
The Hedgehog Signaling Pathway in Idiopathic Pulmonary Fibrosis: Resurrection Time


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hh Signal Transduction
2.1. Element of Hh Signal Transduction
2.1.1. Hh Ligand
2.1.2. Ptch
2.1.3. Smo
2.1.4. Gli
2.1.5. Sufu
2.1.6. Kif7
2.2. Hh Signaling Pathway
2.2.1. Canonical Pathway
2.2.2. Non-Canonical Pathway
3. Hh Signaling in Lung Development
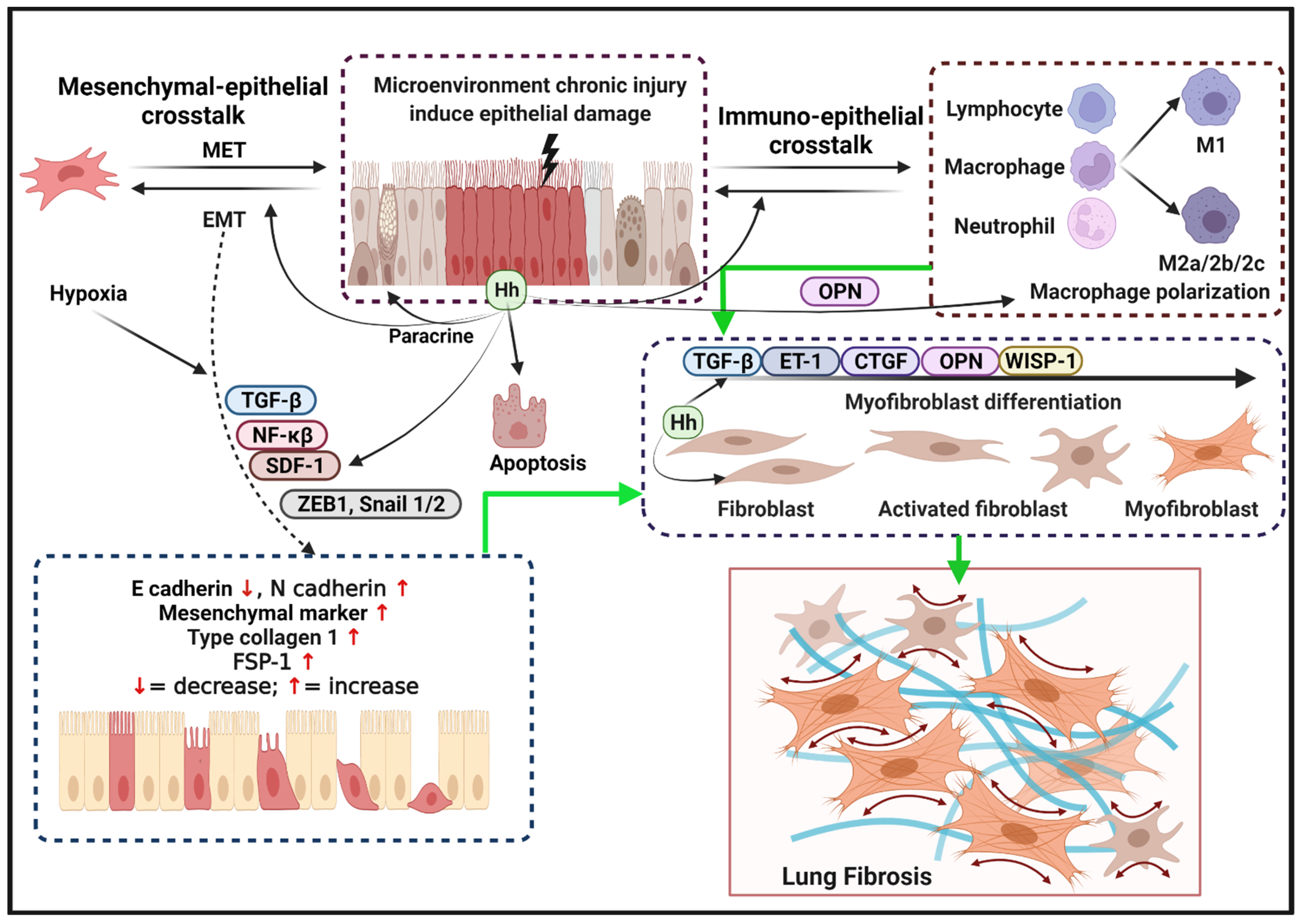
4. Reactivation of the Hh Pathway in IPF
4.1. Hh Signaling Regulates Lung Epithelial Repairment
4.2. Hh Signaling Controls Fibroblast Activation and Myofibroblast Differentiation
4.3. Hh Signaling Regulates EMT
4.4. Hh Signaling Regulates Macrophage Activation and Polarization
5. Targeting the Hh Signaling Pathway as Therapy for Pulmonary Fibrosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nair, A.; Chauhan, P.; Saha, B.; Kubatzky, K.F. Conceptual Evolution of Cell Signaling. Int. J. Mol. Sci. 2019, 20, 3292. [Google Scholar] [CrossRef] [Green Version]
- Di-Bella, J.P.; Colman-Lerner, A.; Ventura, A.C. Properties of cell signaling pathways and gene expression systems operating far from steady-state. Sci. Rep. 2018, 8, 1–14. [Google Scholar]
- Handly, L.N.; Yao, J.; Wollman, R. Signal Transduction at the Single-Cell Level: Approaches to Study the Dynamic Nature of Signaling Networks. J. Mol. Biol. 2016, 428, 3669–3682. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.E.; Chiang, C. Hedgehog secretion and signal transduction in vertebrates. J. Biol. Chem. 2012, 287, 17905–17913. [Google Scholar] [CrossRef] [Green Version]
- Groves, I.; Placzek, M.; Fletcher, A.G. Of mitogens and morphogens: Modelling Sonic Hedgehog mechanisms in vertebrate development. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190660. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [Green Version]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Domenech, M.; Bjerregaard, R.; Bushman, W.; Beebe, D.J. Hedgehog signaling in myofibroblasts directly promotes prostate tumor cell growth. Integr. Biol. 2012, 4, 142–152. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, J.; Zeng, X.; Nie, M.; Luan, J.; Wang, Y.; Ju, D.; Yin, K. Hedgehog signaling in gastrointestinal carcinogenesis and the gastrointestinal tumor microenvironment. Acta Pharm. Sin. B 2021, 11, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Kugler, M.C.; Joyner, A.L.; Loomis, C.A.; Munger, J.S. Sonic hedgehog signaling in the lung. From development to disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 1–13. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; Cao, H.; Chen, L.; Hou, J.; Xiang, Z.; Hu, K.; Han, X. The hedgehog and Wnt/β-catenin system machinery mediate myofibroblast differentiation of LR-MSCs in pulmonary fibrogenesis. Cell Death Dis. 2018, 9, 639. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Wang, Y.; Xie, J. The Hedgehog pathway: Role in cell differentiation, polarity and proliferation. Arch. Toxicol. 2015, 89, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Bürglin, T.R. The Hedgehog protein family. Genome Biol. 2008, 9, 241. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog signaling. J. Cell Sci. 2007, 120, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Buglino, J.A.; Resh, M.D. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J. Biol. Chem. 2008, 283, 22076–22088. [Google Scholar] [CrossRef] [Green Version]
- Nieuwenhuis, E.; Hui, C.C. Hedgehog signaling and congenital malformations. Clin. Genet. 2005, 67, 193–208. [Google Scholar] [CrossRef]
