
Emerging Roles of Calcium Signaling in the Development of Non-Alcoholic Fatty Liver Disease

 ,
,
Abstract
1. Introduction
2. Alterations in Ca2+ Homeostasis and Organelle Dysfunction in Development of NAFLD
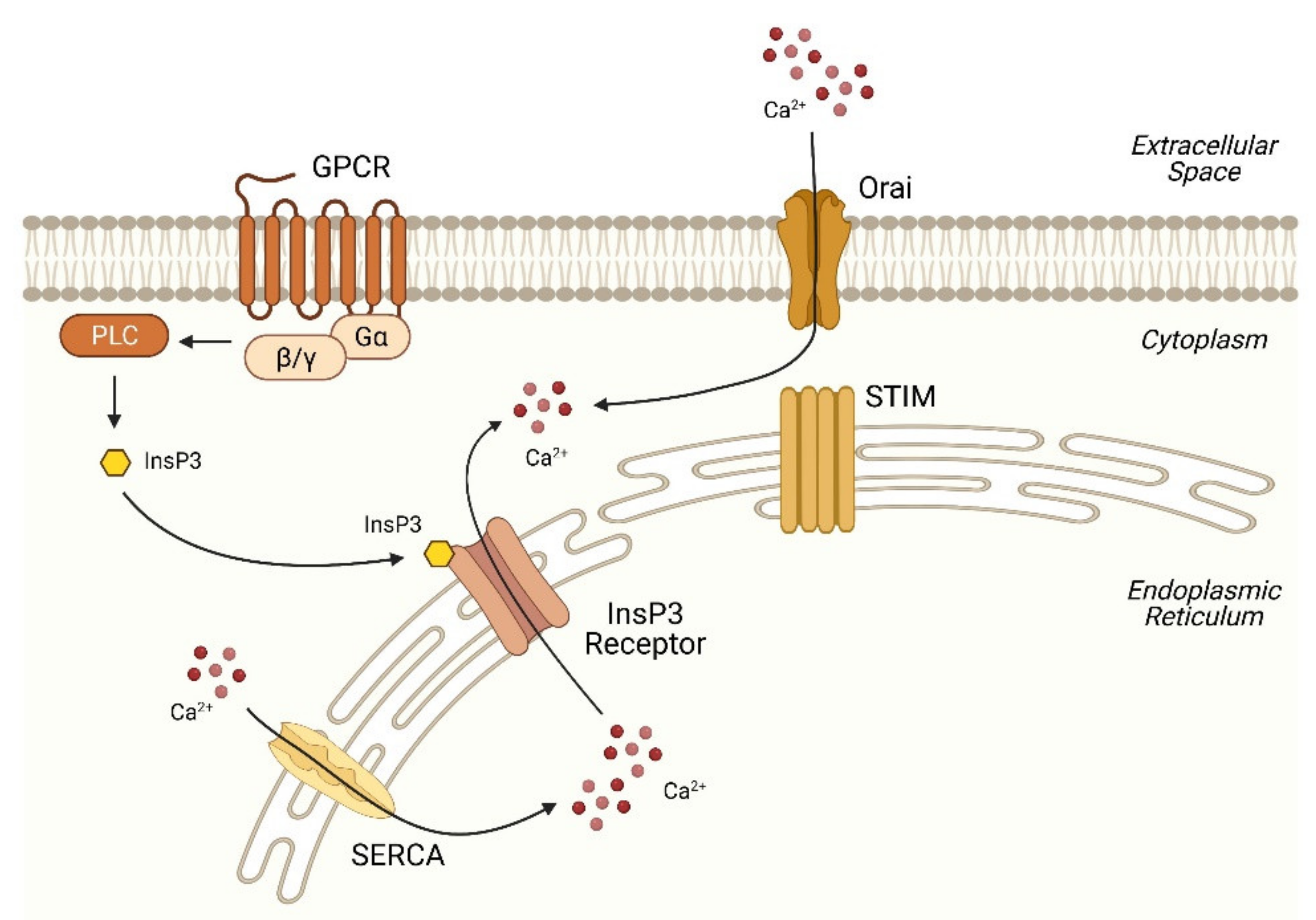
2.1. Basic Machinery of Ca2+ Signaling
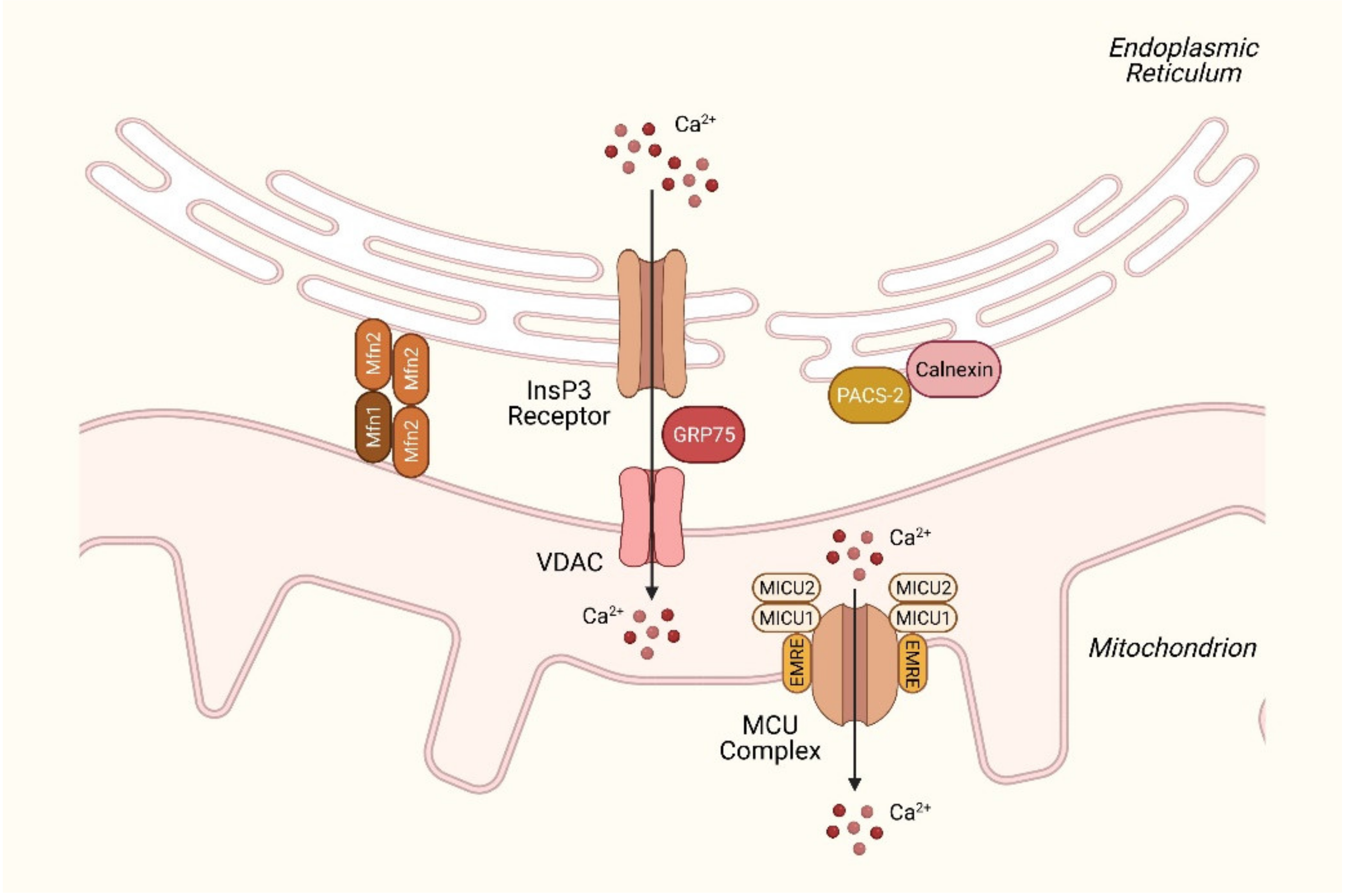
2.2. Disruption of Ca2+ Homeostasis in the ER in NAFLD
2.3. Disturbances in Mitochondrial Ca2+ Homeostasis in NAFLD
2.4. Dysregulation in Lysosomal Ca2+ Signaling in NAFLD
2.5. Aberrant Ca2+ Signaling in Hepatic Nonparenchymal Cells in NAFLD
3. Ca2+ Signaling as a Therapeutic Target for NAFLD
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Fossi, C.; Quattrini, S.; Guasti, L.; Pampaloni, B.; Gronchi, G.; Giusti, F.; Romagnoli, C.; Cianferotti, L.; Marcucci, G.; et al. Calcium Intake in Bone Health: A Focus on Calcium-Rich Mineral Waters. Nutrients 2018, 10, 1930. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca2+ handling in excitable cells in health and disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef]
- Amaya, M.J.; Nathanson, M.H. Calcium signaling in the liver. Compr. Physiol. 2013, 3, 515–539. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, T.; Yoshikawa, S.; Miyawaki, A.; Wada, K.; Maeda, N.; Mikoshiba, K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature 1989, 342, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Niinobe, M.; Mikoshiba, K. A cerebellar Purkinje cell marker P400 protein is an inositol 1,4,5-trisphosphate (InsP3) receptor protein. Purification and characterization of InsP3 receptor complex. EMBO J. 1990, 9, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef]
- Dupont, G.; Combettes, L.; Bird, G.S.; Putney, J.W. Calcium oscillations. Cold Spring Harb. Perspect. Biol. 2011, 3, a004226. [Google Scholar] [CrossRef] [PubMed]
- Westerlund, A.M.; Delemotte, L. Effect of Ca2+ on the promiscuous target-protein binding of calmodulin. PLoS Comput. Biol. 2018, 14, e1006072. [Google Scholar] [CrossRef]
- Burgoyne, R.D.; Helassa, N.; McCue, H.V.; Haynes, L.P. Calcium Sensors in Neuronal Function and Dysfunction. Cold Spring Harb. Perspect. Biol. 2019, 11, a035154. [Google Scholar] [CrossRef] [PubMed]
- Haiech, J.; Moreau, M.; Leclerc, C.; Kilhoffer, M.C. Facts and conjectures on calmodulin and its cousin proteins, parvalbumin and troponin C. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1046–1053. [Google Scholar] [CrossRef]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Calcium pumps: Why so many? Compr. Physiol. 2012, 2, 1045–1060. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Bugrim, A.E. Regulation of Ca2+ release by cAMP-dependent protein kinase. A mechanism for agonist-specific calcium signaling? Cell Calcium 1999, 25, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, G.; Goode, J.; Paz, J.C.; Ouyang, K.; Screaton, R.; Fischer, W.H.; Chen, J.; Tabas, I.; Montminy, M. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 2012, 485, 128–132. [Google Scholar] [CrossRef]
