
Interleukin-32θ Triggers Cellular Senescence and Reduces Sensitivity to Doxorubicin-Mediated Cytotoxicity in MDA-MB-231 Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. IL-32θ Reduced MDA-MB 231 Cell Proliferation
2.2. IL-32θ Induced Cellular Senescence but Not Senescence-Associated Secretory Phenotype (SASP)
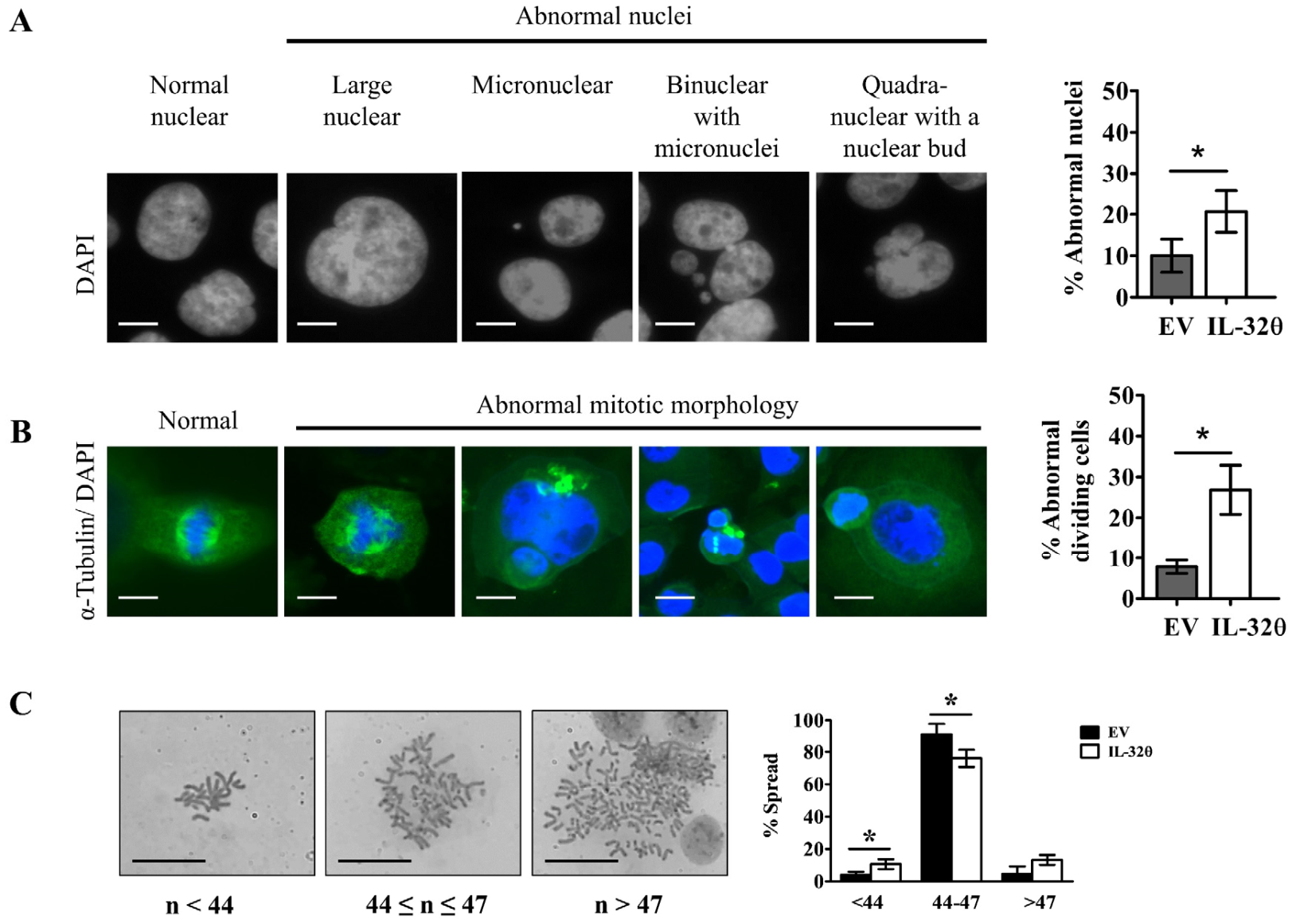
2.3. Interleukin-32θ Induced Genome Instability, Abnormal Nuclear Morphology, and Aberrant Cell Division
2.4. Interleukin-32θ Induced G1 Arrest, but Not Apoptosis or Autophagy
2.5. IL-32θ Maintained Senescence and Resisted against DR-Induced Cell Death
2.6. Interleukin-32θ Supports Ploidy Formation and G2/M Arrest in Cells Incubated with 0.1 μM DR
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Clonogenic Assay
4.3. Cell Viability Assay
4.4. Reverse Transcription Quantitative PCR (RT-qPCR)
4.5. Western Blotting
4.6. Cell Cycle Analysis and Apoptosis Detection by Flow Cytometry
4.7. Cell Proliferation Assay (BrdU)
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. Immunofluorescence Assay
4.10. Senescence-Associated β-Galactosidase (SA-β-gal) Assay
4.11. Chromosome Spread Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DR | Doxorubicin |
| EV | Empty vector |
| MOMP | Mitochondrial outer membrane permeabilization |
| PGCCs | Polyploid giant cancer cells |
| SASP | Senescence-associated secretory phenotype |
| TIS | Therapy-induced senescence |
| TNBC | Triple-negative breast cancer |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2020, 70, 313. [Google Scholar]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J. Triple-negative breast cancer: Role of specific chemotherapy agents. Cancer J. 2010, 16, 53–61. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef]
- Kang, J.W.; Park, Y.S.; Lee, D.H.; Kim, M.S.; Bak, Y.; Ham, S.Y.; Park, S.H.; Kim, H.; Ahn, J.H.; Hong, J.T.; et al. Interaction network mapping among IL-32 isoforms. Biochimie 2014, 101, 248–251. [Google Scholar] [CrossRef]
- Park, J.S.; Choi, S.Y.; Lee, J.H.; Lee, M.; Nam, E.S.; Jeong, A.L.; Lee, S.; Han, S.; Lee, M.S.; Lim, J.S.; et al. Interleukin-32 beta stimulates migration of MDA-MB-231 and MCF-7cells via the VEGF-STAT3 signaling pathway. Cell Oncol. 2013, 36, 493–503. [Google Scholar] [CrossRef]
- Pham, T.H.; Bak, Y.; Kwon, T.; Kwon, S.B.; Oh, J.W.; Park, J.H.; Choi, Y.K.; Hong, J.T.; Yoon, D.Y. Interleukin-32theta inhibits tumor-promoting effects of macrophage-secreted CCL18 in breast cancer. Cell Commun. Signal. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Bak, Y.; Kwon, T.; Bak, I.S.; Hong, J.; Yu, D.Y.; Yoon, D.Y. IL-32theta inhibits stemness and epithelial-mesenchymal transition of cancer stem cells via the STAT3 pathway in colon cancer. Oncotarget 2016, 7, 7307–7317. [Google Scholar] [CrossRef]
