
Neurodegeneration and Astrogliosis in the Human CA1 Hippocampal Subfield Are Related to hsp90ab1 and bag3 in Alzheimer’s Disease

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
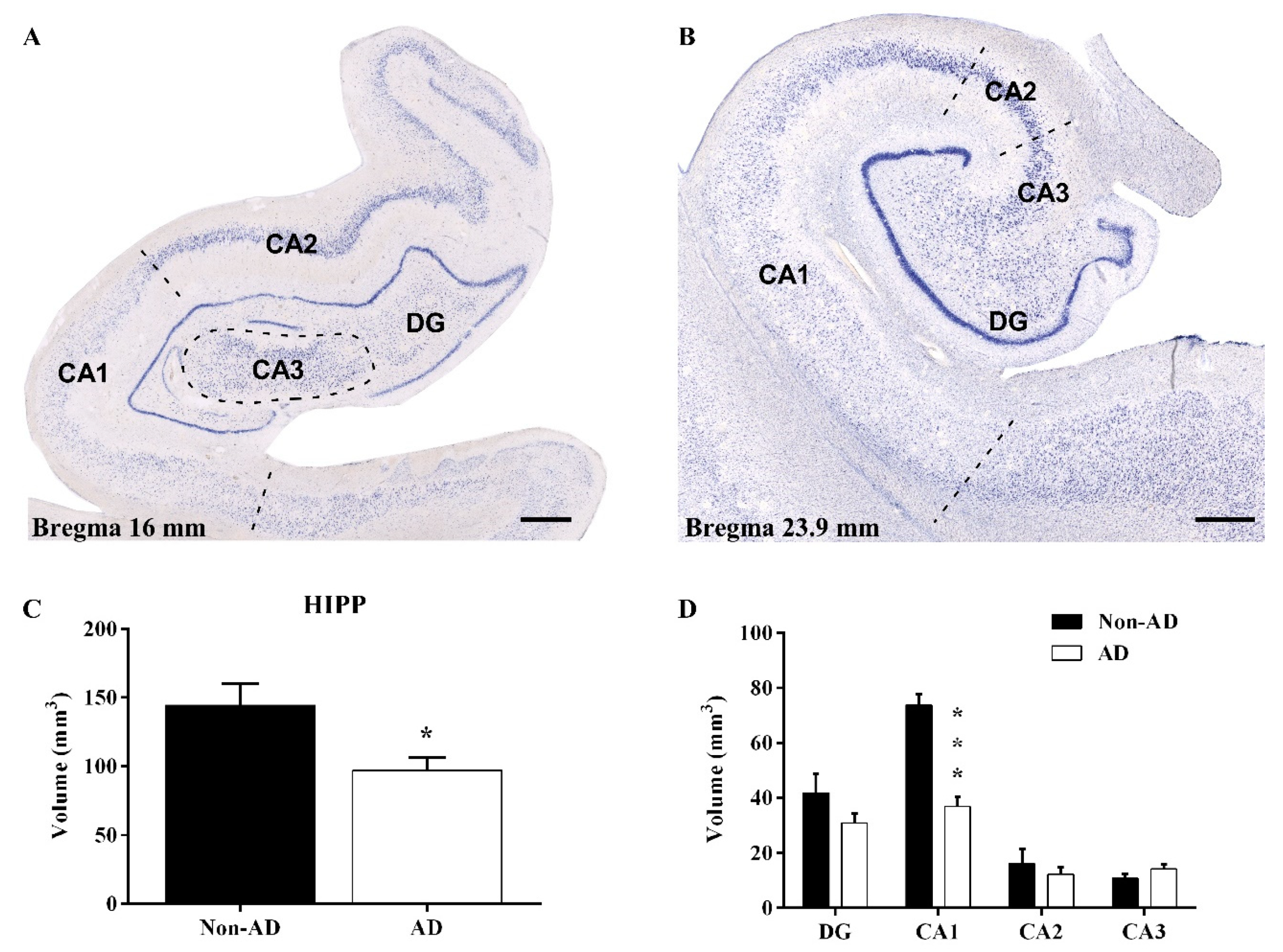
2.1. Reduction in the Hippocampal Volume
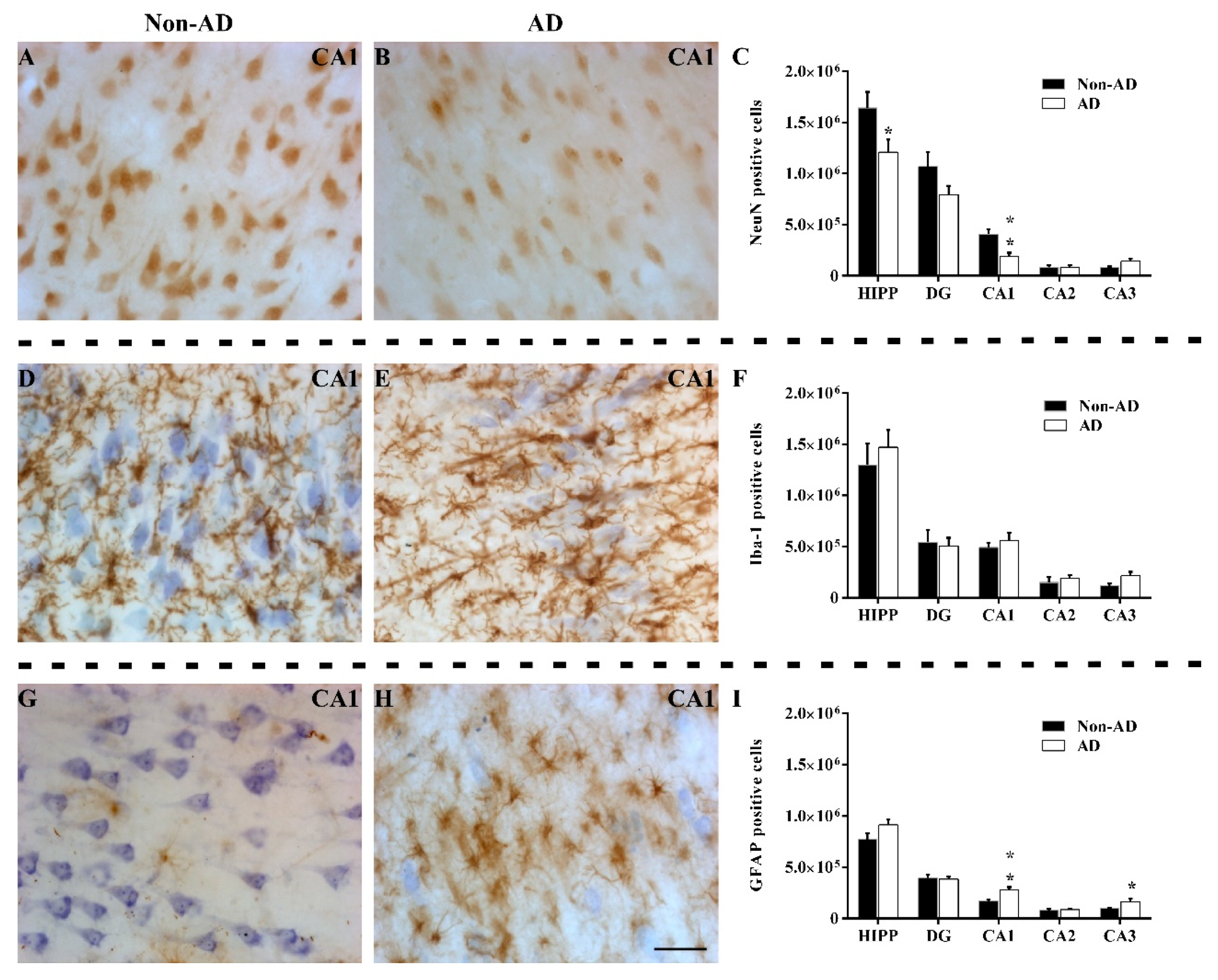
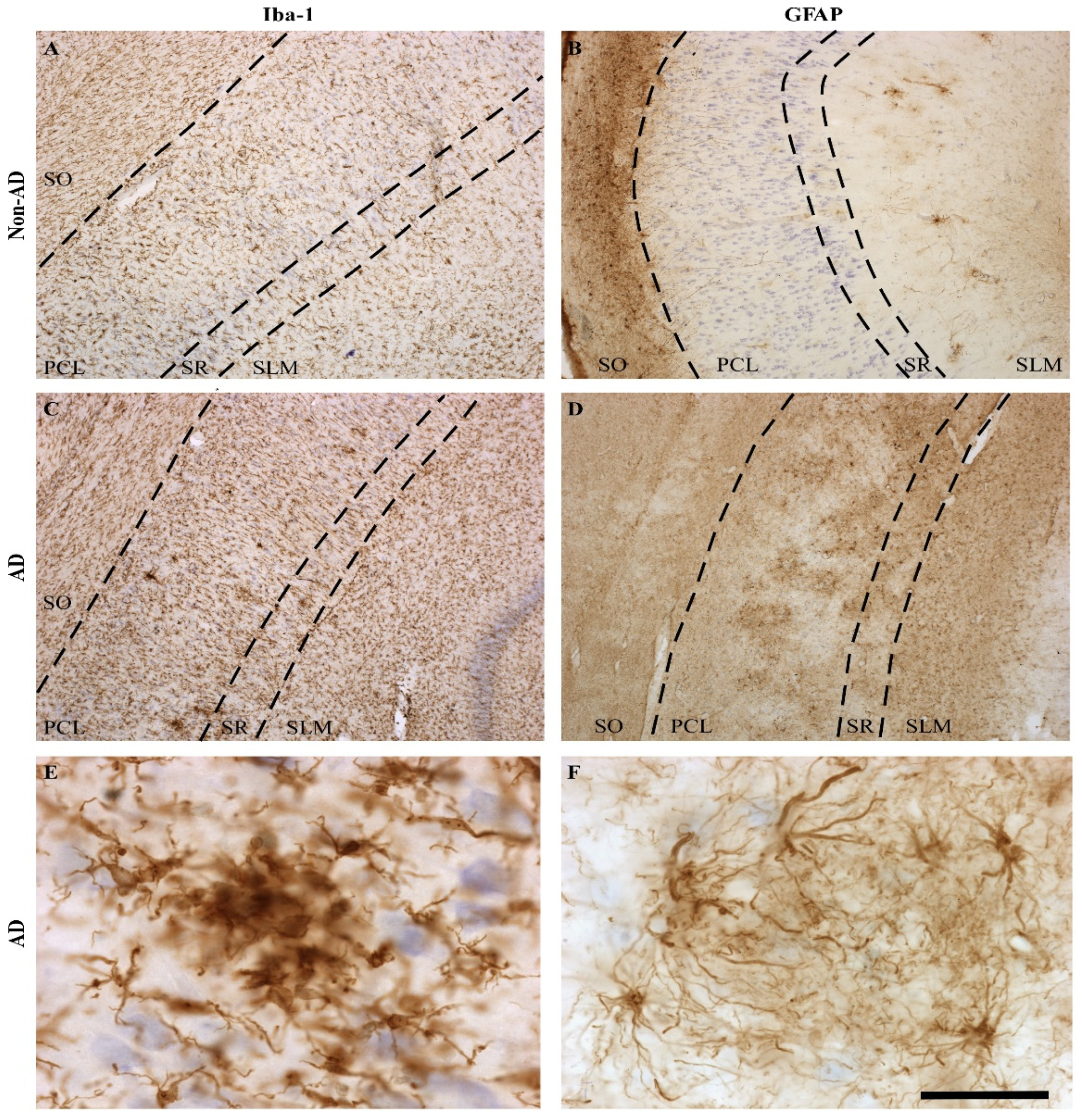
2.2. The Extent of Neurodegeneration, Microgliosis and Astrogliosis Differs among Hippocampal Fields
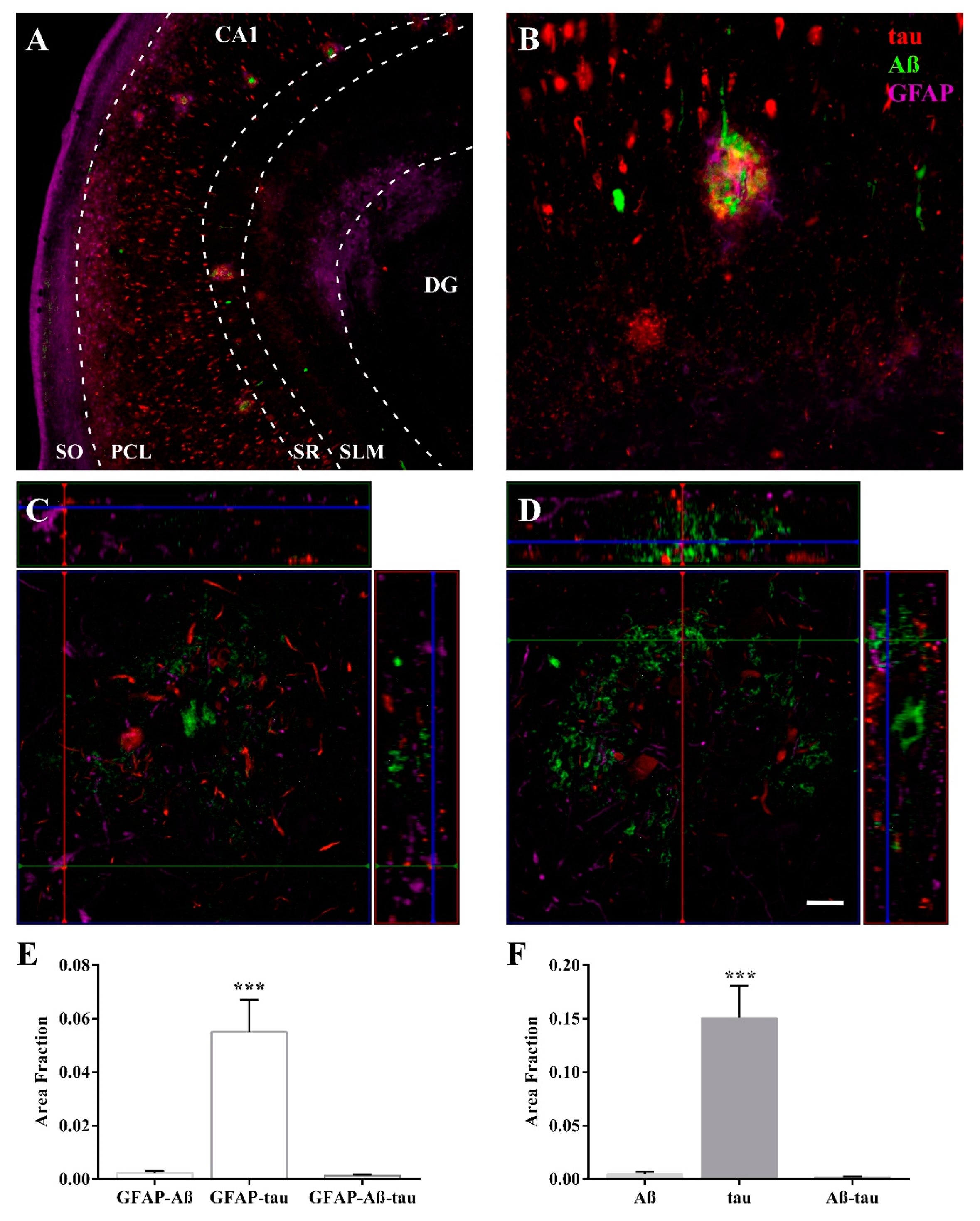
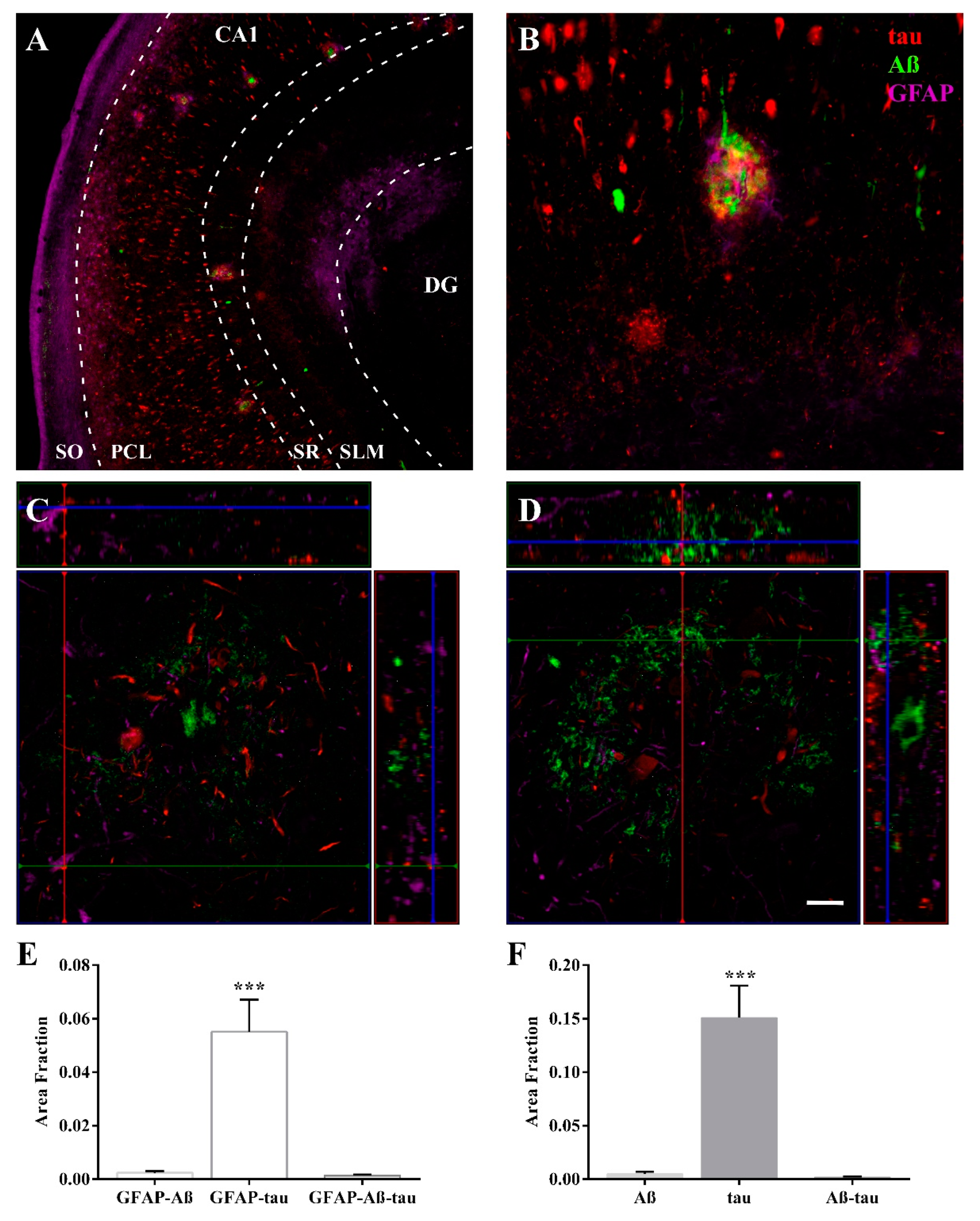
2.3. Expression of Astroglia and Tau in CA1
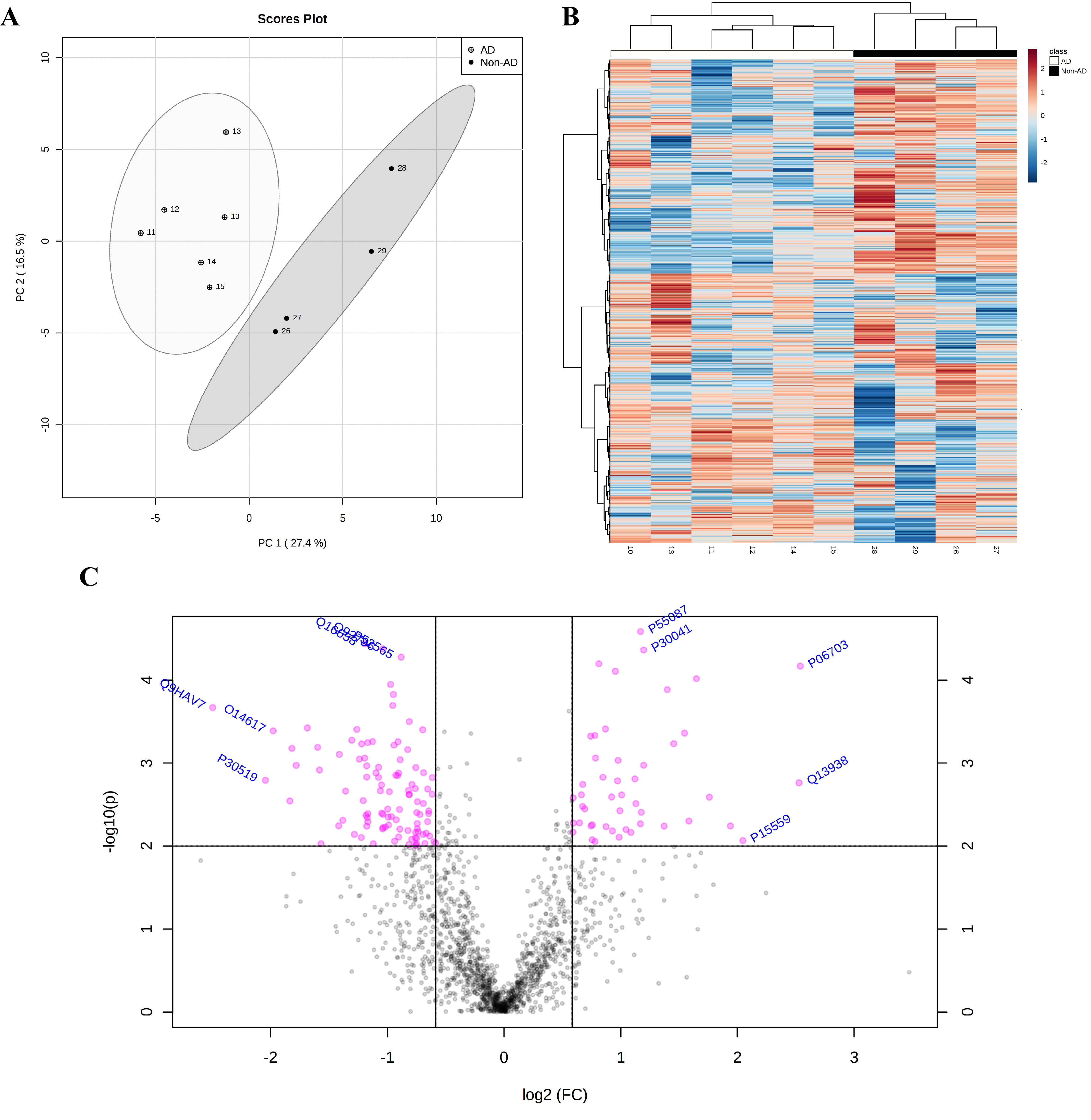
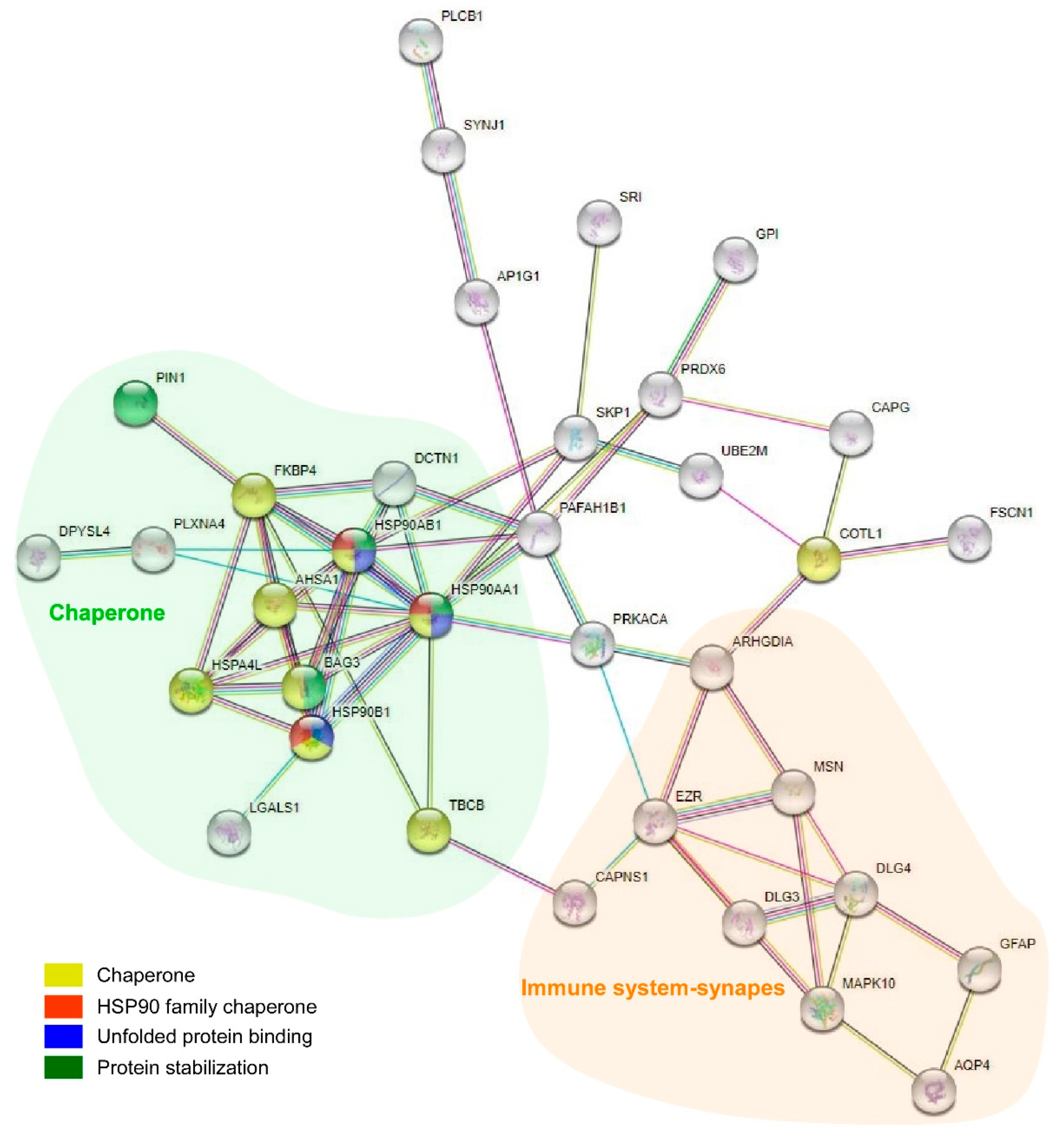
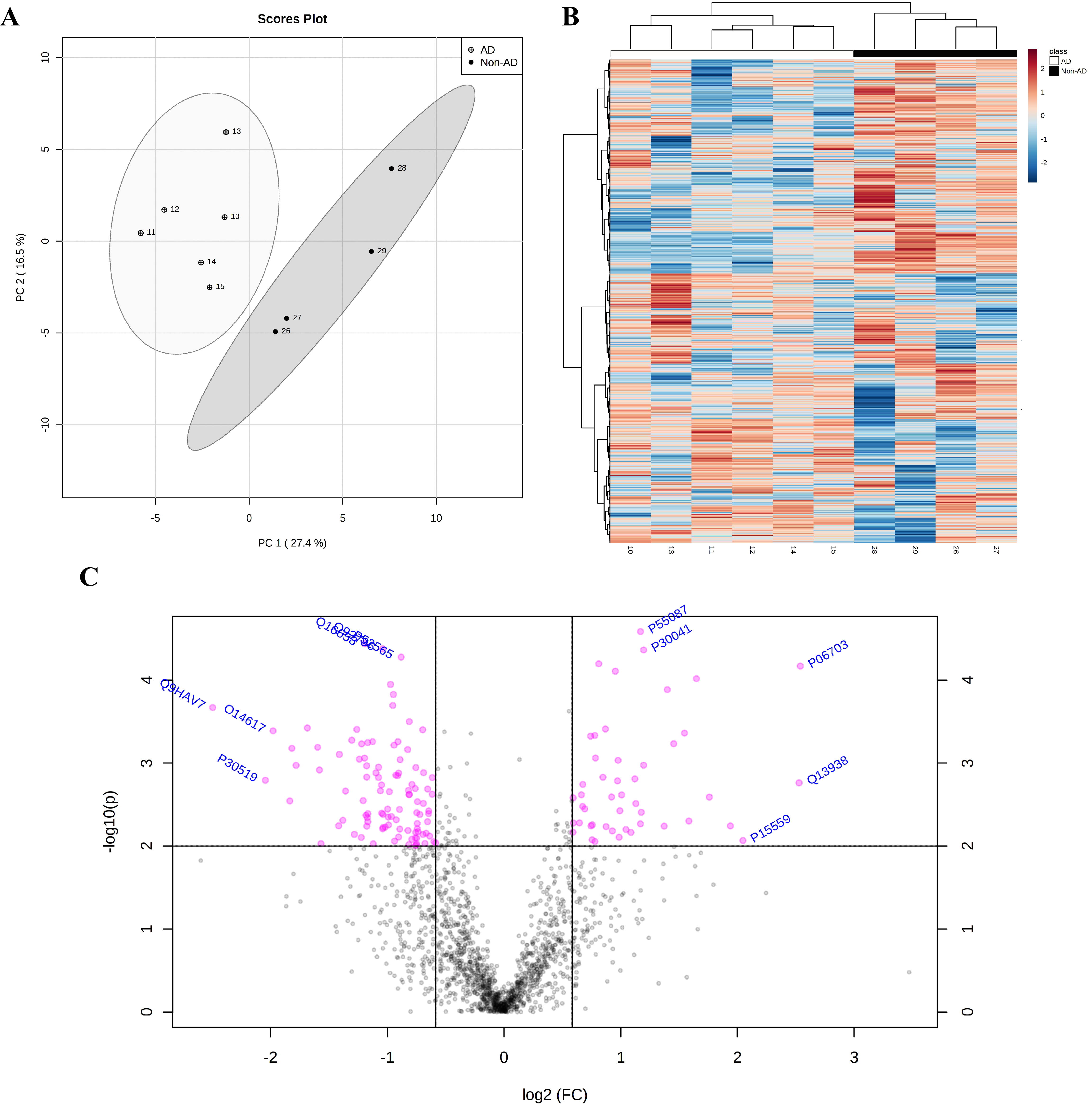
2.4. Proteomic Analysis Highlights the Involvement of HSP90 Family cChaperones
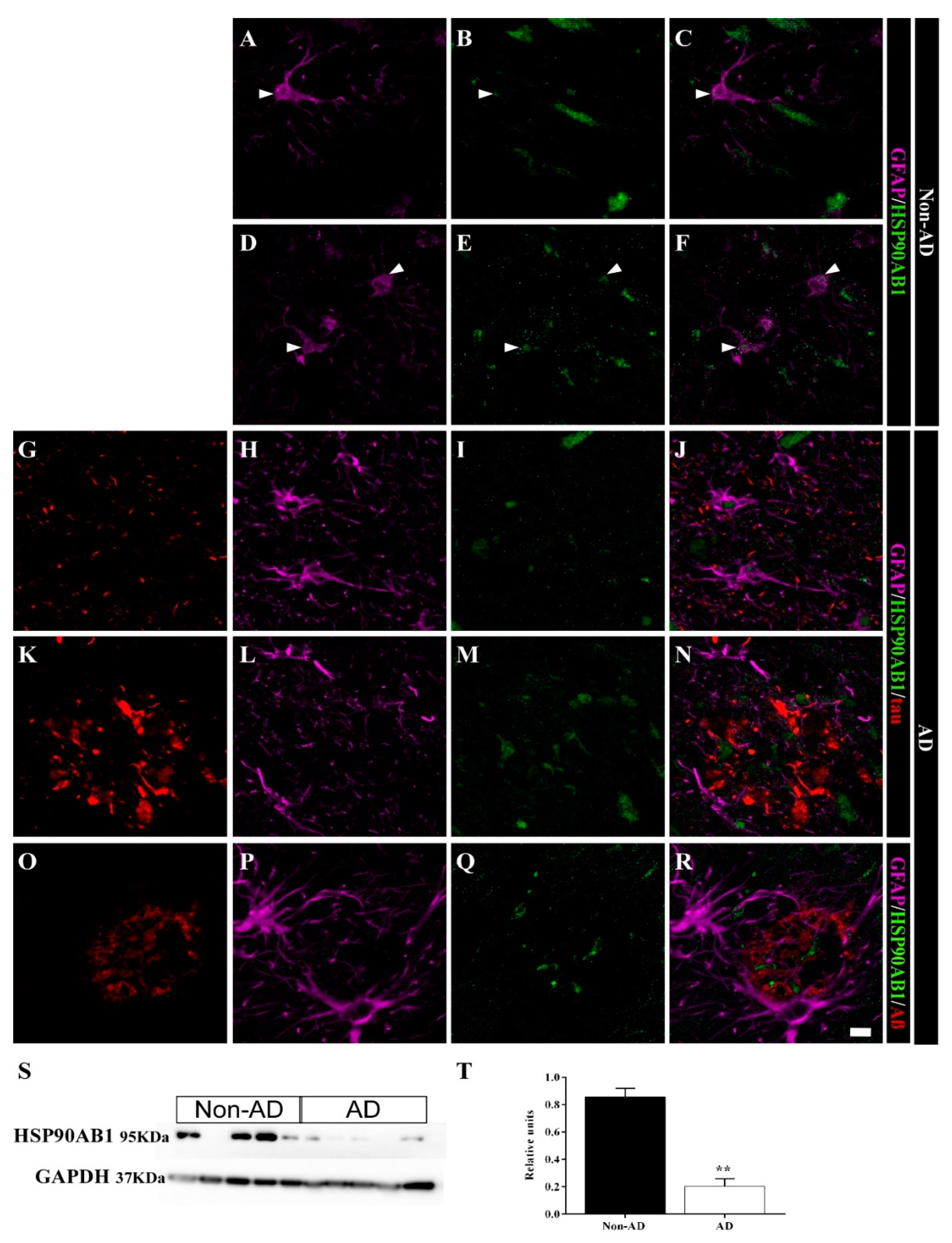
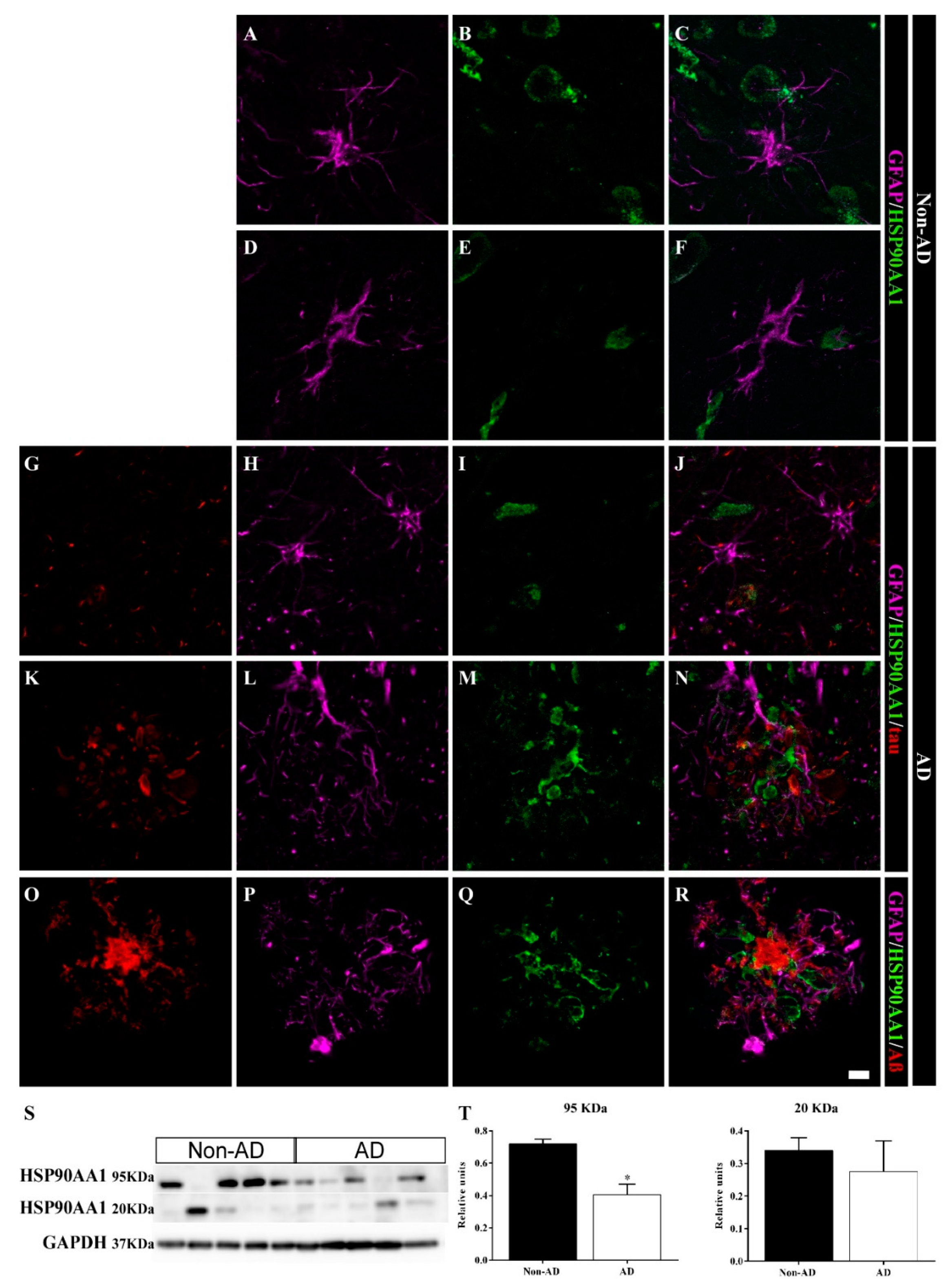
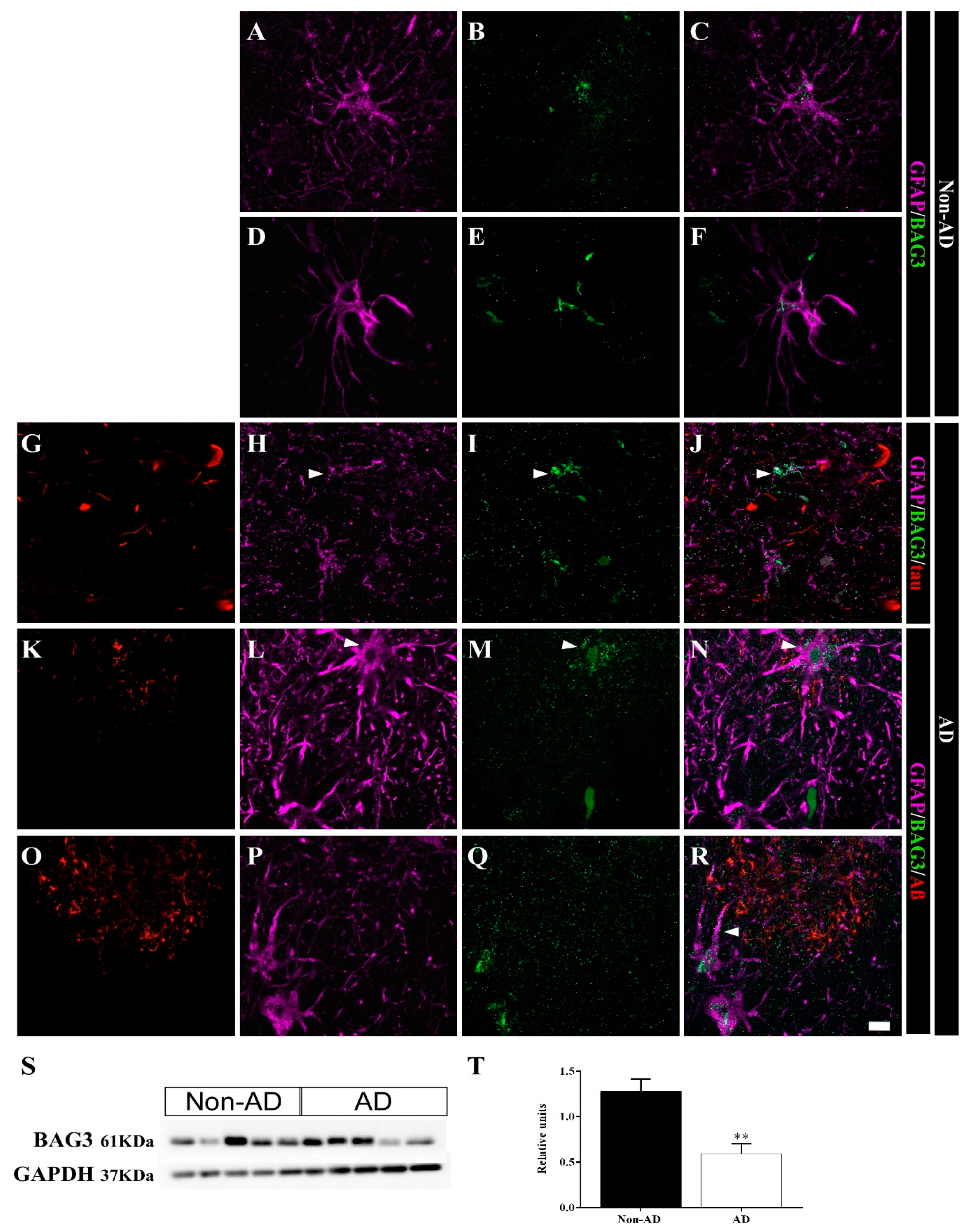
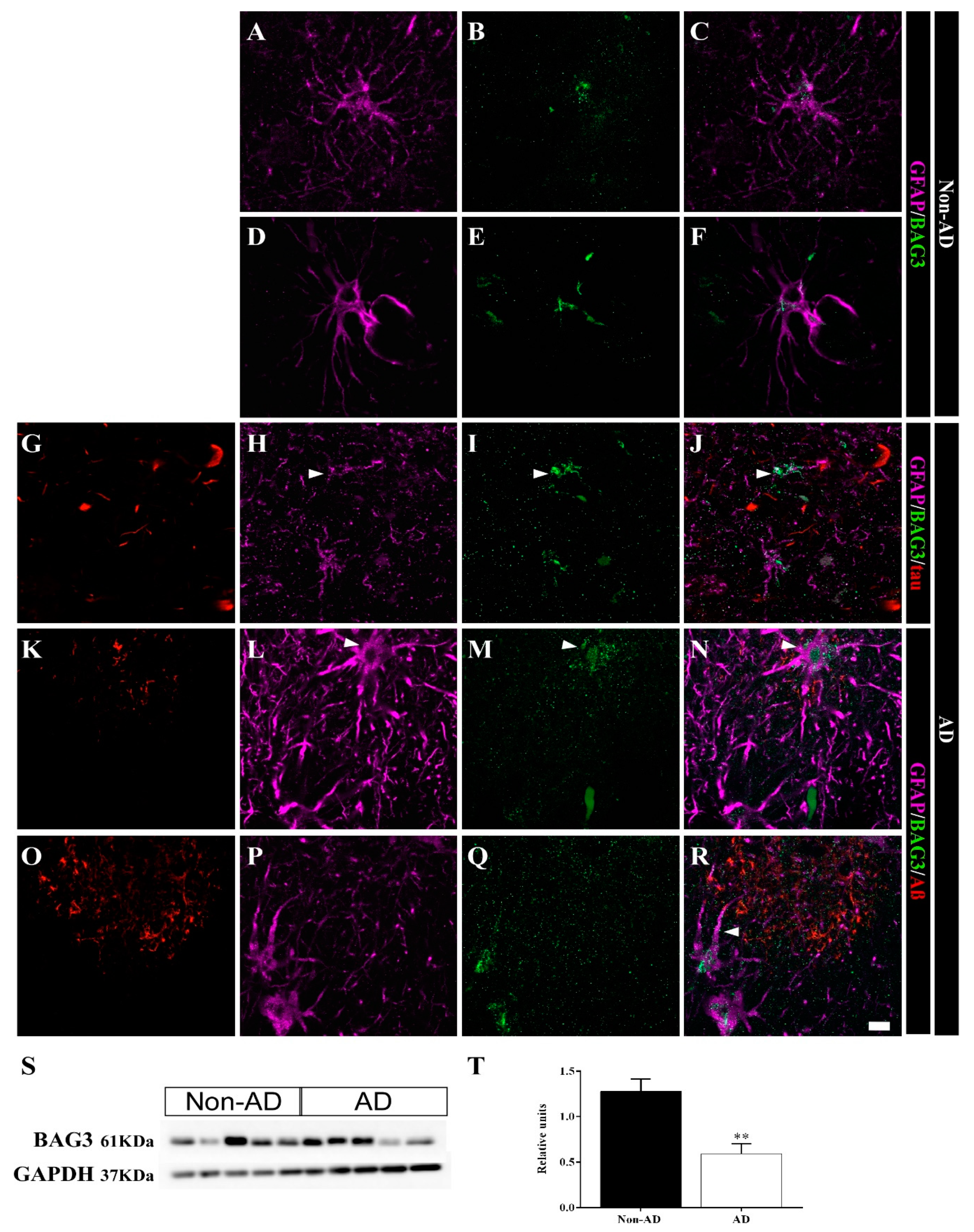
2.5. Differential Colocalization of Chaperones with Astrocytes
3. Discussion
3.1. Volume Reduction, Neurodegeneration and Astrogliosis Occur Specifically in the CA1 Region
3.2. HSP90AB1 and BAG3 Expression in Astrocytes Indicates Possible Roles in Aβ and Tau Homeostasis
4. Materials and Methods
4.1. Human Samples
4.2. Immunoperoxidase Immunohistochemistry
4.3. Immunofluorescence Immunohistochemistry
4.4. Stereological Quantifications
4.5. Immunoblotting
4.6. Proteomic Analysis
4.6.1. Sample Preparation
4.6.2. Protein-Peptide-MS/MS Library Building
4.6.3. SWATH Data Acquisition and Analysis
4.7. Pathway Analysis
4.8. Confocal Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021. [Google Scholar] [CrossRef]
- Tromp, D.; Dufour, A.; Lithfous, S.; Pebayle, T.; Despres, O. Episodic memory in normal aging and Alzheimer disease: Insights from imaging and behavioral studies. Ageing Res. Rev. 2015, 24, 232–262. [Google Scholar] [CrossRef]
- Rahman, M.H.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh Thi, T.; Akter, R.; Goh, S.H.; Kim, C.-S.; Lee, K.-J. Redox Effects of Molecular Hydrogen and Its Therapeutic Efficacy in the Treatment of Neurodegenerative Diseases. Processes 2021, 9, 308. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Kate, M.; Barkhof, F.; Boccardi, M.; Visser, P.J.; Jack, C.R., Jr.; Lovblad, K.O.; Frisoni, G.B.; Scheltens, P.; Geneva Task Force for the Roadmap of Alzheimer’s, B. Clinical validity of medial temporal atrophy as a biomarker for Alzheimer’s disease in the context of a structured 5-phase development framework. Neurobiol. Aging 2017, 52, 167–182.e161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, L.N.; Day, G.S.; Salter, A.; Keefe, S.; Marple, L.M.; Long, J.; LaMontagne, P.; Massoumzadeh, P.; Snider, B.J.; Kanthamneni, M.; et al. Select Atrophied Regions in Alzheimer disease (SARA): An improved volumetric model for identifying Alzheimer disease dementia. Neuroimage Clin. 2020, 26, 102248. [Google Scholar] [CrossRef] [PubMed]
