coupled by a Single Rate-Limiting Enzyme, PRPS2, to Drive Cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef]
- Lv, Y.; Wang, X.; Li, X.; Xu, G.; Bai, Y.; Wu, J.; Piao, Y.; Shi, Y.; Xiang, R.; Wang, L. Nucleotide de novo synthesis increases breast cancer stemness and metastasis via cGMP-PKG-MAPK signaling pathway. PLoS Biol. 2020, 18, e3000872. [Google Scholar] [CrossRef] [PubMed]
- Jadeski, L.C.; Chakraborty, C.; Lala, P.K. Nitric oxide-mediated promotion of mammary tumour cell migration requires sequential activation of nitric oxide synthase, guanylate cyclase and mitogen-activated protein kinase. Int. J. Cancer 2003, 106, 496–504. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Wang, W.-Z.; Wang, D.; Skaggs, K.; Zhang, H.-T. Phosphodiesterase 7(PDE7): A unique drug target for central nervous system diseases. Neuropharmacology 2021, 196, 108694. [Google Scholar] [CrossRef]
- Deguchi, A. Activation of Protein Kinase G Is Sufficient to Induce Apoptosis and Inhibit Cell Migration in Colon Cancer Cells. Cancer Res. 2004, 64, 3966–3973. [Google Scholar] [CrossRef]
- Fujimoto, H.; Ando, Y.; Yamashita, T.; Terazaki, H.; Tanaka, Y.; Sasaki, J.; Matsumoto, M.; Suga, M.; Ando, M. Nitric Oxide Synthase Activity in Human Lung Cancer. Jpn. J. Cancer Res. 1997, 88, 1190–1198. [Google Scholar] [CrossRef]
- Gallo, O.; Fini-Storchi, I.; Vergari, W.A.; Masini, E.; Morbidelli, L.; Ziche, M.; Franchi, A. Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J. Natl. Cancer Inst. 1998, 90, 587–596. [Google Scholar] [CrossRef]
- Tuttle, T.R.; Mierzwa, M.L.; Wells, S.I.; Fox, S.R.; Ben-Jonathan, N. The cyclic GMP/protein kinase G pathway as a therapeutic target in head and neck squamous cell carcinoma. Cancer Lett. 2015, 370, 279–285. [Google Scholar] [CrossRef]
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320. [Google Scholar] [CrossRef]
- Florio, T.; Arena, S.; Pattarozzi, A.; Thellung, S.; Corsaro, A.; Villa, V.; Massa, A.; Diana, F.; Spoto, G.; Forcella, S.; et al. Basic fibroblast growth factor activates endothelial nitric-oxide synthase in CHO-K1 cells via the activation of ceramide synthesis. Mol. Pharmacol. 2003, 63, 297–310. [Google Scholar] [CrossRef]
- Thomsen, L.L.; Miles, D.W.; Happerfield, L.; Bobrow, L.G.; Knowles, R.G.; Moncada, S. Nitric oxide synthase activity in human breast cancer. Br. J. Cancer 1995, 72, 41–44. [Google Scholar] [CrossRef]
- Tse, G.M.K.; Wong, F.C.; Tsang, A.K.H.; Lee, C.S.; Lui, P.C.W.; Lo, A.; Law, B.K.B.; Scolyer, R.A.; Karim, R.Z.; Putti, T.C. Stromal nitric oxide synthase (NOS) expression correlates with the grade of mammary phyllodes tumour. J. Clin. Pathol. 2005, 58, 600–604. [Google Scholar] [CrossRef]
- Singh, R.; Pervin, S.; Karimi, A.; Cederbaum, S.; Chaudhuri, G. Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000, 60, 3305–3312. [Google Scholar]
- Mortensen, K.; Holck, S.; Christensen, I.J.; Skouv, J.; Hougaard, D.M.; Blom, J.; Larsson, L.I. Endothelial cell nitric oxide synthase in peritumoral microvessels is a favorable prognostic indicator in premenopausal breast cancer patients. Clin. Cancer Res. 1999, 5, 1093–1097. [Google Scholar]
- Mujoo, K.; Sharin, V.G.; Martin, E.; Choi, B.-K.; Sloan, C.; Nikonoff, L.E.; Kots, A.; Murad, F. Role of soluble guanylyl cyclase–cyclic GMP signaling in tumor cell proliferation. Nitric Oxide 2010, 22, 43–50. [Google Scholar] [CrossRef]