- Park, C.W.; Bak, Y.; Kim, M.J.; Srinivasrao, G.; Hwang, J.; Sung, N.K.; Kim, B.Y.; Yu, J.H.; Hong, J.T.; Yoon, D.Y. The Novel Small Molecule STK899704 Promotes Senescence of the Human A549 NSCLC Cells by Inducing DNA Damage Responses and Cell Cycle Arrest. Front. Pharm. 2018, 9, 163. [Google Scholar] [CrossRef]
- Buchwalter, A.; Hetzer, M.W. Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 2017, 8, 328. [Google Scholar] [CrossRef]
- Gisselsson, D.; Bjork, J.; Hoglund, M.; Mertens, F.; Dal Cin, P.; Akerman, M.; Mandahl, N. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am. J. Pathol. 2001, 158, 199–206. [Google Scholar] [CrossRef]
- Neumuller, R.A.; Knoblich, J.A. Dividing cellular asymmetry: Asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009, 23, 2675–2699. [Google Scholar] [CrossRef]
- Masamha, C.P.; Benbrook, D.M. Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res. 2009, 69, 6565–6572. [Google Scholar] [CrossRef]
- Mazumder, S.; DuPree, E.L.; Almasan, A. A dual role of cyclin E in cell proliferation and apoptosis may provide a target for cancer therapy. Curr. Cancer Drug Targets 2004, 4, 65–75. [Google Scholar] [CrossRef][Green Version]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef]
- Aydinlik, S.; Erkisa, M.; Cevatemre, B.; Sarimahmut, M.; Dere, E.; Ari, F.; Ulukaya, E. Enhanced cytotoxic activity of doxorubicin through the inhibition of autophagy in triple negative breast cancer cell line. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Tormo, E.; Ballester, S.; Adam-Artigues, A.; Burgues, O.; Alonso, E.; Bermejo, B.; Menendez, S.; Zazo, S.; Madoz-Gurpide, J.; Rovira, A.; et al. The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Sci. Rep. 2019, 9, 5316. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef]
- Shu, Z.; Row, S.; Deng, W.M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef]
- Bar-On, O.; Shapira, M.; Hershko, D.D. Differential effects of doxorubicin treatment on cell cycle arrest and Skp2 expression in breast cancer cells. Anticancer Drugs 2007, 18, 1113–1121. [Google Scholar] [CrossRef]
- Demirci, D.; Dayanc, B.; Mazi, F.A.; Senturk, S. The Jekyll and Hyde of Cellular Senescence in Cancer. Cells 2021, 10, 208. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- He, Q.; Au, B.; Kulkarni, M.; Shen, Y.; Lim, K.J.; Maimaiti, J.; Wong, C.K.; Luijten, M.N.H.; Chong, H.C.; Lim, E.H.; et al. Chromosomal instability-induced senescence potentiates cell non-autonomous tumourigenic effects. Oncogenesis 2018, 7, 62. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Kim, M.S.; Kang, J.W.; Jeon, J.S.; Kim, J.K.; Kim, J.W.; Hong, J.; Yoon, D.Y. IL-32theta gene expression in acute myeloid leukemia suppresses TNF-alpha production. Oncotarget 2015, 6, 40747–40761. [Google Scholar] [CrossRef][Green Version]