- Delli Pizzi, S.; Franciotti, R.; Bubbico, G.; Thomas, A.; Onofrj, M.; Bonanni, L. Atrophy of hippocampal subfields and adjacent extrahippocampal structures in dementia with Lewy bodies and Alzheimer’s disease. Neurobiol. Aging 2016, 40, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, K.; Hu, X.; Guo, Q. Differential Atrophy in the Hippocampal Subfield Volumes in Four Types of Mild Dementia. Front. Neurosci. 2020, 14, 699. [Google Scholar] [CrossRef] [PubMed]
- Simic, G.; Kostovic, I.; Winblad, B.; Bogdanovic, N. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J. Comp. Neurol. 1997, 379, 482–494. [Google Scholar] [CrossRef]
- Bobinski, M.; De Leon, M.J.; Wegiel, J.; Desanti, S.; Convit, A.; Saint Louis, L.A.; Rusinek, H.; Wisniewski, H.M. The histological validation of post mortem Magnetic Resonance Imaging-Determined hippocampal volume in Alzheimer’s disease. Neuroscience 1999, 95, 721–725. [Google Scholar] [CrossRef]
- Kril, J.J.; Hodges, J.; Halliday, G. Relationship between hippocampal volume and CA1 neuron loss in brains of humans with and without Alzheimer’s disease. Neurosci. Lett. 2004, 361, 9–12. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Coleman, P.D.; Flood, D.G.; Troncoso, J.C. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 1994, 344, 769–772. [Google Scholar] [CrossRef]
- West, M.J.; Kawas, C.H.; Martin, L.J.; Troncoso, J.C. The CA1 region of the human hippocampus is a hot spot in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 908, 255–259. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Kawas, C.H.; Stewart, W.F.; Rudow, G.L.; Troncoso, J.C. Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol. Aging 2004, 25, 1205–1212. [Google Scholar] [CrossRef] [Green Version]
- Vinters, H.V. Emerging concepts in Alzheimer’s disease. Annu. Rev. Pathol. 2015, 10, 291–319. [Google Scholar] [CrossRef]
- Rahman, M.H.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh, T.T.; Akter, R.; Jeong, Y.J.; Goh, S.H.; Kim, C.S.; Lee, K.J. Therapeutic Potential of Natural Products in Treating Neurodegenerative Disorders and Their Future Prospects and Challenges. Molecules 2021, 26, 5327. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.C. Prion-like mechanisms in Alzheimer disease. Handb. Clin. Neurol. 2018, 153, 303–319. [Google Scholar] [CrossRef]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 2015, 138, 2814–2833. [Google Scholar] [CrossRef]
- Cope, T.E.; Rittman, T.; Borchert, R.J.; Jones, P.S.; Vatansever, D.; Allinson, K.; Passamonti, L.; Vazquez Rodriguez, P.; Bevan-Jones, W.R.; O’Brien, J.T.; et al. Tau burden and the functional connectome in Alzheimer’s disease and progressive supranuclear palsy. Brain 2018, 141, 550–567. [Google Scholar] [CrossRef]