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical mechanisms of vertebrate hedgehog signaling. Development 2019, 146, dev166892. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, S.; Hervold, K.; Ramirez-Weber, F.A.; Kornberg, T.B. Two patched protein subtypes and a conserved domain of group I proteins that regulates turnover. J. Biol. Chem. 2008, 283, 30964–30969. [Google Scholar] [CrossRef] [Green Version]
- Martín, V.; Carrillo, G.; Torroja, C.; Guerrero, I. The sterol-sensing domain of patched protein seems to control smoothened activity through patched vesicular trafficking. Curr. Biol. 2001, 11, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Arensdorf, A.M.; Marada, S.; Ogden, S.K. Smoothened Regulation: A Tale of Two Signals. Trends Pharmacol. Sci. 2016, 37, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koebernick, K.; Pieler, T. Gli-type zinc finger proteins as bipotential transducers of Hedgehog signaling. Differentiation 2002, 70, 69–76. [Google Scholar] [CrossRef]
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli Proteins: Regulation in Development and Cancer. Cells 2019, 8, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle 2006, 5, 2457–2463. [Google Scholar] [CrossRef] [PubMed]
- Hatayama, M.; Aruga, J. Gli Protein Nuclear Localization Signal. In Vitamins and Hormones; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 88, pp. 73–89. [Google Scholar]
- Cherry, A.L.; Finta, C.; Karlström, M.; Jin, Q.; Schwend, T.; Astorga-Wells, J.; Zubarev, R.A.; Del Campo, M.; Criswell, A.R.; De Sanctis, D.; et al. Structural basis of SUFU-GLI interaction in human Hedgehog signalling regulation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2563–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Shi, Q.; Jiang, J. Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 6383–6388. [Google Scholar] [CrossRef] [Green Version]
- Liem, K.F.; He, M.; Ocbina, P.J.R.; Anderson, K.V. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13377–13382. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.H.C.; Zhang, X.; Yu, C.; Li, Z.J.; Wunder, J.S.; Hui, C.C.; Alman, B.A. Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 2011, 138, 3791–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurya, A.K.; Ben, J.; Zhao, Z.; Lee, R.T.H.; Niah, W.; Ng, A.S.M.; Iyu, A.; Yu, W.; Elworthy, S.; van Eeden, F.J.M.; et al. Positive and Negative Regulation of Gli Activity by Kif7 in the Zebrafish Embryo. PLoS Genet. 2013, 9, e1003955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijlsma, M.F.; Spek, C.A.; Zivkovic, D.; van de Water, S.; Rezaee, F.; Peppelenbosch, M.P. Repression of Smoothened by Patched-Dependent (Pro-)Vitamin D3 Secretion. PLoS Biol. 2006, 4, e232. [Google Scholar] [CrossRef] [Green Version]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Myers, B.R.; Neahring, L.; Zhang, Y.; Roberts, K.J.; Beachy, P.A. Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl. Acad. Sci. USA 2017, 114, E11141–E11150. [Google Scholar] [CrossRef] [Green Version]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of smoothened. Nature 2002, 418, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberger, F.; Ruiz i Altaba, A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.; Segal, R.A. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepny, A.; Rogers, S.; Jayasekara, W.S.N.; Park, K.; McCloy, R.A.; Cochrane, C.R.; Ganju, V.; Cooper, W.A.; Sage, J.; Peacock, C.D.; et al. The role of canonical and non-canonical Hedgehog signaling in tumor progression in a mouse model of small cell lung cancer. Oncogene 2017, 36, 5544–5550. [Google Scholar] [CrossRef]
