1. Introduction
Cancer is a disease that seriously threatens human life and health worldwide [
1]. The occurrence and development of cancer is a complex multistep process involving the accumulation of multiple genetic and epigenetic changes that lead to alterations in a large number of tumor-related genes, such as the activation of oncogenes and the inactivation of tumor suppressor genes [
2,
3]. However, cancer is still insufficiently understood due to its complexity, which hinders efforts to achieve a complete cure of cancer. Therefore, further explorations of the molecular mechanism underlying the occurrence and development of cancer, such as discovering the role of more tumor-related genes in the progression of cancer, are still indispensable.
KIAA1217, the human homolog of murine Skt (sickle tail), was identified in the large HUGE (human unidentified gene-encoded) protein database [
4] and is a macromolecular protein with an unknown function. To date, only a few articles have reported KIAA1217 [
5,
6]. It is ubiquitously expressed in the cytoplasm [
5]. Because it is specifically expressed in the nucleus pulposus of intervertebral discs in humans and mice, KIAA1217 is presumed to be necessary for the normal development of intervertebral discs and is a good candidate gene for lumbar disc herniations in humans [
6]. In particular, a novel KIAA1217-RET fusion gene was identified in lung adenocarcinomas [
5]. This fusion gene, resulting from the rearrangement of chromosome 10, is produced by the fusion of KIAA1217 (NM_001282767.1) exons 1–11 and RET exons 11–20 in a patient with non-small cell lung cancer [
5]. The expression of the KIAA1217-RET fusion gene increases cell proliferation and invasion through the activation of the PI3K/AKT and ERK signaling pathways and ultimately leads to the oncogenic transformation of lung cells [
5]. However, the potential role and mechanism of KIAA1217 in the progression of cancer remains unclear.
Liver cancer, of which hepatocellular carcinoma (HCC) accounts for 75–85% of cases, is the sixth most commonly diagnosed cancer (4.7% of the total cases) and the third leading cause of cancer-related death (8.3%) [
1]. In particular, HCC is one of the malignant tumors with a high incidence and the worst survival rate in China [
7]. In recent years, with the continuous development of HCC diagnostic and treatment methods, the overall survival rate has been improved to a large extent [
8,
9,
10]. However, the prognosis of patients with HCC is still unfavorable, with only an 18% 5-year survival rate from 2009–2015 [
11]. This low survival rate is largely attributed to insidious onset, rapid progression, strong infiltration, and early metastasis of HCC [
12,
13]. Indeed, most patients with liver cancer are diagnosed at an advanced stage, and many have already developed intrahepatic metastasis or even systemic metastases in other tissues [
8,
9,
10,
14,
15]. Thus, metastasis is one of the key factors affecting the survival and prognosis of patients with HCC [
12,
13], and the identification of potential key targets and elucidation of the molecular mechanism of HCC metastasis is very important.
Notably, epithelial-mesenchymal transition (EMT) plays a critical role in the invasion and metastasis cascades of cancer [
16,
17,
18], including HCC [
19,
20,
21]. EMT is a reversible biological process in which epithelial cells transform into mesenchymal cell phenotypes under certain conditions [
16]. Cancer cells undergoing EMT lose cell polarity and cell-cell adhesions, reorganize the cytoskeleton, downregulate epithelial markers, and upregulate mesenchymal markers [
16]. These changes lead to the increased migration and invasion of cancer cells [
22]. Studies have shown that EMT is precisely regulated by a complex and dynamic mechanism involving the regulation of multiple signaling pathways and multiple factors, including transcription factors, microRNAs, and growth factors [
23,
24]. Hence, the mechanism of EMT induction in the invasion and metastasis of HCC must be further explored.