- Kim, M.S.; Kang, J.W.; Lee, D.H.; Bak, Y.; Park, Y.S.; Song, Y.S.; Ham, S.Y.; Oh, D.K.; Hong, J.; Yoon, D.Y. IL-32theta negatively regulates IL-1beta production through its interaction with PKCdelta and the inhibition of PU.1 phosphorylation. FEBS Lett. 2014, 588, 2822–2829. [Google Scholar] [CrossRef]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintas-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef]
- Chen, T.; Wang, C.; Liu, Q.; Meng, Q.; Sun, H.; Huo, X.; Sun, P.; Peng, J.; Liu, Z.; Yang, X.; et al. Dasatinib reverses the multidrug resistance of breast cancer MCF-7 cells to doxorubicin by downregulating P-gp expression via inhibiting the activation of ERK signaling pathway. Cancer Biol. Ther. 2015, 16, 106–114. [Google Scholar] [CrossRef]
- Park, H.K.; Lee, J.E.; Lim, J.; Jo, D.E.; Park, S.A.; Suh, P.G.; Kang, B.H. Combination treatment with doxorubicin and gamitrinib synergistically augments anticancer activity through enhanced activation of Bim. BMC Cancer 2014, 14, 431. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Teissier, S.; Pang, C.L.; Thierry, F. The E2F5 repressor is an activator of E6/E7 transcription and of the S-phase entry in HPV18-associated cells. Oncogene 2010, 29, 5061–5070. [Google Scholar] [CrossRef]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc 2006, 1, 1559–1582. [Google Scholar] [CrossRef]
- Shen, Y.; Vignali, P.; Wang, R. Rapid Profiling Cell Cycle by Flow Cytometry Using Concurrent Staining of DNA and Mitotic Markers. Bio Protoc. 2017, 7, e2517. [Google Scholar] [CrossRef]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY) 2013, 5, 37–50. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, T.-H.; Park, H.-M.; Kim, J.; Hong, J.-T.; Yoon, D.-Y. Interleukin-32θ Triggers Cellular Senescence and Reduces Sensitivity to Doxorubicin-Mediated Cytotoxicity in MDA-MB-231 Cells. Int. J. Mol. Sci. 2021, 22, 4974. https://doi.org/10.3390/ijms22094974
Pham T-H, Park H-M, Kim J, Hong J-T, Yoon D-Y. Interleukin-32θ Triggers Cellular Senescence and Reduces Sensitivity to Doxorubicin-Mediated Cytotoxicity in MDA-MB-231 Cells. International Journal of Molecular Sciences. 2021; 22(9):4974. https://doi.org/10.3390/ijms22094974
Chicago/Turabian StylePham, Thu-Huyen, Hyo-Min Park, Jinju Kim, Jin-Tae Hong, and Do-Young Yoon. 2021. "Interleukin-32θ Triggers Cellular Senescence and Reduces Sensitivity to Doxorubicin-Mediated Cytotoxicity in MDA-MB-231 Cells" International Journal of Molecular Sciences 22, no. 9: 4974. https://doi.org/10.3390/ijms22094974
APA StylePham, T.-H., Park, H.-M., Kim, J., Hong, J.-T., & Yoon, D.-Y. (2021). Interleukin-32θ Triggers Cellular Senescence and Reduces Sensitivity to Doxorubicin-Mediated Cytotoxicity in MDA-MB-231 Cells. International Journal of Molecular Sciences, 22(9), 4974. https://doi.org/10.3390/ijms22094974