- Incardona, J.P.; Gruenberg, J.; Roelink, H. Sonic hedgehog induces the segregation of patched and smoothened in endosomes. Curr. Biol. 2002, 12, 983–995. [Google Scholar] [CrossRef]
- He, M.; Subramanian, R.; Bangs, F.; Omelchenko, T.; Liem, K.F.; Kapoor, T.M.; Anderson, K.V. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol. 2014, 16, 663–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.; Kicheva, A.; Ribeiro, A.; Blassberg, R.; Page, K.M.; Barnes, C.P.; Briscoe, J. Ptch1 and Gli regulate Shh signalling dynamics via multiple mechanisms. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; De Lopes, G.P.F.; Spohr, T.C.L.D.S.E. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 1–15. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical hedgehog signaling pathway in cancer: Activation of GLI transcription factors beyond smoothened. Front. Genet. 2019, 10, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, K.S.; Chang, C.F.; Lin, S.S. Sonic hedgehog signaling in organogenesis, tumors, and tumor microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef] [Green Version]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Cassandras, M.; Peng, T. The role of Hedgehog signaling in adult lung regeneration and maintenance. J. Dev. Biol. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulley, D.; Wienhold, M.; Sun, X. The pulmonary mesenchyme directs lung development. Curr. Opin. Genet. Dev. 2015, 32, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Silva, H.; Correia-Pinto, J.; Moura, R.S. Canonical Sonic Hedgehog Signaling in Early Lung Development. J. Dev. Biol. 2017, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Ahlbrecht, K.; Seeger, W.; Voswinckel, R. The role of Sonic Hedgehog in postnatal mouse lung development. Pneumologie 2012, 66, A404. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, H.; Teng, H.; Shi, J.; Zhang, Y. Expression of SHH signaling pathway components in the developing human lung. Histochem. Cell Biol. 2010, 134, 327–335. [Google Scholar] [CrossRef]
- Giroux-Leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Huang, M.; Sun, S.; Wu, Y.; Lin, X. Epithelial heparan sulfate regulates Sonic Hedgehog signaling in lung development. PLoS Genet. 2017, 13, 1–22. [Google Scholar] [CrossRef]
- Burri, P.H. Structural aspects of postnatal lung development-alveolar formation and growth. Biol. Neonate 2006, 89, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Loomis, C.A.; Zhao, Z.; Cushman, J.C.; Liu, L.; Munger, J.S. Sonic Hedgehog Signaling Regulates Myofibroblast Function during Alveolar Septum Formation in Murine Postnatal Lung. Am. J. Respir. Cell Mol. Biol. 2017, 57, 280–293. [Google Scholar] [CrossRef]
- Belgacemi, R.; Danopoulos, S.; Deslée, G.; Dormoy, V.; Al Alam, D. Hedgehog signalling crosstalks orchestrate human lung development. Eur. Respir. J. 2020, 56, 596. [Google Scholar]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarelli, A.V.; Masciale, V.; Aramini, B.; Coló, G.P.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; et al. Molecular Mechanisms and Cellular Contribution from Lung Fibrosis to Lung Cancer Development. Int. J. Mol. Sci. 2021, 22, 12179. [Google Scholar] [CrossRef]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-J.; Tao, H.; Li, J. Hedgehog signaling pathway as key player in liver fibrosis: New insights and perspectives. Expert Opin. Ther. Targets 2014, 18, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Peng, Y.; Li, H. The Injury-Related Activation of Hedgehog Signaling Pathway Modulates the Repair-Associated Inflammation in Liver Fibrosis. Front. Immunol. 2017, 8, 1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Zhang, Z.; Zhang, P.; Yu, M.; Yang, T. Role of canonical Hedgehog signaling pathway in liver. Int. J. Biol. Sci. 2018, 14, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Tan, R.J.; Liu, Y. Sonic hedgehog signaling in kidney fibrosis: A master communicator. Sci. China Life Sci. 2016, 59, 920–929. [Google Scholar] [CrossRef] [Green Version]