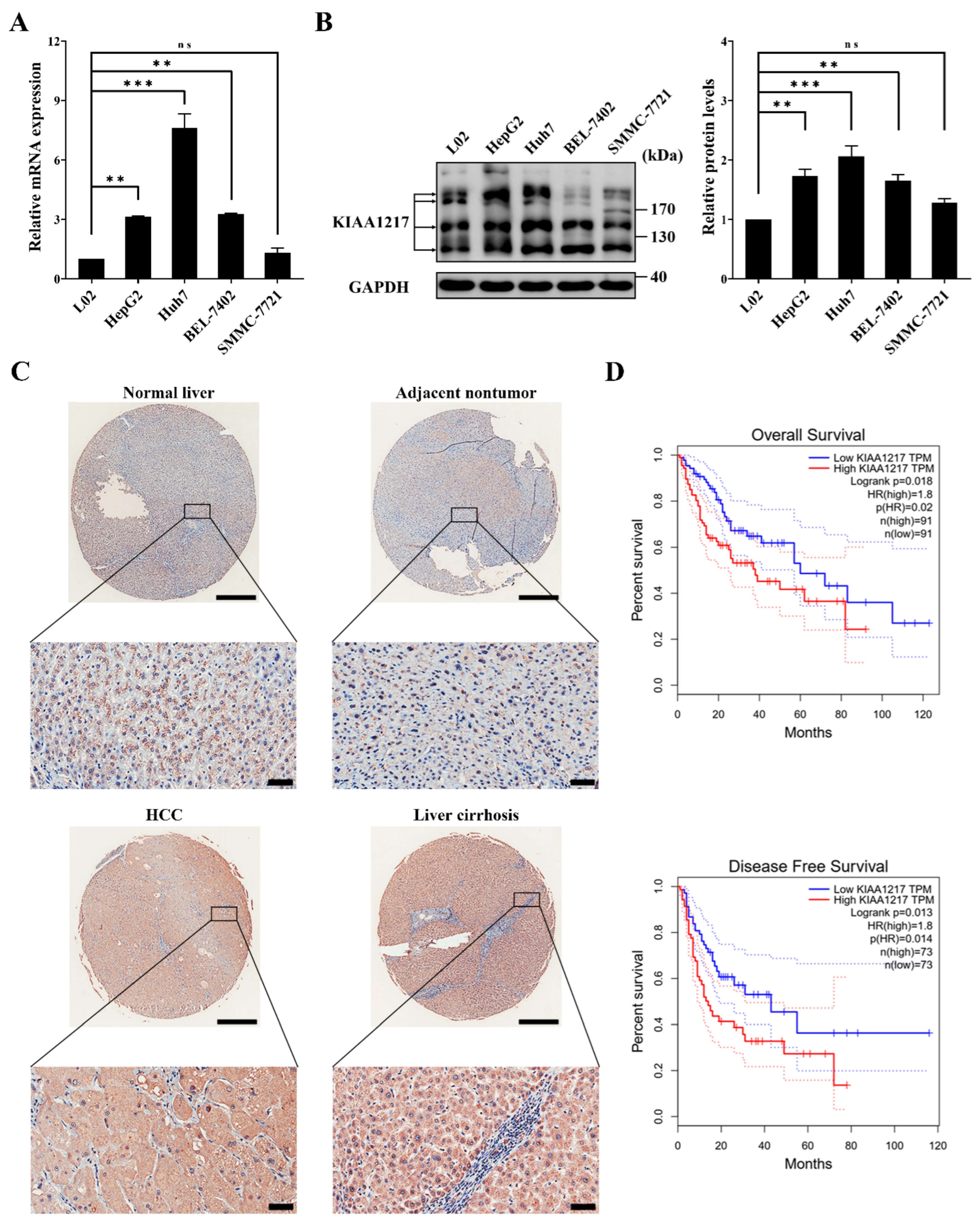
In this study, we found that KIAA1217 expression was frequently upregulated in HCC cell lines and tissues, and high KIAA1217 expression was closely associated with shorter survival of patients with HCC. Then, we described a critical role for KIAA1217 in EMT induction and HCC metastasis and elucidated the underlying molecular mechanisms. We identified KIAA1217 as an oncogenic protein during HCC progression for the first time and suggested it as a potential antimetastatic target for HCC treatment.