- Fraser, M.; Chan, S.L.; Chan, S.S.L.; Fiscus, R.R.; Tsang, B.K. Regulation of p53 and suppression of apoptosis by the soluble guanylyl cyclase/cGMP pathway in human ovarian cancer cells. Oncogene 2005, 25, 2203–2212. [Google Scholar] [CrossRef]
- Sen, S.; Kawahara, B.; Chaudhuri, G. Mitochondrial-associated nitric oxide synthase activity inhibits cytochrome c oxidase: Implications for breast Cancer. Free. Radic. Biol. Med. 2013, 57, 210–220. [Google Scholar] [CrossRef]
- Losenkova, K.; Zuccarini, M.; Karikoski, M.; Laurila, J.; Boison, D.; Jalkanen, S.; Yegutkin, G.G. Compartmentalization of adenosine metabolism in cancer cells and its modulation during acute hypoxia. J. Cell Sci. 2020, 133, jcs241463. [Google Scholar] [CrossRef]
- Taylor, C.; Moncada, S. Nitric Oxide, Cytochrome C Oxidase, and the Cellular Response to Hypoxia. Arter. Thromb. Vasc. Biol. 2010, 30, 643–647. [Google Scholar] [CrossRef]
- Wielinga, P.R.; van der Heijden, I.; Reid, G.; Beijnen, J.H.; Wijnholds, J.; Borst, P. Characterization of the MRP4- and MRP5-mediated Transport of Cyclic Nucleotides from Intact Cells. J. Biol. Chem. 2003, 278, 17664–17671. [Google Scholar] [CrossRef]
- Honorat, M.; Mesnier, A.; Vendrell, J.; Guitton, J.; Bieche, I.; Lidereau, R.; Kruh, G.D.; Dumontet, C.; Cohen, P.; Payen, L. ABCC11 expression is regulated by estrogen in MCF7 cells, correlated with estrogen receptor expression in postmenopausal breast tumors and overexpressed in tamoxifen-resistant breast cancer cells. Endocr.-Relat. Cancer 2008, 15, 125–138. [Google Scholar] [CrossRef]
- Cheepala, S.; Hulot, J.-S.; Morgan, J.A.; Sassi, Y.; Zhang, W.; Naren, A.P.; Schuetz, J.D. Cyclic Nucleotide Compartmentalization: Contributions of Phosphodiesterases and ATP-Binding Cassette Transporters. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 231–253. [Google Scholar] [CrossRef]
- Haider, M.; Elsherbeny, A.; Pittalà, V.; Fallica, A.; Alghamdi, M.; Greish, K. The Potential Role of Sildenafil in Cancer Management through EPR Augmentation. J. Pers. Med. 2021, 11, 585. [Google Scholar] [CrossRef]
- Martinez, S.E. GAF Domains: Two-Billion-Year-Old Molecular Switches that Bind Cyclic Nucleotides. Mol. Interv. 2002, 2, 317–323. [Google Scholar] [CrossRef]
- Marino, N.; Collins, J.W.; Shen, C.; Caplen, N.; Merchant, A.S.; Gokmen-Polar, Y.; Goswami, C.P.; Hoshino, T.; Qian, Y.; Sledge, G.W.; et al. Identification and validation of genes with expression patterns inverse to multiple metastasis suppressor genes in breast cancer cell lines. Clin. Exp. Metastasis 2014, 31, 771–786. [Google Scholar] [CrossRef]
- Andersson, K.-E. PDE5 inhibitors-pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 2018, 175, 2554–2565. [Google Scholar] [CrossRef]
- Cannon, J.E.; Pepke-Zaba, J. Riociguat for pulmonary hypertension. Expert Rev. Clin. Pharmacol. 2014, 7, 259–270. [Google Scholar] [CrossRef]
- Edwards, C.J.; Blanco, F.J.; Crowley, J.; Birbara, C.A.; Jaworski, J.; Aelion, J.; Stevens, R.M.; Vessey, A.; Zhan, X.; Bird, P. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis and current skin involvement: A phase III, randomised, controlled trial (PALACE 3). Ann. Rheum. Dis. 2016, 75, 1065–1073. [Google Scholar] [CrossRef]
- Kreisel, W.; Lazaro, A.; Trebicka, J.; Perdekamp, M.G.; Schmitt-Graeff, A.; Deibert, P. Cyclic GMP in Liver Cirrhosis—Role in Pathophysiology of Portal Hypertension and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 10372. [Google Scholar] [CrossRef]
- Mónica, F.Z.; Antunes, E. Stimulators and activators of soluble guanylate cyclase for urogenital disorders. Nat. Rev. Urol. 2017, 15, 42–54. [Google Scholar] [CrossRef]