- Froidure, A.; Marchal-Duval, E.; Homps-Legrand, M.; Ghanem, M.; Justet, A.; Crestani, B.; Mailleux, A. Chaotic activation of developmental signalling pathways drives idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2020, 29, 190140. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhao, X.; Jiang, R.; Chen, X.; Zhu, X.; Chen, K.; Chen, S.; Zhang, X.; Qin, Y.; Liu, Y.; et al. Increased expression of Sonic hedgehog restores diabetic endothelial progenitor cells and improves cardiac repair after acute myocardial infarction in diabetic mice. Int. J. Mol. Med. 2019, 44, 1091–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. 2015, 24, 102–114. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Idiopathic pulmonary fibrosis: An epithelial/fibroblastic cross-talk disorder. Respir. Res. 2001, 3, 1–8. [Google Scholar]
- Maher, T.M.; Wells, A.U.; Laurent, G.J. Idiopathic pulmonary fibrosis: Multiple causes and multiple mechanisms? Eur. Respir. J. 2007, 30, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.; López-Otín, C.; Pardo, A. Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 48, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Rabeyrin, M.; Thivolet, F.; Ferretti, G.R.; Chalabreysse, L.; Jankowski, A.; Cottin, V.; Pison, C.; Cordier, J.-F.; Lantuejoul, S. Usual interstitial pneumonia end-stage features from explants with radiologic and pathological correlations. Ann. Diagn. Pathol. 2015, 19, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Zissel, G.; Watz, H.; Drömann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human alveolar epithelial cells type II are capable of TGFβ-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Barratt, S.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [Green Version]
- Winters, N.I.; Burman, A.; Kropski, J.A.; Blackwell, T.S. Epithelial Injury and Dysfunction in the Pathogenesis of Idiopathic PulmonaryFibrosis. Am. J. Med. Sci. 2019, 357, 374–378. [Google Scholar] [CrossRef] [Green Version]
- Pardo, A.; Selman, M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann. Am. Thorac. Soc. 2016, 13, S417–S421. [Google Scholar] [CrossRef]
- Willis, B.C.; duBois, R.M.; Borok, Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc. Am. Thorac. Soc. 2006, 3, 377–382. [Google Scholar] [CrossRef]
- Li, M.; Luan, F.; Zhao, Y.; Hao, H.; Zhou, Y.; Han, W.; Fu, X. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp. Biol. Med. (Maywood) 2016, 241, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartis, D.; Mise, N.; Mahida, R.Y.; Eickelberg, O.; Thickett, D.R. Epithelia-mesenchymal transition in lung development and disease: Does it exist and is it important? Thorax 2014, 69, 760–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olajuyin, A.M.; Zhang, X.; Ji, H.L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 2019, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta-Mol. Basis Dis. 2013, 1832, 911–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef]
- Tan, S.; Li, Q.; Duan, F.; Yuan, Q.; Deng, G. Mechanism of Tripchlorolide Inhibiting Hedgehog Signaling Pathway to Delay Lung Fibrosis. J. Biomater. Tissue Eng. 2020, 10, 992–998. [Google Scholar] [CrossRef]