3. Discussion
Although tumor invasion and metastasis are the main causes of a poor prognosis and cancer-related death, effective strategies that suppress cancer metastasis are still lacking in the clinic [
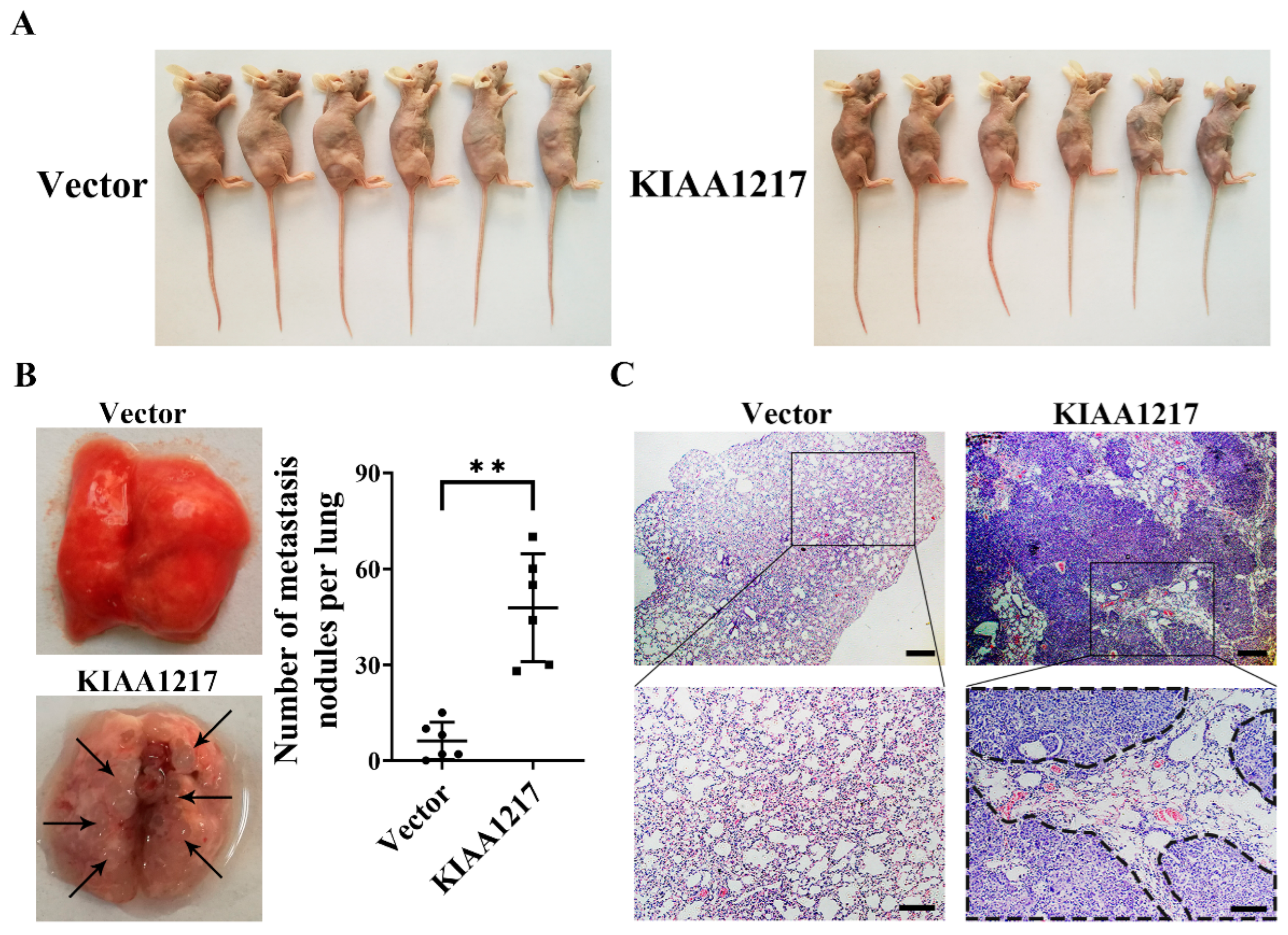
30]. Therefore, analyses of the molecular drivers of tumor metastasis, such as aberrant actions of oncogenes or tumor suppressor genes, may help researchers identify therapeutic target genes to suppress metastasis. In this study, we showed for the first time that KIAA1217, a macromolecular protein with an unknown function, significantly promoted HCC metastasis by inducing EMT. We also revealed that high KIAA1217 expression was closely related to shorter survival of patients with HCC, indicating its potential as a therapeutic target for the suppression of HCC metastasis.