- Ubeda-Banon, I.; Saiz-Sanchez, D.; Flores-Cuadrado, A.; Rioja-Corroto, E.; Gonzalez-Rodriguez, M.; Villar-Conde, S.; Astillero-Lopez, V.; Cabello-de la Rosa, J.P.; Gallardo-Alcaniz, M.J.; Vaamonde-Gamo, J.; et al. The human olfactory system in two proteinopathies: Alzheimer’s and Parkinson’s diseases. Transl. Neurodegener. 2020, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Vogels, T.; Murgoci, A.N.; Hromadka, T. Intersection of pathological tau and microglia at the synapse. Acta Neuropathol. Commun. 2019, 7, 109. [Google Scholar] [CrossRef]
- Wilton, D.K.; Dissing-Olesen, L.; Stevens, B. Neuron-Glia Signaling in Synapse Elimination. Annu. Rev. Neurosci. 2019, 42, 107–127. [Google Scholar] [CrossRef]
- Lee, S.J.; Seo, B.R.; Koh, J.Y. Metallothionein-3 modulates the amyloid beta endocytosis of astrocytes through its effects on actin polymerization. Mol. Brain 2015, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Bolos, M.; Llorens-Martin, M.; Jurado-Arjona, J.; Hernandez, F.; Rabano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimers Dis. 2016, 50, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Martini-Stoica, H.; Cole, A.L.; Swartzlander, D.B.; Chen, F.; Wan, Y.W.; Bajaj, L.; Bader, D.A.; Lee, V.M.Y.; Trojanowski, J.Q.; Liu, Z.; et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 2018, 215, 2355–2377. [Google Scholar] [CrossRef]
- Hervas, R.; Oroz, J. Mechanistic Insights into the Role of Molecular Chaperones in Protein Misfolding Diseases: From Molecular Recognition to Amyloid Disassembly. Int. J. Mol. Sci. 2020, 21, 9186. [Google Scholar] [CrossRef] [PubMed]
- Sturner, E.; Behl, C. The Role of the Multifunctional BAG3 Protein in Cellular Protein Quality Control and in Disease. Front. Mol. Neurosci. 2017, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Narasimhan, S.; Changolkar, L.; Riddle, D.M.; Kats, A.; Stieber, A.; Weitzman, S.A.; Zhang, B.; Li, Z.; Roberson, E.D.; Trojanowski, J.Q.; et al. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Pedrero-Prieto, C.M.; Flores-Cuadrado, A.; Saiz-Sanchez, D.; Ubeda-Banon, I.; Frontinan-Rubio, J.; Alcain, F.J.; Mateos-Hernandez, L.; de la Fuente, J.; Duran-Prado, M.; Villar, M.; et al. Human amyloid-beta enriched extracts: Evaluation of in vitro and in vivo internalization and molecular characterization. Alzheimers Res. Ther. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Hondius, D.C.; van Nierop, P.; Li, K.W.; Hoozemans, J.J.; van der Schors, R.C.; van Haastert, E.S.; van der Vies, S.M.; Rozemuller, A.J.; Smit, A.B. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimers Dement. 2016, 12, 654–668. [Google Scholar] [CrossRef]
- Haytural, H.; Mermelekas, G.; Emre, C.; Nigam, S.M.; Carroll, S.L.; Winblad, B.; Bogdanovic, N.; Barthet, G.; Granholm, A.C.; Orre, L.M.; et al. The Proteome of the Dentate Term.minal Zone of the Perforant Path Indicates Presynaptic Impairment in Alzheimer Disease. Mol. Cell Proteom. 2020, 19, 128–141. [Google Scholar] [CrossRef] [Green Version]