- Jia, G.; Chandriani, S.; Abbas, A.R.; DePianto, D.J.; N’Diaye, E.N.; Yaylaoglu, M.B.; Moore, H.M.; Peng, I.; Devoss, J.; Collard, H.R.; et al. CXCL14 is a candidate biomarker for Hedgehog signalling in idiopathic pulmonary fibrosis. Thorax 2017, 72, 780–787. [Google Scholar] [CrossRef]
- Kugler, M.C.; Yie, T.-A.; Cai, Y.; Berger, J.Z.; Loomis, C.A.; Munger, J.S. The Hedgehog target Gli1 is not required for bleomycin-induced lung fibrosis. Exp. Lung Res. 2019, 45, 22–29. [Google Scholar] [CrossRef]
- Prasse, A.; Ramaswamy, M.; Mohan, S.; Pan, L.; Kenwright, A.; Neighbors, M.; Belloni, P.; LaCamera, P.P. A Phase 1b Study of Vismodegib with Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. Pulm. Ther. 2019, 5, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Noble, P.W. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123–129. [Google Scholar] [CrossRef]
- Kolb, M.; Borensztajn, K.; Crestani, B.; Kolb, M. Idiopathic Pulmonary Fibrosis: From Epithelial Injury to Biomarkers-Insights from the Bench Side. Respiration 2013, 86, 441–452. [Google Scholar]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef] [PubMed]
- Kage, H.; Borok, Z. EMT and interstitial lung disease: A mysterious relationship. Curr. Opin. Pulm. Med. 2012, 18, 517–523. [Google Scholar] [CrossRef]
- Rybinski, B.; Franco-Barraza, J.; Cukierman, E. The wound healing, chronic fibrosis, and cancer progression triad. Physiol. Genomics 2014, 46, 223–244. [Google Scholar] [CrossRef]
- Shh Expression in Pulmonary Injury and Disease-Madame Curie Bioscience Database-NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6249/ (accessed on 16 December 2021).
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.A.; Hoyne, G.F.; Ahmad, S.A.; Jarman, E.; Wallace, W.A.H.; Harrison, D.J.; Haslett, C.; Lamb, J.R.; Howie, S.E.M. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J. Pathol. 2003, 199, 488–495. [Google Scholar] [CrossRef]
- Fitch, P.M.; Howie, S.E.M.; Wallace, W.A.H. Oxidative damage and TGF-β differentially induce lung epithelial cell sonic hedgehog and tenascin-C expression: Implications for the regulation of lung remodelling in idiopathic interstitial lung disease. Int. J. Exp. Pathol. 2011, 92, 8–17. [Google Scholar] [CrossRef]
- Marasciulo, F.L.; Montagnani, M.; Potenza, M.A. Endothelin-1: The yin and yang on vascular function. Curr. Med. Chem. 2006, 13, 1655–1665. [Google Scholar] [CrossRef]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Mora, A.; Shim, H.; Stecenko, A.; Brigham, K.L.; Rojas, M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 291–299. [Google Scholar] [CrossRef]
- Jaffar, J.; Griffiths, K.; Oveissi, S.; Duan, M.; Foley, M.; Glaspole, I.; Symons, K.; Organ, L.; Westall, G. CXCR4+ cells are increased in lung tissue of patients with idiopathic pulmonary fibrosis. Respir. Res. 2020, 21, 221. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Arora, S.; Bhardwaj, A.; Srivastava, S.K.; Kadakia, M.P.; Wang, B.; Grizzle, W.E.; Owen, L.B.; Singh, S. CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated kinase- and Akt kinase-mediated activation of nuclear factor κB: Implications for bidirectional tumor-stromal interactions. J. Biol. Chem. 2012, 287, 39115–39124. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ma, Q.; Xu, Q.; Liu, H.; Lei, J.; Duan, W.; Bhat, K.; Wang, F.; Wu, E.; Wang, Z. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012, 322, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neurohr, C.; Nishimura, S.L.; Sheppard, D. Activation of transforming growth factor-beta by the integrin alphavbeta8 delays epithelial wound closure. Am. J. Respir. Cell Mol. Biol. 2006, 35, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhu, X.; Hua, Y.; Zhao, Q.; Wang, K.; Zhen, L.; Wang, G.; Lv, J.; Luo, A.; Cho, W.C.; et al. YY1 mediates TGF-β1-induced EMT and pro-fibrogenesis in alveolar epithelial cells. Respir. Res. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X.-F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef]