- Ribaudo, G.; Pagano, M.A.; Bova, S.; Zagotto, G. New Therapeutic Applications of Phosphodiesterase 5 Inhibitors (PDE5-Is). Curr. Med. Chem. 2016, 23, 1239–1249. [Google Scholar] [CrossRef]
- Yu, H.M.; Chung, H.K.; Park, K.S. The PDE5 inhibitor udenafil ameliorates nonalcoholic fatty liver disease by improving mitochondrial function. Biochem. Biophys. Res. Commun. 2021, 558, 57–63. [Google Scholar] [CrossRef]
- Bian, K.; Murad, F. sGC-cGMP Signaling: Target for Anticancer Therapy. Adv. Exp. Med. Biol. 2014, 814, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Wadsten, P.; Su, S.; Rawlinson, N.; Hofman, F.M.; Hill, C.K.; Schonthal, A.H. The Type IV Phosphodiesterase Inhibitor Rolipram Induces Expression Inhibitors p21Cip1 and p27Kip1, Resulting in Growth Inhibition, Increased Differentiation, and Subsequent Apoptosis of Malignant A-172 Glioma Cells. Cancer Biol. Ther. 2002, 1, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Burgos, M.; Losada-Garcia, A.; Cruz-Hernández, C.D.; Cortés-Ramírez, S.A.; Camacho-Arroyo, I.; Gonzalez-Covarrubias, V.; Morales-Pacheco, M.; Trujillo-Bornios, S.I.; Rodríguez-Dorantes, M. New Approaches in Oncology for Repositioning Drugs: The Case of PDE5 Inhibitor Sildenafil. Front. Oncol. 2021, 11, 627229. [Google Scholar] [CrossRef]
- Peak, T.; Richman, A.; Gur, S.; Yafi, F.A.; Hellstrom, W.J. The Role of PDE5 Inhibitors and the NO/cGMP Pathway in Cancer. Sex. Med. Rev. 2016, 4, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; Jiang, C.; Lu, J.; Mehta, R.G.; Piazza, G.A.; Paranka, N.S.; Pamukcu, R.; Ahnen, D.J. Sulfone Metabolite of Sulindac Inhibits Mammary Carcinogenesis. Cancer Res. 1997, 57, 267–271. [Google Scholar]
- Alshafie, G.A.; Abou-Issa, H.M.; Seibert, K.; Harris, R.E. Chemotherapeutic evaluation of Celecoxib, a cyclooxygenase-2 inhibitor, in a rat mammary tumor model. Oncol. Rep. 2000, 7, 1377–1458. [Google Scholar] [CrossRef]
- Cuzick, J.; Otto, F.; Baron, J.A.; Brown, P.H.; Burn, J.; Greenwald, P.; Jankowski, J.; La Vecchia, C.; Meyskens, F.; Senn, H.J.; et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: An international consensus statement. Lancet Oncol. 2009, 10, 501–507. [Google Scholar] [CrossRef]
- Dannenberg, A.J.; Altorki, N.K.; Boyle, J.O.; Dang, C.; Howe, L.R.; Weksler, B.B.; Subbaramaiah, K. Cyclo-oxygenase 2: A pharmacological target for the prevention of cancer. Lancet Oncol. 2001, 2, 544–551. [Google Scholar] [CrossRef]
- Kwan, M.L.; Habel, L.A.; Slattery, M.L.; Caan, B. NSAIDs and breast cancer recurrence in a prospective cohort study. Cancer Causes Control. 2007, 18, 613–620. [Google Scholar] [CrossRef]
- Plastaras, J.; Guengerich, F.P.; Nebert, D.W.; Marnett, L.J. Xenobiotic-metabolizing Cytochromes P450 Convert Prostaglandin Endoperoxide to Hydroxyheptadecatrienoic Acid and the Mutagen, Malondialdehyde. J. Biol. Chem. 2000, 275, 11784–11790. [Google Scholar] [CrossRef]
- Tuli, H.S.; Joshi, R.; Aggarwal, D.; Kaur, G.; Kaur, J.; Kumar, M.; Parashar, N.C.; Khan, A.; Sak, K. Molecular mechanisms underlying chemopreventive potential of butein: Current trends and future perspectives. Chem. Interact. 2021, 350, 109699. [Google Scholar] [CrossRef]
- Liu, C.H.; Chang, S.-H.; Narko, K.; Trifan, O.C.; Wu, M.-T.; Smith, E.; Haudenschild, C.; Lane, T.F.; Hla, T. Overexpression of Cyclooxygenase-2 Is Sufficient to Induce Tumorigenesis in Transgenic Mice. J. Biol. Chem. 2001, 276, 18563–18569. [Google Scholar] [CrossRef]