- Ho Kim, J.; Franck, J.; Kang, T.; Heinsen, H.; Ravid, R.; Ferrer, I.; Hee Cheon, M.; Lee, J.Y.; Shin Yoo, J.; Steinbusch, H.W.; et al. Proteome-wide characterization of signalling interactions in the hippocampal CA4/DG subfield of patients with Alzheimer’s disease. Sci. Rep. 2015, 5, 11138. [Google Scholar] [CrossRef] [Green Version]
- Schrotter, A.; Oberhaus, A.; Kolbe, K.; Seger, S.; Mastalski, T.; El Magraoui, F.; Hoffmann-Posorske, E.; Bohnert, M.; Deckert, J.; Braun, C.; et al. LMD proteomics provides evidence for hippocampus field-specific motor protein abundance changes with relevance to Alzheimer’s disease. Biochim. Biophys. Acta Proteins. Proteom. 2017, 1865, 703–714. [Google Scholar] [CrossRef]
- Xu, B.; Gao, Y.; Zhan, S.; Xiong, F.; Qiu, W.; Qian, X.; Wang, T.; Wang, N.; Zhang, D.; Yang, Q.; et al. Quantitative protein profiling of hippocampus during human aging. Neurobiol. Aging 2016, 39, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Patassini, S.; Rustogi, N.; Riba-Garcia, I.; Hale, B.D.; Phillips, A.M.; Waldvogel, H.; Haines, R.; Bradbury, P.; Stevens, A.; et al. Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun. Biol. 2019, 2, 43. [Google Scholar] [CrossRef]
- Park, S.A.; Jung, J.M.; Park, J.S.; Lee, J.H.; Park, B.; Kim, H.J.; Park, J.H.; Chae, W.S.; Jeong, J.H.; Choi, S.H.; et al. SWATH-MS analysis of cerebrospinal fluid to generate a robust battery of biomarkers for Alzheimer’s disease. Sci. Rep. 2020, 10, 7423. [Google Scholar] [CrossRef]
- Chang, R.Y.; Etheridge, N.; Nouwens, A.S.; Dodd, P.R. SWATH analysis of the synaptic proteome in Alzheimer’s disease. Neurochem. Int. 2015, 87, 1–12. [Google Scholar] [CrossRef]
- Mai, J.K.; Paxinos, G.; Voss, T. Atlas of the Human Brain; Academic Press: New York, NY, USA, 2007. [Google Scholar]
- Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat Shock Proteins in Alzheimer’s Disease: Role and Targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef] [Green Version]
- Mungas, D.; Reed, B.R.; Jagust, W.J.; DeCarli, C.; Mack, W.J.; Kramer, J.H.; Weiner, M.W.; Schuff, N.; Chui, H.C. Volumetric MRI predicts rate of cognitive decline related to AD and cerebrovascular disease. Neurology 2002, 59, 867–873. [Google Scholar] [CrossRef]
- Price, J.L.; Ko, A.I.; Wade, M.J.; Tsou, S.K.; McKeel, D.W.; Morris, J.C. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch. Neurol. 2001, 58, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejias, E.; Navarro, V.; Jimenez, S.; Sanchez-Mico, M.; Sanchez-Varo, R.; Nunez-Diaz, C.; Trujillo-Estrada, L.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 2016, 132, 897–916. [Google Scholar] [CrossRef] [Green Version]