KIAA1217 is present in the HUGE protein database. The database was established by the Kazusa Gene Research Center in 1994 [
4]. It is mainly used to analyze the proteins (>50 kD) encoded by newly recognized large cDNAs (>4 kb), and the genes encoding these proteins are uniformly named by KIAA plus four numbers [
4]. To date, few reports have investigated KIAA1217, of which only a few studies have described its relation to the development of cancer [
5,
31,
32,
33,
34]. Based on genetic association studies in pancreatic cancer databases, KIAA1217 is upregulated, suggesting that KIAA1217 may be a putative target for pancreatic cancer [
32]. Copy number variation of KIAA1217 is also frequently observed in HCC [
31] or breast cancer [
34]. However, the potential role and mechanism of KIAA1217 in the progression of cancer remains unclear. In the present study, KIAA1217 was found to be frequently overexpressed in HCC cell lines and tissues. It is worth noting that KIAA1217 was low expressed in SMMC-7721 cells. We speculate that this is due to the extraordinary heterogeneity of HCC [
14]. Furthermore, overexpression and knockdown experiments indicated that KIAA1217 promoted cell migration and invasion by inducing EMT in vitro. Nevertheless, it had no effect on HCC proliferation. Consistently, a metastasis assay in vivo showed that the mice intravenously injected with HCC cells overexpressing KIAA1217 exhibited more severe cachexia and a greater number of HCC nodules on the surface of the lungs, indicating that KIAA1217 significantly enhanced lung metastasis in vivo. Clinically, patients with HCC presenting high KIAA1217 expression experienced shorter OS and DFS times than patients with low expression. In addition, in contrast to the matched nontumor tissues, KIAA1217 expression was increased in the 18 other types of cancers, which was analyzed using the GEPIA web server [
25] (
Figure S5). This finding suggests that KIAA1217 may also play a certain role in other tumors, which deserves further investigation. Taken together, our study shows that KIAA1217 may function as an oncogene and promote HCC invasion and metastasis in vitro and in vivo.
In addition, a novel KIAA1217-RET fusion gene was identified in lung adenocarcinomas and revealed that the KIAA1217-RET fusion gene functions as an oncogenic driver gene [
5]. Interestingly, another study by our team also observed a truncated transcript of KIAA1217 expressed in ovarian cancer cell lines (unpublished data). This finding suggests that a fusion gene consisting of part of the KIAA1217 sequence and another gene exists in ovarian cancer; however, this requires further validation. The occurrence of genomic instability, including gene fusion or rearrangement, is one of the important mechanisms inducing cancer [
35]. Therefore, KIAA1217, an oncogene, also appears to participate in the occurrence and development of cancer in the form of fusion genes via genome rearrangements.
Mechanistic studies revealed that KIAA1217 induced EMT by interacting with and activating STAT3 and that KIAA1217 mediated the activation of STAT3 by recruiting and activating JAK1/2. The STAT3 pathway is a classic signaling pathway in cells that is also closely related to the progression of cancer [
28,
36]. Our study further confirmed an essential role for the STAT3 pathway in EMT and metastasis of HCC. Notably, the KIAA1217-induced expression of N-cadherin, an EMT-related marker, did not appear to be affected by STAT3 to the same extent as the expression of other markers. Therefore, we considered that other pathways might also be involved in KIAA1217-induced expression of N-cadherin. Interestingly, the interaction of KIAA1217 and p-STAT3 prevented activated p-STAT3 from being transported to the nucleus, as most p-STAT3 was retained in the cytoplasm. Activated p-STAT3 is widely considered functional by the activation of transcription after transport to the nucleus [
28]; however, our study suggested a different functional pattern of STAT3, in which most p-STAT3 remained in the cytoplasm and coordinated the Notch and Wnt/β-catenin pathways to facilitate EMT induction and HCC metastasis triggered by KIAA1217. Considering the structural features of the KIAA1217 protein predicted by the Pfam website and its ubiquitous expression in the cytoplasm, we speculate that KIAA1217 may function as an adaptor protein or scaffold protein to regulate the signaling pathway network.