- Tinsley, H.; Gary, B.D.; Keeton, A.B.; Zhang, W.; Abadi, A.; Reynolds, R.C.; Piazza, G.A. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol. Cancer Ther. 2009, 8, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Lu, W.; Li, Y.; Piazza, G.A. Inhibition of PDE5 by Sulindac Sulfide Selectively Induces Apoptosis and Attenuates Oncogenic Wnt/β-Catenin–Mediated Transcription in Human Breast Tumor Cells. Cancer Prev. Res. 2011, 4, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, F.; Tabrizi, A.T.; Hashemian, P.; Alijannejad, S.; Rahdar, H.A.; A Ferns, G.; Hassanian, S.M.; Shahidsales, S.; Avan, A. Role of regulatory miRNAs of the Wnt/ β-catenin signaling pathway in tumorigenesis of breast cancer. Gene 2020, 754, 144892. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, H.; Underwood, T.; Lloyd, M.; David, M.; Sperl, G.; Pamukcu, R.; Thompson, W.J. Cyclic GMP-dependent protein kinase activation and induction by exisulind and CP461 in colon tumor cells. J. Pharmacol. Exp. Ther. 2001, 299, 583–592. [Google Scholar]
- Chen, Y.; Lan, T.; Sang, J.; Wu, Y.; Wang, Y.; Jiang, L.; Tao, Y. Type II cGMP-dependent protein kinase inhibits EGF-induced MAPK/JNK signal transduction in breast cancer cells. Oncol. Rep. 2012, 27, 2039–2044. [Google Scholar] [CrossRef]
- Fallahian, F.; Karami-Tehrani, F.; Salami, S.; Aghaei, M. Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J. 2011, 278, 3360–3369. [Google Scholar] [CrossRef]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Andò, S.; Catalano, S. Phosphodiesterase type 5 and cancers: Progress and challenges. Oncotarget 2017, 8, 99179–99202. [Google Scholar] [CrossRef]
- Saravani, R.; Karami-Tehrani, F.; Hashemi, M.; Aghaei, M.; Edalat, R. Inhibition of phosphodiestrase 9 induces c GMP accumulation and apoptosis in human breast cancer cell lines, MCF -7 and MDA - MB -468. Cell Prolif. 2012, 45, 199–206. [Google Scholar] [CrossRef]
- Catalano, S.; Campana, A.; Giordano, C.; Győrffy, B.; Tarallo, R.; Rinaldi, A.; Bruno, G.; Ferraro, A.; Romeo, F.; Lanzino, M.; et al. Expression and Function of Phosphodiesterase Type 5 in Human Breast Cancer Cell Lines and Tissues: Implications for Targeted Therapy. Clin. Cancer Res. 2015, 22, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hay, S.O.; Sohn, J.; Edirisinghe, P.D.; Chandrasena, R.E.P.; Wang, Z.; Li, Q.; Thatcher, G.R.J. Anti-Inflammatory, Antiproliferative, and Cytoprotective Activity of NO Chimera Nitrates of Use in Cancer Chemoprevention. Mol. Pharmacol. 2008, 74, 1381–1391. [Google Scholar] [CrossRef]
- Rigas, B.; Williams, J.L. NO-donating NSAIDs and cancer: An overview with a note on whether NO is required for their action. Nitric Oxide 2008, 19, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Black, K.L.; Yin, D.; Ong, J.M.; Hu, J.; Konda, B.M.; Wang, X.; Ko, M.K.; Bayan, J.-A.; Sacapano, M.R.; Espinoza, A.; et al. PDE5 inhibitors enhance tumor permeability and efficacy of chemotherapy in a rat brain tumor model. Brain Res. 2008, 1230, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; Gennings, C.; Bear, H.D.; Graham, L.J.; Sheth, C.M.; White, K.L.; Gewirtz, D.A. Influence of the phosphodiesterase-5 inhibitor, sildenafil, on sensitivity to chemotherapy in breast tumor cells. Breast Cancer Res. Treat. 2010, 124, 349–360. [Google Scholar] [CrossRef]
- Marques, J.G.; Gaspar, V.; Markl, D.; Costa, E.; Gallardo, E.; Correia, I.J. Co-delivery of Sildenafil (Viagra®) and Crizotinib for Synergistic and Improved Anti-tumoral Therapy. Pharm. Res. 2014, 31, 2516–2528. [Google Scholar] [CrossRef]