- Hansen, L.H.; Gromada, J.; Bouchelouche, P.; Whitmore, T.; Jelinek, L.; Kindsvogel, W.; Nishimura, E. Glucagon-mediated Ca2+ signaling in BHK cells expressing cloned human glucagon receptors. Am. J. Physiol. 1998, 274, C1552–C1562. [Google Scholar] [CrossRef] [PubMed]
- Hardie, R.C.; Muallem, S. Lipids in Ca2+ signalling—An introduction. Cell Calcium 2009, 45, 517–520. [Google Scholar] [CrossRef]
- Hisatsune, C.; Nakamura, K.; Kuroda, Y.; Nakamura, T.; Mikoshiba, K. Amplification of Ca2+ signaling by diacylglycerol-mediated inositol 1,4,5-trisphosphate production. J. Biol. Chem. 2005, 280, 11723–11730. [Google Scholar] [CrossRef] [PubMed]
- Galsgaard, K.D.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Wewer Albrechtsen, N.J. Glucagon Receptor Signaling and Lipid Metabolism. Front. Physiol. 2019, 10, 413. [Google Scholar] [CrossRef]
- Shi, Y.; Shu, Z.J.; Wang, H.; Barnes, J.L.; Yeh, C.K.; Ghosh, P.M.; Katz, M.S.; Kamat, A. Altered expression of hepatic beta-adrenergic receptors in aging rats: Implications for age-related metabolic dysfunction in liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R574–R583. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Takabe, K.; Liu, R.; Peng, K.; Wang, X.; Wang, Y.; Hait, N.C.; Wang, X.; Allegood, J.C.; Yamada, A.; et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology 2015, 61, 1216–1226. [Google Scholar] [CrossRef]
- Secor, J.D.; Fligor, S.C.; Tsikis, S.T.; Yu, L.J.; Puder, M. Free Fatty Acid Receptors as Mediators and Therapeutic Targets in Liver Disease. Front. Physiol. 2021, 12, 656441. [Google Scholar] [CrossRef]
- Arruda, A.P.; Pers, B.M.; Parlakgul, G.; Guney, E.; Goh, T.; Cagampan, E.; Lee, G.Y.; Goncalves, R.L.; Hotamisligil, G.S. Defective STIM-mediated store operated Ca2+ entry in hepatocytes leads to metabolic dysfunction in obesity. eLife 2017, 6, e29968. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Kretsinger, R.H. Structural and functional diversity of EF-hand proteins: Evolutionary perspectives. Protein Sci. 2017, 26, 1898–1920. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Zhang, B.; Li, M.; Zou, Y.; Guo, H.; Zhang, B.; Xia, C.; Zhang, H.; Yang, W.; Xu, C. NFkappaB/Orai1 Facilitates Endoplasmic Reticulum Stress by Oxidative Stress in the Pathogenesis of Non-alcoholic Fatty Liver Disease. Front. Cell Dev. Biol. 2019, 7, 202. [Google Scholar] [CrossRef]