Notably, we occasionally observed that higher expression of KIAA1217 was significantly more common in cirrhotic liver tissues than in normal liver, adjacent nontumor, and HCC tissues when performing IHC analyses of the TMA slide to compare the KIAA1217 expression levels in adjacent nontumor and HCC tissues (
Figure 1C). Liver cirrhosis is the final pathological result of liver damage resulting from various chronic liver diseases and is characterized by tissue fibrosis and the conversion of the normal liver architecture into structurally abnormal nodules [
37]. Liver cirrhosis is also a major risk factor for the development of HCC, and almost all patients with HCC are preceded by cirrhosis [
27]. Although liver cirrhosis is caused by many factors, liver fibrosis, which is the precursor of cirrhosis, is a key pathological process of all chronic liver diseases evolving into cirrhosis [
37,
38,
39]. Myofibroblasts are a critical cell type involved in liver fibrosis [
38]. According to recent studies, the transdifferentiation of hepatic stellate cells is the major source of myofibroblasts [
24,
26,
37,
39]. Moreover, accumulating evidence indicates that myofibroblasts are also derived from portal fibroblasts, circulating fibrocytes, bone marrow, and EMT of hepatocytes and cholangiocytes [
38,
39]. Since we documented that KIAA1217 induced EMT in HCC cells and was highly expressed in cirrhotic liver tissues, we speculate that KIAA1217 may play a certain role in the progression of liver fibrosis and liver cirrhosis, which requires further investigation. Moreover, further studies are needed to determine whether KIAA1217 represents a potential biomarker for the prediction of disease progression or a therapeutic target in cirrhosis in the future.
In conclusion, KIAA1217, which may function as an adaptor protein or scaffold protein in the cytoplasm, forms a complex with JAK1/2 and STAT3 and facilitates STAT3 activation. Then, activated STAT3 remains in the cytoplasm to further activate the Notch and Wnt/β-catenin pathways, through which KIAA1217 induces EMT and accordingly is involved in promoting HCC metastasis (
Figure 8E). Therefore, KIAA1217 is identified as an oncogenic protein during HCC progression and may serve as a putative antimetastatic target for HCC treatment.
4. Materials and Methods
4.1. Cell Culture
Four HCC cell lines (HepG2, Huh7, SMMC-7721, and BEL-7402), the human normal hepatocyte cell line L02, and the human embryonic kidney cell line HEK-293T were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HEK-293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, 10-013-CVRC, Corning, Suzhou, Jiangsu, China) supplemented with 10% fetal bovine serum (FBS, VS500T, Ausbian, Sydney, Australia) and a 1% penicillin-streptomycin solution (15070063, Gibco, Waltham, MA, USA), and other cell lines were maintained in RPMI-1640 medium (10-040-CVRC, Corning, Suzhou, Jiangsu, China) supplemented with 10% FBS and a 1% penicillin-streptomycin solution. All cell lines were cultured at 37 °C with 5% CO2 in an incubator.
4.2. Tissue Microarray (TMA)
HCC TMA slide (HlivH060CD03) was purchased from Outdo Biotechnology Co., Ltd. (Shanghai, China). All patients provided consent for the use of their tissue samples and clinical data. This study was approved by the Committees for Ethical Review of Research at Northeast Normal University.