- Greish, K.; Fateel, M.; Abdelghany, S.; Rachel, N.; Alimoradi, H.; Bakhiet, M.; Alsaie, A. Sildenafil citrate improves the delivery and anticancer activity of doxorubicin formulations in a mouse model of breast cancer. J. Drug Target. 2017, 26, 610–615. [Google Scholar] [CrossRef]
- Pourghadamyari, H.; Hassanvand, F.; Mohammadi, T.; Ayoubzadeh, N.; Tavakoli, A.; Hassanzadeh, N.; Sanikhani, N.S.; Azimi, A.I.; Mirzaei, H.R.; Khodamoradi, M.; et al. Sildenafil enhances cisplatin-induced apoptosis in human breast adenocarcinoma cells. J. Cancer Res. Ther. 2020, 16, 1412–1418. [Google Scholar] [CrossRef]
- El-Naa, M.M.; Othman, M.; Younes, S. Sildenafil potentiates the antitumor activity of cisplatin by induction of apoptosis and inhibition of proliferation and angiogenesis. Drug Des. Dev. Ther. 2016, 10, 3661–3672. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Zhong, X.; Kang, Z.; Chen, R. PDE-7 Inhibitor BRL-50481 Reduces Neurodegeneration and Long-Term Memory Deficits in Mice Following Sevoflurane Exposure. ACS Chem. Neurosci. 2020, 11, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Mitchell, C.; Mayton, E.; Hoke, N.N.; Salloum, F.; Park, M.A.; Qureshi, I.; Lee, R.; Dent, P.; et al. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 18202–18207. [Google Scholar] [CrossRef] [PubMed]
- Poklepovic, A.; Qu, Y.; Dickinson, M.; Kontos, M.C.; Kmieciak, M.; Schultz, E.; Bandopadhyay, D.; Deng, X.; Kukreja, R.C. Randomized study of doxorubicin-based chemotherapy regimens, with and without sildenafil, with analysis of intermediate cardiac markers. Cardio-Oncology 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Webb, T.; Carter, J.; Roberts, J.L.; Poklepovic, A.; McGuire, W.P.; Booth, L.; Dent, P. Celecoxib enhances [sorafenib + sildenafil] lethality in cancer cells and reverts platinum chemotherapy resistance. Cancer Biol. Ther. 2015, 16, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Cruickshanks, N.; Tavallai, S.; Webb, T.; Samuel, P.; Conley, A.; Binion, B.; Young, H.F.; Poklepovic, A.; et al. PDE5 inhibitors enhance celecoxib killing in multiple tumor types. J. Cell. Physiol. 2014, 230, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Klutzny, S.; Anurin, A.; Nicke, B.; Regan, J.; Lange, M.; Schulze, L.; Parczyk, K.; Steigemann, P. PDE5 inhibition eliminates cancer stem cells via induction of PKA signaling. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.; Becher, A.; Diepold, K.; Schmid, E.; Fehn, A.; Brunner, C.; Rouhi, A.; Chiosis, G.; Cronauer, M.; et al. Sildenafil triggers tumor lethality through altered expression of HSP90 and degradation of PKD2. Carcinogenesis 2020, 41, 1421–1431. [Google Scholar] [CrossRef]
- Mehrotra, N.; Gupta, M.; Kovar, A.; Meibohm, B. The role of pharmacokinetics and pharmacodynamics in phosphodiesterase-5 inhibitor therapy. Int. J. Impot. Res. 2006, 19, 253–264. [Google Scholar] [CrossRef]
- Pantziarka, P.; Sukhatme, V.; Crispino, S.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)—selective PDE5 inhibitors as. Ecancermedicalscience 2018, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Forgue, S.T.; Patterson, B.E.; Bedding, A.W.; Payne, C.D.; Phillips, D.L.; Wrishko, R.E.; Mitchell, M.I. Tadalafil pharmacokinetics in healthy subjects. Br. J. Clin. Pharmacol. 2005, 61, 280–288. [Google Scholar] [CrossRef]