- Wilson, C.H.; Ali, E.S.; Scrimgeour, N.; Martin, A.M.; Hua, J.; Tallis, G.A.; Rychkov, G.Y.; Barritt, G.J. Steatosis inhibits liver cell store-operated Ca2+ entry and reduces ER Ca2+ through a protein kinase C-dependent mechanism. Biochem. J. 2015, 466, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Park, S.W.; Zhou, Y.; Lee, J.; Lee, J.; Ozcan, U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 19320–19325. [Google Scholar] [CrossRef]
- Lai, S.; Li, Y.; Kuang, Y.; Cui, H.; Yang, Y.; Sun, W.; Liu, K.; Chen, D.; Yan, Q.; Wen, L. PKCdelta silencing alleviates saturated fatty acid induced ER stress by enhancing SERCA activity. Biosci. Rep. 2017, 37, BSR20170869. [Google Scholar] [CrossRef]
- Ogino, N.; Miyagawa, K.; Kusanaga, M.; Hayashi, T.; Minami, S.; Oe, S.; Honma, Y.; Harada, M. Involvement of sarco/endoplasmic reticulum calcium ATPase-mediated calcium flux in the protective effect of oleic acid against lipotoxicity in hepatocytes. Exp. Cell Res. 2019, 385, 111651. [Google Scholar] [CrossRef]
- Subramanian, M.; Metya, S.K.; Sadaf, S.; Kumar, S.; Schwudke, D.; Hasan, G. Altered lipid homeostasis in Drosophila InsP3 receptor mutants leads to obesity and hyperphagia. Dis. Models Mech. 2013, 6, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef]
- Prole, D.L.; Taylor, C.W. Structure and Function of IP3 Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035063. [Google Scholar] [CrossRef]
- Iwai, M.; Michikawa, T.; Bosanac, I.; Ikura, M.; Mikoshiba, K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2007, 282, 12755–12764. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Pusl, T.; O’Neill, A.F.; Dranoff, J.A.; Nathanson, M.H. The type II inositol 1,4,5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology 2002, 122, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.T.; Florentino, R.M.; Franca, A.; Lima Filho, A.C.; Dos Santos, M.L.; Fonseca, R.C.; Lemos, F.O.; Fonseca, M.C.; Kruglov, E.; Mennone, A.; et al. Expression of the type 3 InsP3 receptor is a final common event in the development of hepatocellular carcinoma. Gut 2019, 68, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Feriod, C.N.; Oliveira, A.G.; Guerra, M.T.; Nguyen, L.; Richards, K.M.; Jurczak, M.J.; Ruan, H.B.; Camporez, J.P.; Yang, X.; Shulman, G.I.; et al. Hepatic Inositol 1,4,5 Trisphosphate Receptor Type 1 Mediates Fatty Liver. Hepatol. Commun. 2017, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zheng, E.; Li, Y.; Li, Y.; Xia, J.; Ding, Q.; Hou, Z.; Ruan, X.Z.; Zhao, L.; Chen, Y. Src-mediated Tyr353 phosphorylation of IP3R1 promotes its stability and causes apoptosis in palmitic acid-treated hepatocytes. Exp. Cell Res. 2021, 399, 112438. [Google Scholar] [CrossRef]