4.3. Animals
Female athymic BALB/c nude mice (4–6 weeks old) were purchased from Changsheng Biotechnology Co., Ltd. (Benxi, Liaoning, China) and raised under specific pathogen-free conditions. Animal care and experimental protocols were conducted with approval from the Northeast Normal University Experimental Animal Care Commission of China and performed following established guidelines.
4.4. Plasmids and siRNA Transfection
The KIAA1217 expression plasmid was constructed and synthesized by GenScript Biotechnology Co., Ltd. (Nanjing, Jiangsu, China), in which the full-length ORF of the human KIAA1217 gene (NM_019590.5) was cloned into the pcDNA3.1-DYK vector. The pCMV-DYK-STAT3 expression plasmid was constructed and maintained in our laboratory. The empty vector, named Vector, served as the negative control.
pGreenPuro lentiviral vectors containing short hairpin RNAs (shRNAs) targeting KIAA1217 were constructed and synthesized by GENEWIZ Biotechnology Co., Ltd. (Suzhou, Jiangsu, China) and designated shKIAA1217-1 and shKIAA1217-2. The lentiviral vector containing the nontargeting shRNA named shNC was used as the negative control. The designed target sequences are described in
Supplementary Table S1.
The siRNA targeting STAT3 was designed and synthesized by GenePharma (Suzhou, Jiangsu, China). The sequences of the siRNAs are listed in
Supplementary Table S1.
Lipofectamine 2000 Transfection Reagent (11668019, Thermo Fisher Scientific, Waltham, MA, USA) or X-tremeGENE HP DNA Transfection Reagent (6366236001, Roche, Mannheim, Germany) was used for plasmid transfection. Lipofectamine 2000 Transfection Reagent or X-tremeGENE siRNA Transfection Reagent (4476093001, Roche, Mannheim, Germany) was used for siRNA transfection. Transfections were performed according to the manufacturer’s protocols.
4.5. Establishment of Stable Cell Lines
The HepG2 cell line stably overexpressing KIAA1217 was obtained by selection for G418 (10131027, Gibco, Waltham, MA, USA) resistance. KIAA1217, or empty expression plasmids, were transfected into HepG2 cells using Lipofectamine 2000 Transfection Reagent according to the manufacturer’s protocols, and the transfected cells were subsequently selected with G418 (200 μg/mL) for 2 weeks to generate stable cell lines. The stably transfected cells were validated by performing RT–qPCR and Western blot analyses. The stable cell lines were then maintained in culture medium supplemented with 200 μg/mL G418.
Stable KIAA1217 knockdown in HepG2 cells was generated by lentivirus-mediated shRNA transduction. For lentiviral packaging, HEK-293T cells were cotransfected with psPAX2, pMD2.G, and pGreenPuro (shKIAA1217-1, shKIAA1217-2, or shNC) using Lipofectamine 2000 Transfection Reagent according to the manufacturer’s protocols. 48 h after transfection, the viral supernatant was collected. HepG2 cells were incubated with virus-containing supernatant for 12–16 h in the presence of 10 μg/mL polybrene (C0351, Beyotime, Shanghai, China). The infected cells were then selected with puromycin (2 μg/mL) (ST551, Beyotime, Shanghai, China) for 48 h. Stable knockdown cells were confirmed using RT–qPCR and Western blot analyses.
4.6. RNA Isolation and Real-Time Quantitative PCR (RT–qPCR)
Total RNA was isolated using TRIzol reagent (15596026, Invitrogen, Waltham, MA, USA) and reverse-transcribed into cDNAs with EasyScript® First-Strand cDNA Synthesis SuperMix (AE301-02, TransGen Biotech, Beijing, China) according to the manufacturer’s protocols.
The cDNA templates were then subjected to RT–qPCR using the SYBR Green PCR Kit (04913914001, Roche, Mannheim, Germany) according to the manufacturer’s instructions. The assay was performed with a PikoReal 96 PCR System (Thermo Fisher Scientific, Waltham, MA, USA). β-Actin was used as an internal control. The relative expression levels were quantified and analyzed using PikoReal 2.1 software (Thermo Fisher Scientific, Waltham, MA, USA). The relative expression levels of the target genes were determined using the 2
−ΔΔCt method. Each sample was analyzed in triplicate. The primers were all synthesized and purchased from GENEWIZ Biotechnology Co., Ltd. (Suzhou, Jiangsu, China). The primer sequences are listed in
Supplementary Table S2.