- Huang, W.; Sundquist, J.; Sundquist, K.; Ji, J. Phosphodiesterase-5 inhibitors use and risk for mortality and metastases among male patients with colorectal cancer. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Tang, H.; Wu, W.; Fu, S.; Zhai, S.; Song, Y.; Han, J. Phosphodiesterase type 5 inhibitors and risk of melanoma: A meta-analysis. J. Am. Acad. Dermatol. 2017, 77, 480–488.e9. [Google Scholar] [CrossRef][Green Version]
- Browning, D.D. The enduring promise of phosphodiesterase 5 inhibitors for colon cancer prevention. Transl. Gastroenterol. Hepatol. 2019, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Islam, B.N.; Browning, D.D. Phosphodiesterase-5 inhibitors for colon cancer chemoprevention. Aging 2018, 10, 2216–2217. [Google Scholar] [CrossRef] [PubMed]
- Islam, B.N.; Sharman, S.K.; Hou, Y.; Bridges, A.E.; Singh, N.; Kim, S.; Kolhe, R.; Trillo-Tinoco, J.; Rodriguez, P.C.; Berger, F.G.; et al. Sildenafil Suppresses Inflammation-Driven Colorectal Cancer in Mice. Cancer Prev. Res. 2017, 10, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.L.; Poklepovic, A.; Booth, L. Curcumin interacts with sildenafil to kill GI tumor cells via endoplasmic reticulum stress and reactive oxygen/ nitrogen species. Oncotarget 2017, 8, 99451–99469. [Google Scholar] [CrossRef] [PubMed]
- Chhonker, S.K.; Rawat, D.; Koiri, R.K. Protective and therapeutic effects of sildenafil and tadalafil on aflatoxin B1-induced hepatocellular carcinoma. Mol. Cell. Biochem. 2020, 476, 1195–1209. [Google Scholar] [CrossRef]
- Kong, D.; Jiang, Y.; Miao, X.; Wu, Z.; Liu, H.; Gong, W. Tadalafil enhances the therapeutic efficacy of BET inhibitors in hepatocellular carcinoma through activating Hippo pathway. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 166267. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Ma, C.; Heinrich, B.; Brown, Z.J.; Sandhu, M.; Zhang, Q.; Fu, Q.; Agdashian, D.; Rosato, U.; Korangy, F.; et al. Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in hepatocellular carcinoma. J. Hepatol. 2018, 70, 449–457. [Google Scholar] [CrossRef]
- Kumazoe, M.; Tsukamoto, S.; Lesnick, C.; Kay, N.E.; Yamada, K.; Shanafelt, T.D.; Tachibana, H. Vardenafil, a clinically available phosphodiesterase inhibitor, potentiates the killing effect of EGCG on CLL cells. Br. J. Haematol. 2014, 168, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Sarfati, M.; Mateo, V.; Baudet, S.; Rubio, M.; Fernandez, C.; Davi, F.; Binet, J.-L.; Delic, J.; Merle-Béral, H. Sildenafil and vardenafil, types 5 and 6 phosphodiesterase inhibitors, induce caspase-dependent apoptosis of B-chronic lymphocytic leukemia cells. Blood 2003, 101, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Tseng, Y.-L.; Leu, W.-J.; Du, C.-M.; Jiang, Y.-H.; Hsu, L.-C.; Hsu, J.-L.; Hou, D.-R.; Guh, J.-H. Discovery of Novel Agents on Spindle Assembly Checkpoint to Sensitize Vinorelbine-Induced Mitotic Cell Death Against Human Non-Small Cell Lung Cancers. Int. J. Mol. Sci. 2020, 21, 5608. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Y.; Zhang, W.; Li, C.; Zhu, Y.; Wang, S. Tadalafil Reverses the Effect of Three-Dimensional Cell Culture System on Stem Cell Features in A549 and SK-MES-1. DNA Cell Biol. 2021, 40, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, W.S.; Park, C. Sildenafil prevents HDACi-induced Epstein-Barr virus reactivation through the PKG pathway in NK/T cell lymphoma; potential implications for HDACi-mediated fatal complications. Antivir. Res. 2021, 189, 105063. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, W.; Zhang, Q.; Liu, Y.; Qiao, X.; Meng, K.; Mao, Y. Phosphodiesterase type 5 inhibitor Tadalafil increases Rituximab treatment efficacy in a mouse brain lymphoma model. J. Neuro-Oncol. 2014, 122, 35–42. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Dent, P. [pemetrexed + sildenafil], via autophagy-dependent HDAC downregulation, enhances the immunotherapy response of NSCLC cells. Cancer Biol. Ther. 2017, 18, 705–714. [Google Scholar] [CrossRef]