- Feriod, C.N.; Nguyen, L.; Jurczak, M.J.; Kruglov, E.A.; Nathanson, M.H.; Shulman, G.I.; Bennett, A.M.; Ehrlich, B.E. Inositol 1,4,5-trisphosphate receptor type II (InsP3R-II) is reduced in obese mice, but metabolic homeostasis is preserved in mice lacking InsP3R-II. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1057–E1064. [Google Scholar] [CrossRef]
- Arruda, A.P.; Pers, B.M.; Parlakgul, G.; Guney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef]
- Khamphaya, T.; Chukijrungroat, N.; Saengsirisuwan, V.; Mitchell-Richards, K.A.; Robert, M.E.; Mennone, A.; Ananthanarayanan, M.; Nathanson, M.H.; Weerachayaphorn, J. Nonalcoholic fatty liver disease impairs expression of the type II inositol 1,4,5-trisphosphate receptor. Hepatology 2018, 67, 560–574. [Google Scholar] [CrossRef]
- Townsend, L.K.; Brunetta, H.S.; Mori, M.A.S. Mitochondria-associated ER membranes in glucose homeostasis and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E1053–E1060. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell. Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; De, S.; Meir, A. The Mitochondrial Voltage-Dependent Anion Channel 1, Ca2+ Transport, Apoptosis, and Their Regulation. Front. Oncol. 2017, 7, 60. [Google Scholar] [CrossRef]
- Bononi, A.; Missiroli, S.; Poletti, F.; Suski, J.M.; Agnoletto, C.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Patergnani, S.; et al. Mitochondria-associated membranes (MAMs) as hotspot Ca2+ signaling units. Adv. Exp. Med. Biol. 2012, 740, 411–437. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernandez-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef]
- Hernandez-Alvarez, M.I.; Sebastian, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernandez, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.L.; Zhao, Y.; Koonen, D.P.; Sletten, T.; Su, B.; Lingrell, S.; Cao, G.; Peake, D.A.; Kuo, M.S.; Proctor, S.D.; et al. Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem. 2010, 285, 22403–22413. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; de Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef]
- Murphy, E.; Pan, X.; Nguyen, T.; Liu, J.; Holmstrom, K.M.; Finkel, T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 2014, 449, 384–385. [Google Scholar] [CrossRef]
- Tomar, D.; Jana, F.; Dong, Z.; Quinn, W.J., 3rd; Jadiya, P.; Breves, S.L.; Daw, C.C.; Srikantan, S.; Shanmughapriya, S.; Nemani, N.; et al. Blockade of MCU-Mediated Ca2+ Uptake Perturbs Lipid Metabolism via PP4-Dependent AMPK Dephosphorylation. Cell Rep. 2019, 26, 3709–3725.e7. [Google Scholar] [CrossRef]
- Antony, A.N.; Paillard, M.; Moffat, C.; Juskeviciute, E.; Correnti, J.; Bolon, B.; Rubin, E.; Csordas, G.; Seifert, E.L.; Hoek, J.B.; et al. MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat. Commun. 2016, 7, 10955. [Google Scholar] [CrossRef]