4.7. Western Blot Assay
Total proteins and nuclear/cytoplasmic proteins were extracted with RIPA lysis buffer (P0013B, Beyotime, Shanghai, China) and a Nuclear-Cytoplasmic Extraction Kit (P0028, Beyotime, Shanghai, China), respectively. Protein lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (88518, Thermo Fisher Scientific, Waltham, MA, USA). The membranes were blocked with TBST (ST673, Beyotime, Shanghai, China) containing 5% skim milk (P0216, Beyotime, Shanghai, China) for 1 hr at room temperature and then incubated with appropriate antibodies at 4 °C overnight. After washing, the membranes were incubated with HRP-conjugated anti-IgG (Proteintech, Wuhan, Hubei, China) for 2 h at room temperature. The antigen-antibody complexes on the membranes were detected with High-sig ECL reagent (180-5001, Tanon, Shanghai, China). The images were acquired using the MicroChemi system (70-25-00, NDR Bio-Imaging Systems, Jerusalem, Israel) and quantified with ImageJ software (ImageJ 1.50i, National Institutes of Health, Bethesda, MD, USA). GAPDH and Histone H3 served as internal controls. The antibodies used in this study are listed in
Supplementary Table S3.
4.8. Wound-Healing Assay
The wound-healing assay was used to evaluate cell migration. Transfected cells were cultured on 6-well plates until reaching confluence, then a line was scraped in the cellular monolayer using sterile 200 μL pipette tips, and then the cells were cultured in a serum-free medium. Images of cell migration toward the wound were captured under an inverted microscope (BX50, Olympus, Tokyo, Japan) at 0, 24, 48, and 72 h after scratching. The migration area was quantified using ImageJ software to assess the rate of wound closure.
4.9. Transwell Migration Assay
The cell migration capacity was also assessed using the Transwell migration assay. Transwell chambers (3422, Corning, New York, NY, USA) with an 8 μm pore size polycarbonate membrane were used to perform the 2-chamber migration assay. Transfected cells (5 × 104) suspended in 200 μL of serum-free medium were seeded into the upper chambers of 24-well plates, and 600 μL of medium containing 10% FBS were added into the lower chambers as the attractant. After incubation at 37 °C for 48 h, non migrated cells that remained on the upper surface of the membranes were removed with cotton swabs, and the cells that had migrated to the lower surface of the membranes were fixed with 4% paraformaldehyde (P0099, Beyotime, Shanghai, China) and stained with 0.1% crystal violet (C0121, Beyotime, Shanghai, China). Ultimately, the migrated cells were captured and counted in five random fields per well under an inverted microscope to assess the cell migration rate.
4.10. Cancer Cell Spheroid Invasion Assay in a Three-Dimensional (3D) Setting
Tumor cell invasion was assessed using a 3D cancer cell spheroid invasion assay [
40]. The cells inoculated on the lid of 10 cm dishes were cultured in hanging drops for 72 h to form spheroids (approximately 1 × 10
3 cells per droplet). The spheroids were collected from the lid and mixed with Matrigel (356234, Corning, New York, NY, USA) and collagen type I (354236, Corning, New York, NY, USA). Then, the polymers were embedded in 24-well plates for 30 min at 37 °C to generate 3D culture systems, followed by submergence of the 3D cultures in the cell culture medium. After 48 h, images of invading cells were captured using an inverted microscope. The invasion area was quantified using ImageJ software to assess the invasion capacity of tumor cells.