- Muramori, F.; Kobayashi, K.; Nakamura, I. A quantitative study of neurofibrillary tangles, senile plaques and astrocytes in the hippocampal subdivisions and entorhinal cortex in Alzheimer’s disease, normal controls and non-Alzheimer neuropsychiatric diseases. Psychiatry Clin. Neurosci. 1998, 52, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Vehmas, A.K.; Kawas, C.H.; Stewart, W.F.; Troncoso, J.C. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Mielke, M.L.; Gomez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Pike, C.J.; Cummings, B.J.; Cotman, C.W. Early association of reactive astrocytes with senile plaques in Alzheimer’s disease. Exp. Neurol. 1995, 132, 172–179. [Google Scholar] [CrossRef]
- Perea, J.R.; Lopez, E.; Diez-Ballesteros, J.C.; Avila, J.; Hernandez, F.; Bolos, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleeman, R.M.; Proctor, E.A. Astrocytic Propagation of Tau in the Context of Alzheimer’s Disease. Front. Cell Neurosci. 2021, 15, 645233. [Google Scholar] [CrossRef] [PubMed]
- Witter, M.P. The perforant path: Projections from the entorhinal cortex to the dentate gyrus. Prog. Brain Res. 2007, 163, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Squire, L.R.; Stark, C.E.; Clark, R.E. The medial temporal lobe. Annu. Rev. Neurosci. 2004, 27, 279–306. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [Green Version]
- Kreiser, R.P.; Wright, A.K.; Block, N.R.; Hollows, J.E.; Nguyen, L.T.; LeForte, K.; Mannini, B.; Vendruscolo, M.; Limbocker, R. Therapeutic Strategies to Reduce the Toxicity of Misfolded Protein Oligomers. Int. J. Mol. Sci. 2020, 21, 8651. [Google Scholar] [CrossRef]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [Green Version]
- Criado-Marrero, M.; Rein, T.; Binder, E.B.; Porter, J.T.; Koren, J., 3rd; Blair, L.J. Hsp90 and FKBP51: Complex regulators of psychiatric diseases. Philos. Trans. R Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [Green Version]
- Ou, J.R.; Tan, M.S.; Xie, A.M.; Yu, J.T.; Tan, L. Heat shock protein 90 in Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 796869. [Google Scholar] [CrossRef] [PubMed]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, K.; Kitamura, Y.; Tsuchiya, D.; Kawasaki, T.; Taniguchi, T.; Shimohama, S. Heat shock protein-90-induced microglial clearance of exogenous amyloid-beta1-42 in rat hippocampus in vivo. Neurosci. Lett. 2003, 344, 87–90. [Google Scholar] [CrossRef]
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, L.J.; Sabbagh, J.J.; Dickey, C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 1219–1232. [Google Scholar] [CrossRef] [Green Version]
- Gamerdinger, M.; Carra, S.; Behl, C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: Focus on BAG proteins. J. Mol. Med. 2011, 89, 1175–1182. [Google Scholar] [CrossRef]
- Ji, C.; Tang, M.; Zeidler, C.; Hohfeld, J.; Johnson, G.V. BAG3 and SYNPO (synaptopodin) facilitate phospho-MAPT/Tau degradation via autophagy in neuronal processes. Autophagy 2019, 15, 1199–1213. [Google Scholar] [CrossRef]