- Hesketh, G.G.; Wartosch, L.; Davis, L.J.; Bright, N.A.; Luzio, J.P. The Lysosome and Intracellular Signalling. In Progress in Molecular and Subcellular Biology; Springer: Berlin/Heidelberg, Germany, 2018; Volume 57, pp. 151–180. [Google Scholar] [CrossRef]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.; Griffiths, G.M. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef]
- Marchant, J.S.; Patel, S. Two-pore channels at the intersection of endolysosomal membrane traffic. Biochem. Soc. Trans. 2015, 43, 434–441. [Google Scholar] [CrossRef]
- Brailoiu, E.; Hooper, R.; Cai, X.; Brailoiu, G.C.; Keebler, M.V.; Dun, N.J.; Marchant, J.S.; Patel, S. An ancestral deuterostome family of two-pore channels mediates nicotinic acid adenine dinucleotide phosphate-dependent calcium release from acidic organelles. J. Biol. Chem. 2010, 285, 2897–2901. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef]
- Gerndt, S.; Chen, C.C.; Chao, Y.K.; Yuan, Y.; Burgstaller, S.; Scotto Rosato, A.; Krogsaeter, E.; Urban, N.; Jacob, K.; Nguyen, O.N.P.; et al. Agonist-mediated switching of ion selectivity in TPC2 differentially promotes lysosomal function. eLife 2020, 9, e54712. [Google Scholar] [CrossRef]
- Gunaratne, G.S.; Brailoiu, E.; He, S.; Unterwald, E.M.; Patel, S.; Slama, J.T.; Walseth, T.F.; Marchant, J.S. Essential requirement for JPT2 in NAADP-evoked Ca2+ signaling. Sci. Signal. 2021, 14, eabd5605. [Google Scholar] [CrossRef]
- Roggenkamp, H.G.; Khansahib, I.; Hernandez, C.L.; Zhang, Y.; Lodygin, D.; Kruger, A.; Gu, F.; Mockl, F.; Lohndorf, A.; Wolters, V.; et al. HN1L/JPT2: A signaling protein that connects NAADP generation to Ca2+ microdomain formation. Sci. Signal. 2021, 14, eabd5647. [Google Scholar] [CrossRef] [PubMed]
- Klein-Szanto, A.J.P.; Bassi, D.E. Keep recycling going: New approaches to reduce LDL-C. Biochem. Pharmacol. 2019, 164, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Holdt, L.M.; Chen, C.C.; Hassan, S.; Muller, C.; Jors, S.; Cuny, H.; Kissing, S.; Schroder, B.; Butz, E.; et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat. Commun. 2014, 5, 4699. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef]
- Scotto Rosato, A.; Montefusco, S.; Soldati, C.; di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat. Commun. 2019, 10, 5630. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; McNiven, M.A. Lipid Droplet Formation and Lipophagy in Fatty Liver Disease. Semin. Liver Dis. 2019, 39, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.; Cuk, M.; Patel, B.; Lian, J.; Ouimet, M.; Kaufmann, U.; Yang, J.; Horvath, R.; Hornig-Do, H.T.; Chrzanowska-Lightowlers, Z.M.; et al. Store-Operated Ca2+ Entry Controls Induction of Lipolysis and the Transcriptional Reprogramming to Lipid Metabolism. Cell Metab. 2017, 25, 698–712. [Google Scholar] [CrossRef]