- Rodríguez-Pascual, F.; Busnadiego, O.; González-Santamaría, J. The profibrotic role of endothelin-1: Is the door still open for the treatment of fibrotic diseases? Life Sci. 2014, 118, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, C.; Abraham, D.; Renzoni, E.A. Endothelin in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 44, 1–10. [Google Scholar] [CrossRef]
- Moore, M.W.; Herzog, E.L. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.F. Origin of Myofibroblasts in Lung Fibrosis. Curr. Tissue Microenviron. Rep. 2020, 1, 155–162. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Cigna, N.; Moshai, E.F.; Joëlle, M.-S.; Stervinou-Wémeau, L.; Monique, D.; Soler, P.; Crestani, B.; Mailleux, A. Involvement of the Hedgehog signalling pathway in idiopathic pulmonary fibrosis. Eur. Respir. J. 2011, 38, 3233. [Google Scholar]
- Cigna, N.; Farrokhi Moshai, E.; Brayer, S.; Marchal-Somme, J.; Wémeau-Stervinou, L.; Fabre, A.; Mal, H.; Lesèche, G.; Dehoux, M.; Soler, P.; et al. The Hedgehog System Machinery Controls Transforming Growth Factor-β-Dependent Myofibroblastic Differentiation in Humans. Am. J. Pathol. 2012, 181, 2126–2137. [Google Scholar] [CrossRef]
- Hu, B.; Liu, J.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Ding, L.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailleux, A.A.; Moshai, E.F.; Crestani, B. Sonic Hedgehog signaling in pulmonary fibrosis: A spiky issue? Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L391–L393. [Google Scholar] [CrossRef] [Green Version]
- Horn, A.; Palumbo, K.; Cordazzo, C.; Dees, C.; Akhmetshina, A.; Tomcik, M.; Zerr, P.; Avouac, J.; Gusinde, J.; Zwerina, J.; et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum. 2012, 64, 2724–2733. [Google Scholar] [CrossRef]
- Wasson, C.W.; Ross, R.L.; Wells, R.; Corinaldesi, C.; Georgiou, I.C.; Riobo-Del Galdo, N.A.; Del Galdo, F. Long non-coding RNA HOTAIR induces GLI2 expression through Notch signalling in systemic sclerosis dermal fibroblasts. Arthritis Res. Ther. 2020, 22, 286. [Google Scholar] [CrossRef]
- Moshai, E.F.; Wémeau-Stervinou, L.; Cigna, N.; Brayer, S.; Sommé, J.M.; Crestani, B.; Mailleux, A.A. Targeting the hedgehog-glioma-associated oncogene homolog pathway inhibits bleomycin-induced lung fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2014, 51, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-C.; Wu, S.-B.; Kau, H.-C.; Wei, Y.-H. Essential role of connective tissue growth factor (CTGF) in transforming growth factor-β1 (TGF-β1)-induced myofibroblast transdifferentiation from Graves’ orbital fibroblasts. Sci. Rep. 2018, 8, 7276. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-Regulation and Profibrotic Role of Osteopontin in Human Idiopathic Pulmonary Fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef] [Green Version]
- Syn, W.-K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Ji, J.; Chen, X.; Cao, H.; Tan, Y.; Cui, Y.; Xiang, Z.; Han, X. Alveolar epithelial cell-derived Sonic hedgehog promotes pulmonary fibrosis through OPN-dependent alternative macrophage activation. FEBS J. 2021, 288, 3530–3546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xu, H.; Zhang, Y.; Yi, X.; Zhang, G.; Zhang, X.; Xu, D.; Gao, X.; Li, S.; Zhu, Y.; et al. Targeting the RAS axis alleviates silicotic fibrosis and Ang II-induced myofibroblast differentiation via inhibition of the hedgehog signaling pathway. Toxicol. Lett. 2019, 313, 30–41. [Google Scholar] [CrossRef]