- Lei, Z.; Brizzee, C.; Johnson, G.V. BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol. Aging 2015, 36, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Seidel, K.; Vinet, J.; Dunnen, W.F.; Brunt, E.R.; Meister, M.; Boncoraglio, A.; Zijlstra, M.P.; Boddeke, H.W.; Rub, U.; Kampinga, H.H.; et al. The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol. Appl. Neurobiol. 2012, 38, 39–53. [Google Scholar] [CrossRef]
- Wang, G.; Mao, Z. Chaperone-mediated autophagy: Roles in neurodegeneration. Transl. Neurodegener. 2014, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Villar-Conde, S.; Astillero-Lopez, V.; Gonzalez-Rodriguez, M.; Villanueva-Anguita, P.; Saiz-Sanchez, D.; Martinez-Marcos, A.; Flores-Cuadrado, A.; Ubeda-Banon, I. The Human Hippocampus in Parkinson’s Disease: An Integrative Stereological and Proteomic Study. J. Parkinsons. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold Enrichment | Raw p Value | FDR | |
|---|---|---|---|
| PROTEIN CLASS | |||
| Hsp90 family chaperone (PC00028) | 51.83 | 5.74 × 10−5 | 1.58 × 10−3 |
| Non-motor actin binding protein (PC00165) | 12.57 | 4.56 × 10−7 | 8.80 × 10−5 |
| Isomerase (PC00135) | 10.37 | 3.63 × 10−3 | 4.67 × 10−2 |
| MOLECULAR FUNCTION | |||
| Dihydropyrimidinase activity (GO:0004157) | 69.11 | 2.96 × 10−5 | 2.73 × 10−3 |
| Hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in cyclic amides (GO:0016812) | 51.83 | 5.74 × 10−5 | 3.54 × 10−3 |
| Clathrin binding (GO:0030276) | 11.52 | 5.29 × 10−4 | 1.28 × 10−2 |
| Unfolded protein binding (GO:0051082) | 10.05 | 8.56 × 10−4 | 1.58 × 10−2 |
| BIOLOGICAL PROCESS | |||
| Pyrimidine nucleobase catabolic process (GO:0006208) | 59.24 | 4.20 × 10−5 | 9.16 × 10−3 |
| Pyrimidine nucleobase catabolic process (GO:0006208) | 59.24 | 4.20 × 10−5 | 9.16 × 10−3 |
| Pyrimidine nucleobase metabolic process (GO:0006206) | 37.7 | 1.25 × 10−4 | 1.51 × 10−2 |
| Pyrimidine nucleobase metabolic process (GO:0006206) | 37.7 | 1.25 × 10−4 | 1.51 × 10−2 |
| Protein stabilization (GO:0050821) | 29.1 | 2.04 × 10−5 | 1.48 × 10−2 |
| Regulation of protein stability (GO:0031647) | 27.64 | 2.43 × 10−5 | 1.32 × 10−2 |
| Nucleobase metabolic process (GO:0009112) | 24.04 | 3.95 × 10−5 | 9.56 × 10−3 |
| Pyrimidine-containing compound metabolic process (GO:0072527) | 18.85 | 7.44 × 10−4 | 3.96 × 10−2 |
| Establishment or maintenance of cell polarity (GO:0007163) | 12.8 | 6.55 × 10−5 | 1.02 × 10−2 |
| Identifier | Pathway Name | #Found | #Total | p Value | FDR | Submitted Entities Found |
|---|---|---|---|---|---|---|
| R-HSA-422475 | Axon guidance | 21 | 558 | 1.80 × 10−5 | 0.0056 | AP2A2, HSP90AA1, DAG1, CLASP2, MAP2K1, DPYSL5, AP2A1, EZR, PRKACA, RPL35, CSNK2A1, PLXNA4, DPYSL4, DLG3, DLG4, CRMP1, MSN, RPS25, HSP90AB1, PFN2, RPLP2 |
| R-HSA-69275 | G2/M Transition | 11 | 198 | 7.85 × 10−5 | 0.0065 | PRKACA, DCTN1, HSP90AA1, PPP2R2A, CKAP5, PPME1, SKP1, PAFAH1B1, PPP2R1A, HSP90AB1, PRKAR2B |