- Chen, J.; Deng, X.; Liu, Y.; Tan, Q.; Huang, G.; Che, Q.; Guo, J.; Su, Z. Kupffer Cells in Non-alcoholic Fatty Liver Disease: Friend or Foe? Int. J. Biol. Sci. 2020, 16, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, X.J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef]
- Peters, K.M.; Wilson, R.B.; Borradaile, N.M. Non-parenchymal hepatic cell lipotoxicity and the coordinated progression of non-alcoholic fatty liver disease and atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ye, C.; Cheng, Q.; Zhang, X.; Yao, L.; Li, Q.; Huang, J.; Liu, Y.; Zou, Z.; Wang, H.; et al. Macrophage Raptor Deficiency-Induced Lysosome Dysfunction Exacerbates Nonalcoholic Steatohepatitis. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Park, K.S.; Sin, P.J.; Lee, D.H.; Cha, S.K.; Kim, M.J.; Kim, N.H.; Baik, S.K.; Jeong, S.W.; Kong, I.D. Switching-on of serotonergic calcium signaling in activated hepatic stellate cells. World J. Gastroenterol. 2011, 17, 164–173. [Google Scholar] [CrossRef]
- Bataller, R.; Nicolas, J.M.; Gines, P.; Esteve, A.; Nieves Gorbig, M.; Garcia-Ramallo, E.; Pinzani, M.; Ros, J.; Jimenez, W.; Thomas, A.P.; et al. Arginine vasopressin induces contraction and stimulates growth of cultured human hepatic stellate cells. Gastroenterology 1997, 113, 615–624. [Google Scholar] [CrossRef]
- Kruglov, E.A.; Correa, P.R.; Arora, G.; Yu, J.; Nathanson, M.H.; Dranoff, J.A. Molecular basis for calcium signaling in hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G975–G982. [Google Scholar] [CrossRef]
- Soliman, E.M.; Rodrigues, M.A.; Gomes, D.A.; Sheung, N.; Yu, J.; Amaya, M.J.; Nathanson, M.H.; Dranoff, J.A. Intracellular calcium signals regulate growth of hepatic stellate cells via specific effects on cell cycle progression. Cell Calcium 2009, 45, 284–292. [Google Scholar] [CrossRef]
- Huang, Y.; Li, X.; Wang, Y.; Wang, H.; Huang, C.; Li, J. Endoplasmic reticulum stress-induced hepatic stellate cell apoptosis through calcium-mediated JNK/P38 MAPK and Calpain/Caspase-12 pathways. Mol. Cell Biochem. 2014, 394, 1–12. [Google Scholar] [CrossRef]
- Kang, S.; Dahl, R.; Hsieh, W.; Shin, A.; Zsebo, K.M.; Buettner, C.; Hajjar, R.J.; Lebeche, D. Small Molecular Allosteric Activator of the Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA) Attenuates Diabetes and Metabolic Disorders. J. Biol. Chem. 2016, 291, 5185–5198. [Google Scholar] [CrossRef]
- Park, H.W.; Lee, J.H. Calcium channel blockers as potential therapeutics for obesity-associated autophagy defects and fatty liver pathologies. Autophagy 2014, 10, 2385–2386. [Google Scholar] [CrossRef]