- Bimonte, V.M.; Marampon, F.; Antonioni, A.; Fittipaldi, S.; Ferretti, E.; Pestell, R.G.; Curreli, M.; Lenzi, A.; Vitale, G.; Brunetti, A.; et al. Phosphodiesterase Type-5 Inhibitor Tadalafil Modulates Steroid Hormones Signaling in a Prostate Cancer Cell Line. Int. J. Mol. Sci. 2021, 22, 754. [Google Scholar] [CrossRef]
- Danley, K.T.; Tan, A.; Catalona, W.J.; Leikin, R.; Helenowski, I.; Jovanovic, B.; Gurley, M.; Kuzel, T.M. The association of phosphodiesterase-5 inhibitors with the biochemical recurrence-free and overall survival of patients with prostate cancer following radical prostatectomy. Urol. Oncol. Semin. Orig. Investig. 2021. [Google Scholar] [CrossRef]
- Haseltine, J.M.; Hopkins, M.; Schofield, E.; Kollmeier, M.A.; Shasha, D.; Gorovets, D.; McBride, S.M.; Mulhall, J.P.; Zelefsky, M.J. Sildenafil Citrate and Risk of Biochemical Recurrence in Prostate Cancer Patients Treated With Radiation Therapy: Post-Hoc Analysis of a Randomized Controlled Trial. J. Sex. Med. 2021, 18, 1467–1472. [Google Scholar] [CrossRef]
- Hsu, J.-L.; Leu, W.-J.; Hsu, L.-C.; Ho, C.-H.; Liu, S.-P.; Guh, J.-H. Phosphodiesterase Type 5 Inhibitors Synergize Vincristine in Killing Castration-Resistant Prostate Cancer Through Amplifying Mitotic Arrest Signaling. Front. Oncol. 2020, 10, 1274. [Google Scholar] [CrossRef]
- Muniyan, S.; Rachagani, S.; Parte, S.; Halder, S.; Seshacharyulu, P.; Kshirsagar, P.; Siddiqui, J.A.; Vengoji, R.; Rauth, S.; Islam, R.; et al. Sildenafil potentiates the therapeutic efficacy of docetaxel in advanced prostate cancer by stimulating NO-cGMP signaling. Clin. Cancer Res. 2020, 26, 5720–5734. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef] [PubMed]
- Tzoumas, N.; Farrah, T.E.; Dhaun, N.; Webb, D.J. Established and emerging therapeutic uses of PDE type 5 inhibitors in cardiovascular disease. Br. J. Pharmacol. 2019, 177, 5467–5488. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Tumor Model | Co-Treatment | Effect | Mechanism of Action | Reference |
|---|---|---|---|---|
| Tadalafil (50 μM) + Sulindac Sulfide (COX-inhibitor) (100 μM) | Tumor growth inhibition | PKG-mediated degradation of nuclear β-catenin and suppression of survivin and vasodilator-stimulated phosphoprotein (VASP). | [74] |
| Sildenafil (0.5 μM) + Celecoxib (1 μM) + FTY720 (~50 nM) | Synergistic cytotoxic activity | Suppression of anti-aptotic ERK, AKT, p70 S6K, mTOR, NFκB, activation of JNK, p38 MAPK, ceramide-mediated CD95 activation. | [94] |
|
| Synergistic cytotoxic activity (reduced tumor volume) and reduction of doxorubicin-mediated cardiotoxic effects | Increased EPR-based anticancer drug delivery | [88] |
|
| Suppression of cancer stem cells (CSC) subpopulation | Differentiation of CSCs to non-stem-like tumor cells upon cGMP-mediated cAMP/PKA signaling | [96] |
|
| Synergistic cytotoxic activity | Reduced angiogenesis and proliferation, increased apoptosis: decrease of VEGF, angiogenin and tumor necrosis factor-alpha and Ki-67, increase of caspase-3 expression | [90] |