- Cao, H.; Chen, X.; Hou, J.; Wang, C.; Xiang, Z.; Shen, Y.; Han, X. The Shh/Gli signaling cascade regulates myofibroblastic activation of lung-resident mesenchymal stem cells via the modulation of Wnt10a expression during pulmonary fibrogenesis. Lab. Investig. 2020, 100, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Ma, Q.; Yin, J.; Zhang, H.; Liu, X. WISP-1 induced by mechanical stress contributes to fibrosis and hypertrophy of the ligamentum flavum through Hedgehog-Gli1 signaling. Exp. Mol. Med. 2021, 53, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, Z.; Xu, Z.; Yin, H.; Bai, L.; Ma, Z.; DeCoster, M.A.; Qian, G.; Wu, G. Activation of the sonic hedgehog signaling controls human pulmonary arterial smooth muscle cell proliferation in response to hypoxia. Biochim. Biophys. Acta-Mol. Cell Res. 2010, 1803, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Lin, C.H.; Chen, J.Y.; Li, C.H.; Liu, Y.T.; Chen, B.C. Induction of Connective Tissue Growth Factor Expression by Hypoxia in Human Lung Fibroblasts via the MEKK1/MEK1/ERK1/GLI-1/GLI-2 and AP-1 Pathways. PLoS ONE 2016, 11, e0160593. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.; Jones, M.G.; Davies, D.E.; Wang, Y. Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J. Lung Heal. Dis. 2019, 3, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial-Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina (Kaunas) 2019, 55, 83. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J.; et al. Paracrine signalling during ZEB1-mediated epithelial–mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Song, W.; Zhou, L.; Mao, Y.; Hong, W. House dust mite induces Sonic hedgehog signaling that mediates epithelial-mesenchymal transition in human bronchial epithelial cells. Mol. Med. Rep. 2019, 20, 4674–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrokhi Moshai, E.; Mailleux, A.; Besnard, V.; Brayer, S.; Dehoux, M.; Crestani, B. Inhibition of the sonic hedgehog pathway at the primary cilium prevents the effect of TGF-beta 1 on alveolar epithelial cells. Eur. Respir. J. 2012, 40, P3770. [Google Scholar]
- Xiao, K.; He, W.; Guan, W.; Hou, F.; Yan, P.; Xu, J.; Zhou, T.; Liu, Y.; Xie, L. Mesenchymal stem cells reverse EMT process through blocking the activation of NF-κB and Hedgehog pathways in LPS-induced acute lung injury. Cell Death Dis. 2020, 11, 863. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, A.; Hendee, K.; Muyleart, M.; Mammoto, T. Endothelial Twist1-PDGFB signaling mediates hypoxia-induced proliferation and migration of αSMA-positive cells. Sci. Rep. 2020, 10, 7563. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef]
- Dunaeva, M.; Voo, S.; van Oosterhoud, C.; Waltenberger, J. Sonic hedgehog is a potent chemoattractant for human monocytes: Diabetes mellitus inhibits Sonic hedgehog-induced monocyte chemotaxis. Basic Res. Cardiol. 2009, 105, 61. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, M.A.; Donnelly, J.M.; Engevik, A.C.; Xiao, C.; Yang, L.; Kenny, S.; Varro, A.; Hollande, F.; Samuelson, L.C.; Zavros, Y. Gastric Sonic Hedgehog Acts as a Macrophage Chemoattractant During the Immune Response to Helicobacter pylori. Gastroenterology 2012, 142, 1150–1159.e6. [Google Scholar] [CrossRef] [Green Version]