| R-HSA-3371568 | Attenuation phase | 3 | 14 | 9.22 × 10−4 | 0.0350 | HSP90AA1, HSP90AB1, FKBP4 |
| R-HSA-399954 | Sema3A PAK dependent Axon repulsion | 3 | 16 | 0.00135 | 0.0459 | HSP90AA1, PLXNA4, HSP90AB1 |
| R-HSA-9613829 | Chaperone Mediated Autophagy | 3 | 22 | 0.00331 | 0.0744 | HSP90AA1, HSP90AB1, GFAP |
| R-HSA-168249 | Innate Immune System | 27 | 1187 | 0.00399 | 0.0744 | HMGB1, HSP90AA1, S100A1, PPIA, PRDX6, FABP5, C4B, C4B_2, SUGT1, BAIAP2, SKP1, HEBP2, GSTP1, GPI, AP2A2, PIN1, COTL1, HSP90B1, MGST1, PPP2R1A, MAP2K1, PADI2, MAPK10, UBE2M, PRKACA, HMOX2, CD44, HSP90AB1 |
| R-HSA-5336415 | Uptake and function of diphtheria toxin | 2 | 7 | 0.00405 | 0.0744 | HSP90AA1, HSP90AB1 |
| R-HSA-5339562 | Uptake and actions of bacterial toxins | 4 | 48 | 0.00407 | 0.0744 | HSP90AA1, PDCD6IP, MAP2K1, HSP90AB1 |
| R-HSA-3371571 | HSF1-dependent transactivation | 3 | 24 | 0.00421 | 0.0744 | P07900, P08238, Q02790 |
| R-HSA-3371497 | HSP90 chaperone cycle for steroid hormone receptors (SHR) | 4 | 57 | 0.00921 | 0.0760 | DCTN1, HSP90AA1, HSP90AB1, FKBP4 |
| R-HSA-3371556 | Cellular response to heat stress | 5 | 95 | 0.01141 | 0.0799 | HSP90AA1, HSPA4L, BAG3, HSP90AB1, FKBP4 |
| R-HSA-3371511 | HSF1 activation | 2 | 12 | 0.04212 | 0.1685 | HSP90AA1, HSP90AB1 |
| R-HSA-449147 | Signaling by Interleukins | 11 | 456 | 0.04615 | 0.1728 | HMGB1, PRKACA, HSP90AA1, PPIA, FSCN1, HSP90B1, SKP1, PPP2R1A, MAP2K1, MSN, MAPK10 |
| R-HSA-8953897 | Cellular responses to external stimuli | 13 | 579 | 1.80 × 10−5 | 0.0056 | CSRP1, DCTN1, HSP90AA1, HSPA4L, PRDX6, MAPK10, FKBP4, RPL35, BAG3, GSTP1, RPS25, HSP90AB1, RPLP2 |
| Case | Gender | Age (y) | PMD (h) | Brain Weight (g) | Cause of Death | Braak Stage | Treatment | Assay |
|---|---|---|---|---|---|---|---|---|
| AD cases (n = 16) | ||||||||
| 1 | F | 74 | 4:00 | 1042 | Cardiorespiratory arrest | V | Formalin-fixed | IHC, IFC1 |
| 2 | F | 80 | 4:00 | 910 | Respiratory infection | V | Formalin-fixed | IHC, IFC1, IFC2 |
| 3 | M | 77 | 5:00 | 1330 | Sepsis (respiratory origin) | VI | Formalin-fixed | IHC, IFC1 |
| 4 | F | 84 | 2:00 | 920 | Unknow | V | Formalin-fixed | IHC, IFC1 |
| 5 | M | 77 | 6:00 | 1060 | Acute respiratory infection | VI | Formalin-fixed | IHC, IFC1 |
| 6 | M | 92 | 6:00 | 960 | Unknow | VI | Formalin-fixed | IHC, IFC2 |
| 7 | M | 75 | 3:00 | 1050 | Multiorganic arrest | V | Formalin-fixed | IHC |
| 8 | F | 85 | 2:00 | 1150 | Cardiorespiratory arrest | VI | Formalin-fixed | IHC, IFC2 |
| 9 | F | 83 | 2:00 | 1000 | Respiratory insufficiency | VI | Formalin-fixed | IHC |
| 10 | F | 81 | 6:30 | 935 | Cardiorespiratory arrest | VI | Fresh-frozen | PR, WB |
| 11 | F | 75 | 16:15 | 1200 | Septic shock, sacral ulcer | VI | Fresh-frozen | PR, WB |
| 12 | M | 80 | 21:45 | 1300 | Respiratory insufficiency | VI (LBD) | Fresh-frozen | PR, WB |
| 13 | F | 80 | 5:00 | 1060 | Acute heart failure | VI | Fresh-frozen | PR, WB |