| Sildenafil (50, 100 μM) plus cisplatin (15 μM and 22 μM) | Synergistic cytotoxic activity | Tumor cell sensitization to cisplatin, increase of ROS accumulation into the extracellular environment, increased apoptosis via activation of caspase 3 and BAX, and decreased BCL2) | [89] |
| Sildenafil (40.33 μM)/Crizotinib (55.25 μM)- dual-loaded PEG-PLA micelles | Decrease in cell viability | Caspase-3 and caspase-7 activation | [87] |
| Sildenafil (10–50 µM) + HSP90 inhibitor, PU-H71 (50 nM) | Synergistic cytotoxic activity | Decreased HSP90 expression, degradation of PKD2 and increased apoptosis | [97] |
| Cancer Type | Role of PDE5i | References | PDE Inhibitor | Clinical Trial.Gov |
|---|---|---|---|---|
| Breast |
| [51,64,74,88,89] | Sildenafil (PDE5i) | NCT01375699 |
| Pentoxifylline (non-specific PDEi) | NCT03916068 NCT02898376 NCT00022204 NCT00188669 NCT01082003 NCT00583700 | |||
| Colon |
| [103,104,105,106] | Udenafil (PDE5i) | NCT00607282 |
| Hepatoma |
| [107,108,109] | Tadalafil (PDE5i) | NCT03785210 |
| Pentoxifylline (non-specific PDEi) | NCT01149304 | |||
| Leukemia |
| [110,111] | Sildenafil, Tadalafil, Vardenafil (PDE5i) | NCT01117142 |
| Pentoxifylline (non-specific PDEi) | NCT02451774 | |||
| Theophylline (PDE3i-PDE4i) | NCT00003808 | |||
| Lung |
| [51,112,113] | Papaverine hydrochloride (PDE10i) Sildenafil (PDE5i) | NCT03824327 |
| Pentoxifylline (non-specific PDEi) | NCT00752115 | |||
| Theophylline (PDE3i-PDE4i) Tadalafil (PDE5i) | NCT01871454 | |||
| Caffeine (non-specific PDEi) | NCT01799161 NCT02080078 NCT04069936 NCT01402089 | |||
| Lymphoma |
| [114,115] | Sildenafil, Tadalafil, Vardenafil (PDE5i) | NCT01117142 |
| Pentoxifylline (non-specific PDEi) | NCT02451774 | |||
| Ovarian |
| [94,116] | Caffeine (non-specific PDEi) | NCT04718740 |
| Dipyridamole (PDE3i) | NCT00002487 | |||
| Prostate |
| [117,118,119,120,121] | Sildenafil (PDE5i): | NCT00906269 NCT00142506 NCT01996852 NCT01054001 |
| Tadalafil (PDE5i): | NCT00931528 NCT00215631 NCT00122499 | |||
| Udenafil (PDE5i): | NCT03142542 | |||
| Papaverine hydrochloride (PDE10i): | NCT00080808 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Iorio, P.; Ronci, M.; Giuliani, P.; Caciagli, F.; Ciccarelli, R.; Caruso, V.; Beggiato, S.; Zuccarini, M. Pros and Cons of Pharmacological Manipulation of cGMP-PDEs in the Prevention and Treatment of Breast Cancer. Int. J. Mol. Sci. 2022, 23, 262. https://doi.org/10.3390/ijms23010262
Di Iorio P, Ronci M, Giuliani P, Caciagli F, Ciccarelli R, Caruso V, Beggiato S, Zuccarini M. Pros and Cons of Pharmacological Manipulation of cGMP-PDEs in the Prevention and Treatment of Breast Cancer. International Journal of Molecular Sciences. 2022; 23(1):262. https://doi.org/10.3390/ijms23010262
Chicago/Turabian StyleDi Iorio, Patrizia, Maurizio Ronci, Patricia Giuliani, Francesco Caciagli, Renata Ciccarelli, Vanni Caruso, Sarah Beggiato, and Mariachiara Zuccarini. 2022. "Pros and Cons of Pharmacological Manipulation of cGMP-PDEs in the Prevention and Treatment of Breast Cancer" International Journal of Molecular Sciences 23, no. 1: 262. https://doi.org/10.3390/ijms23010262
APA StyleDi Iorio, P., Ronci, M., Giuliani, P., Caciagli, F., Ciccarelli, R., Caruso, V., Beggiato, S., & Zuccarini, M. (2022). Pros and Cons of Pharmacological Manipulation of cGMP-PDEs in the Prevention and Treatment of Breast Cancer. International Journal of Molecular Sciences, 23(1), 262. https://doi.org/10.3390/ijms23010262