- Pereira, T.A.; Xie, G.; Choi, S.S.; Syn, W.-K.; Voieta, I.; Lu, J.; Chan, I.S.; Swiderska, M.; Amaral, K.B.; Antunes, C.M.; et al. Macrophage-derived Hedgehog ligands promotes fibrogenic and angiogenic responses in human schistosomiasis mansoni. Liver Int. Off. J. Int. Assoc. Study Liver 2013, 33, 149–161. [Google Scholar] [CrossRef]
- Hanna, A.; Metge, B.J.; Bailey, S.K.; Chen, D.; Chandrashekar, D.S.; Varambally, S.; Samant, R.S.; Shevde, L.A. Inhibition of Hedgehog signaling reprograms the dysfunctional immune microenvironment in breast cancer. Oncoimmunology 2019, 8, 1548241. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Su, M.; Tang, D.; Hao, L.; Xun, X.-H.; Huang, Y. Ligustrazin increases lung cell autophagy and ameliorates paraquat-induced pulmonary fibrosis by inhibiting PI3K/Akt/mTOR and hedgehog signalling via increasing miR-193a expression. BMC Pulm. Med. 2019, 19, 35. [Google Scholar] [CrossRef]
- Gu, S.; Yan, M.; Wang, C.; Meng, X.; Xiang, Z.; Qiu, Y.; Han, X. Microcystin-leucine-arginine induces liver fibrosis by activating the Hedgehog pathway in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2020, 533, 770–778. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Song, C.; Zhang, J.; Wang, Y.; Lv, C.; Song, X. Astilbin ameliorates pulmonary fibrosis via blockade of Hedgehog signaling pathway. Pulm. Pharmacol. Ther. 2018, 50, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.C.; Hanke, C.W.; Caro, I. Vismodegib and the Hedgehog Pathway Inhibitors: A Historical Perspective to Current Clinical Application. J. Drugs Dermatol. 2018, 17, 506–508. [Google Scholar] [PubMed]
- Cao, H.-L.; Zhou, J.; Chen, X.-B.; Landeck, L.; Yang, J.-Q.; Chen, J.-Q.; Li, W.; Cai, S.-Q.; Zheng, M.; Man, X.-Y. Inhibition of the hedgehog pathway leads to antifibrotic effects in dermal fibrosis. Discov. Med. 2016, 22, 311–318. [Google Scholar]
- Jin, G.; Sivaraman, A.; Lee, K. Development of taladegib as a sonic hedgehog signaling pathway inhibitor. Arch. Pharm. Res. 2017, 40, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- A Study Evaluating the Safety and Efficacy of ENV-101 in Subjects With Idiopathic Pulmonary Fibrosis (IPF). Available online: https://clinicaltrials.gov/ct2/show/NCT04968574 (accessed on 29 November 2021).
- Farrokhi Moshai, E.; Wemeau-Stervinou, L.; Cigna, N.; Marchal-Somme, J.; Besnard, V.; Brayer, S.; Fabre, A.; Mailleux, A.; Crestani, B. Targeting the hedgehog/GLI pathway decreases bleomycin-induced lung fibrosis. Eur. Respir. J. 2013, 42, 3329. [Google Scholar]
- Xiao, H.; Zhang, G.-F.; Liao, X.-P.; Li, X.-J.; Zhang, J.; Lin, H.; Chen, Z.; Zhang, X. Anti-fibrotic effects of pirfenidone by interference with the hedgehog signalling pathway in patients with systemic sclerosis-associated interstitial lung disease. Int. J. Rheum. Dis. 2018, 21, 477–486. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Effendi, W.I.; Nagano, T. The Hedgehog Signaling Pathway in Idiopathic Pulmonary Fibrosis: Resurrection Time. Int. J. Mol. Sci. 2022, 23, 171. https://doi.org/10.3390/ijms23010171
Effendi WI, Nagano T. The Hedgehog Signaling Pathway in Idiopathic Pulmonary Fibrosis: Resurrection Time. International Journal of Molecular Sciences. 2022; 23(1):171. https://doi.org/10.3390/ijms23010171
Chicago/Turabian StyleEffendi, Wiwin Is, and Tatsuya Nagano. 2022. "The Hedgehog Signaling Pathway in Idiopathic Pulmonary Fibrosis: Resurrection Time" International Journal of Molecular Sciences 23, no. 1: 171. https://doi.org/10.3390/ijms23010171
APA StyleEffendi, W. I., & Nagano, T. (2022). The Hedgehog Signaling Pathway in Idiopathic Pulmonary Fibrosis: Resurrection Time. International Journal of Molecular Sciences, 23(1), 171. https://doi.org/10.3390/ijms23010171