- Park, H.W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.J.; Kim, J.S.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef]
- Ali, E.S.; Girard, D.; Petrovsky, N. Impaired Ca2+ signaling due to hepatic steatosis mediates hepatic insulin resistance in Alstrom syndrome mice that is reversed by GLP-1 analog treatment. Am. J. Physiol. Cell Physiol. 2021, 321, C187–C198. [Google Scholar] [CrossRef]
- Ali, E.S.; Hua, J.; Wilson, C.H.; Tallis, G.A.; Zhou, F.H.; Rychkov, G.Y.; Barritt, G.J. The glucagon-like peptide-1 analogue exendin-4 reverses impaired intracellular Ca2+ signalling in steatotic hepatocytes. Biochim. Biophys. Acta 2016, 1863, 2135–2146. [Google Scholar] [CrossRef]
- Fang, Y.; Ji, L.; Zhu, C.; Xiao, Y.; Zhang, J.; Lu, J.; Yin, J.; Wei, L. Liraglutide Alleviates Hepatic Steatosis by Activating the TFEB-Regulated Autophagy-Lysosomal Pathway. Front. Cell Dev. Biol. 2020, 8, 602574. [Google Scholar] [CrossRef]
- Jung, T.W.; Kim, H.C.; Abd El-Aty, A.M.; Jeong, J.H. Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway. J. Biol. Chem. 2018, 293, 3981–3988. [Google Scholar] [CrossRef]
- Laiglesia, L.M.; Lorente-Cebrian, S.; Martinez-Fernandez, L.; Sainz, N.; Prieto-Hontoria, P.L.; Burrell, M.A.; Rodriguez-Ortigosa, C.M.; Martinez, J.A.; Moreno-Aliaga, M.J. Maresin 1 mitigates liver steatosis in ob/ob and diet-induced obese mice. Int. J. Obes. 2018, 42, 572–579. [Google Scholar] [CrossRef]
- Ouyang, Z.; Li, W.; Meng, Q.; Zhang, Q.; Wang, X.; Elgehama, A.; Wu, X.; Shen, Y.; Sun, Y.; Wu, X.; et al. A natural compound jaceosidin ameliorates endoplasmic reticulum stress and insulin resistance via upregulation of SERCA2b. Biomed. Pharmacother. 2017, 89, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Liu, W.X.; Durnaoglu, S.; Lee, S.K.; Lian, J.; Lehner, R.; Ahnn, J.; Agellon, L.B.; Michalak, M. Loss of Calreticulin Uncovers a Critical Role for Calcium in Regulating Cellular Lipid Homeostasis. Sci. Rep. 2017, 7, 5941. [Google Scholar] [CrossRef]
- Myhill, N.; Lynes, E.M.; Nanji, J.A.; Blagoveshchenskaya, A.D.; Fei, H.; Carmine Simmen, K.; Cooper, T.J.; Thomas, G.; Simmen, T. The subcellular distribution of calnexin is mediated by PACS-2. Mol. Biol. Cell 2008, 19, 2777–2788. [Google Scholar] [CrossRef]
- Gutierrez, T.; Qi, H.; Yap, M.C.; Tahbaz, N.; Milburn, L.A.; Lucchinetti, E.; Lou, P.H.; Zaugg, M.; LaPointe, P.G.; Mercier, P.; et al. The ER chaperone calnexin controls mitochondrial positioning and respiration. Sci. Signal. 2020, 13, eaax6660. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name | Subcellular Localization | Function | Involvement in NAFLD |
|---|---|---|---|