| 14 | F | 72 | 14:00 | 1060 | Cardiorespiratory arrest, Deep venous thrombosis | VI (LBD) | Fresh-frozen | PR, WB |
| 15 | F | 90 | 12:15 | 920 | Respiratory insufficiency, respiratory infection | VI | Fresh-frozen | PR |
| 16 | F | 76 | 11:10 | 900 | Respiratory insufficiency | VI | Fresh-frozen | WB |
| Non-AD cases (n = 16) | ||||||||
| 17 | M | 84 | 3:00 | 1400 | Non filiate miocardiopathy/ Cardiac arrest | - | Formalin-fixed | IHC, IFC2 |
| 18 | F | 81 | 5:00 | 1100 | Pionefritis by E. coli/ Multiorganic arrest | - | Formalin-fixed | IHC |
| 19 | M | 88 | 3:00 | 1285 | Unknow | II | Formalin-fixed | IHC |
| 20 | M | 83 | 4:00 | 1152 | Unknow | II | Formalin-fixed | IHC |
| 21 | F | 62 | 2:00 | 1050 | Myelodysplastic Syndrome | - | Formalin-fixed | IHC, IFC2 |
| 22 | M | 63 | 2:00 | 1400 | Breast adenocarcinoma | - | Formalin-fixed | IHC, IFC2 |
| 23 | M | 58 | 6:00 | 1500 | Acute myocardial infarction | - | Formalin-fixed | IHC |
| 24 | M | 53 | 5:00 | 1300 | Rectum adenocarcinoma | - | Formalin-fixed | IHC |
| 25 | M | 78 | 4:00 | 1100 | Lung carcinoma | - | Formalin-fixed | IHC |
| 26 | F | 83 | 7:20 | 1320 | Intestinal embolism, surgery ischemia | II | Fresh-frozen | PR, WB |
| 27 | M | 83 | 13:00 | 1630 | AgD I, pathology due to argyrophile granules/cardiorespiratory insufficiency | - | Fresh-frozen | PR |
| 28 | F | 37 | 9:00 | 1200 | Refractory septic shock | - | Fresh-frozen | PR, WB |
| 29 | M | 57 | 12:00 | 1560 | Lung Carcinoma | - | Fresh-frozen | PR |
| 30 | M | 68 | 4:10 | 1350 | Sepsis | Fresh-frozen | WB | |
| 31 | F | 82 | 4:00 | 800 | Respiratory insufficiency | - | Fresh-frozen | WB |
| 32 | F | 71 | 7:08 | 975 | Cardiorespiratory arrest | - | Fresh-frozen | WB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Rodriguez, M.; Villar-Conde, S.; Astillero-Lopez, V.; Villanueva-Anguita, P.; Ubeda-Banon, I.; Flores-Cuadrado, A.; Martinez-Marcos, A.; Saiz-Sanchez, D. Neurodegeneration and Astrogliosis in the Human CA1 Hippocampal Subfield Are Related to hsp90ab1 and bag3 in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 165. https://doi.org/10.3390/ijms23010165
Gonzalez-Rodriguez M, Villar-Conde S, Astillero-Lopez V, Villanueva-Anguita P, Ubeda-Banon I, Flores-Cuadrado A, Martinez-Marcos A, Saiz-Sanchez D. Neurodegeneration and Astrogliosis in the Human CA1 Hippocampal Subfield Are Related to hsp90ab1 and bag3 in Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(1):165. https://doi.org/10.3390/ijms23010165
Chicago/Turabian StyleGonzalez-Rodriguez, Melania, Sandra Villar-Conde, Veronica Astillero-Lopez, Patricia Villanueva-Anguita, Isabel Ubeda-Banon, Alicia Flores-Cuadrado, Alino Martinez-Marcos, and Daniel Saiz-Sanchez. 2022. "Neurodegeneration and Astrogliosis in the Human CA1 Hippocampal Subfield Are Related to hsp90ab1 and bag3 in Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 1: 165. https://doi.org/10.3390/ijms23010165