4.11. Immunofluorescence (IF) Staining Assay
Cells were seeded onto coverslips in 24-well plates and then fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 (ST797, Beyotime, Shanghai, China), and blocked with 5% bovine serum albumin (BSA, A8020, Solarbio, Beijing, China). After incubation with appropriate antibodies at 4 °C overnight, cells were incubated with Cy3 or FITC-labeled anti-IgG (Beyotime, Shanghai, China) at room temperature, followed by nuclear counterstaining with DAPI (C1005, Beyotime, Shanghai, China). Images were captured using a laser scanning confocal microscope (LSM 880, ZEISS, Oberkochen, Germany). The antibodies used in this study are shown in
Supplementary Table S3.
4.12. Immunohistochemistry (IHC) Assay
An immunohistochemistry assay was performed to detect KIAA1217 expression in the TMA tissue samples. The TMA slide was dewaxed in xylene and rehydrated with a series of graded alcohol solutions. Then, antigen retrieval was performed with sodium citrate buffer (C1032, Solarbio, Beijing, China). According to the protocols of the two-step detection kit (PV-9001, ZSGB-BIO, Beijing, China), the TMA slide was incubated in an appropriate endogenous peroxidase blocker for 10 min at room temperature, followed by incubation with anti-SKT (KIAA1217 is also known as SKT) antibodies overnight at 4 °C and subsequent incubation with response enhancer and enhanced enzyme-labeled goat anti-rabbit IgG polymer for 20 min at room temperature. A DAB Chromogenic Kit (ZLI-9017, ZSGB-BIO, Beijing, China) was used to detect antibody binding, and the reaction was stopped by immersing the TMAs in running water once a brown color appeared. Finally, the TMA slide was counterstained with hematoxylin (G1121, Solarbio, Beijing, China), dehydrated using a series of graded alcohol solutions, and mounted. Images were photographed with an inverted microscope. Appropriate positive and negative controls were included for each run of the IHC assay. The antibodies used in this study are listed in
Supplementary Table S3.
4.13. Coimmunoprecipitation (Co-IP) Assay
Co-IP was performed using Protein A/G Magnetic Beads (HY-K0202, MedChemExpress, Monmouth Junction, NJ, USA) according to the manufacturer’s protocols to confirm protein-protein interactions. 25 μL of magnetic beads pretreated with PBST (0.5% Triton X-100 in PBS) solution were fully suspended with PBST containing the antibodies at a final concentration of 5 μg/mL and incubated at 4 °C for 2 h in a flip mixer. Proteins extracted from cells using IP lysis solution (87787, Thermo Fisher Scientific, Waltham, MA, USA) were fully suspended with the antibody-magnetic bead complexes and incubated at 4 °C overnight in a flip mixer (88881002, Thermo Fisher Scientific, Waltham, MA, USA). After thorough washing, the antigen-antibody-magnetic bead complexes were boiled in 25 μL of 1 × SDS–PAGE loading buffer, and the supernatant was subjected to Western blot analysis.
4.14. Metastasis Assay In Vivo
Female athymic BALB/c nude mice (4–6 weeks old) were used to establish the metastasis model in vivo. HepG2 cells (1 × 106) stably overexpressing KIAA1217 or the control vector were injected into the tail vein of the mice. After 8 weeks, the mice were sacrificed, and the physical appearance, lungs, and spleens of the mice were photographed. Then, the tumor nodules on the surface of the lungs were separately counted by two researchers. Subsequently, the lungs were excised, fixed with a neutral formalin solution, and embedded in paraffin. The tissues were then serially sectioned and stained with hematoxylin and eosin (H&E) to identify the pathological characteristics. H&E staining was performed using an improved H&E staining kit (G1121, Solarbio, Beijing, China) according to the manufacturer’s protocols.
4.15. Statistical Analysis
Statistical analyses were conducted using SPSS 21.0 software or GraphPad Prism 8 software. The data are presented as the means ± standard errors of the means (SEM) from at least three independent experiments. The differences between groups were analyzed with Student’s t-test when only two groups were compared or using one-way analysis of variance (ANOVA) or two-way ANOVA when more than two groups were compared. Overall survival and disease-free survival curves were obtained using Kaplan–Meier analysis, and differences were compared with the log-rank test. A two-tailed p-value < 0.05 was considered statistically significant.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}