| InsP3R1 | ER membrane | Ca2+ release from ER to cytoplasm | Increased expression is found in NASH patients; hepatic knockout mice are more resistant to lipid accumulation [46]. |
| InsP3R2 | ER membrane | Ca2+ release from ER to cytoplasm | InsP3R2 is down-regulated in NAFLD mouse models and NASH patients; null mice have no apparent lipid metabolic phenotype [51]. |
| STIM1 | ER membrane | Ca2+ sensor that drives Ca2+ entry from extracellular space | STIM1/2 inducible knockout mice have reduced lipolysis but increased lipophagy [95]. |
| Orai | Plasma membrane | Ca2+ entry from extracellular space to cytoplasm | Orai1 is moderately increased in hepatic steatosis [34]; loss-of-function mutations impairs lipolysis but increases lipophagy [95]. |
| SERCA2b | ER membrane | Ca2+ uptake from cytoplasm to ER | Impaired activity is associated with ER stress [36]; overexpression alleviates hepatic steatosis [37]. |
| VDAC | Mitochondrial outer membrane | Entry of Ca2+ and other metabolites | |
| Mfn2 | ER–mitochondria contacts | Forms dimer with Mfn1; ER–mitochondria tethering | Down-regulation is observed in NAFLD mouse models and NASH patients [59]; hepatic ablation results in ER stress and impaired insulin signaling [60]. |
| MCU | Mitochondrial inner membrane | Ca2+ uptake to the mitochondria | Hepatic ablation of MCU delays cytoplasmic Ca2+ clearance and promotes lipid accumulation [71]. |
| TPC2 | Lysosome | Ca2+ release from the lysosome | TPC2-deficient mice are more susceptible to NAFLD when fed with a high-cholesterol diet [84]. |
| Calreticulin | ER lumen | Ca2+ buffering; chaperone | Association with NAFLD is not determined; knockout cells have altered membrane fluidity and ER stress levels [116]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-C.; Hsu, L.-W.; Chen, K.-D.; Chiu, K.-W.; Chen, C.-L.; Huang, K.-T. Emerging Roles of Calcium Signaling in the Development of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 256. https://doi.org/10.3390/ijms23010256
Chen C-C, Hsu L-W, Chen K-D, Chiu K-W, Chen C-L, Huang K-T. Emerging Roles of Calcium Signaling in the Development of Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2022; 23(1):256. https://doi.org/10.3390/ijms23010256
Chicago/Turabian StyleChen, Chien-Chih, Li-Wen Hsu, Kuang-Den Chen, King-Wah Chiu, Chao-Long Chen, and Kuang-Tzu Huang. 2022. "Emerging Roles of Calcium Signaling in the Development of Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 23, no. 1: 256. https://doi.org/10.3390/ijms23010256
APA StyleChen, C.-C., Hsu, L.-W., Chen, K.-D., Chiu, K.-W., Chen, C.-L., & Huang, K.-T. (2022). Emerging Roles of Calcium Signaling in the Development of Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 23(1), 256. https://doi.org/10.3390/ijms23010256