
Chimeric Claudins: A New Tool to Study Tight Junction Structure and Function

 , , ,
, , , 
Abstract
1. Introduction
2. Results and Discussion
2.1. Chimeric CLDN1 Design and Properties
2.2. Surface Plasmon Resonance (SPR) to Determine MBP-CC1 Constant of Affinity
2.3. Surface Plasmon Resonance (SPR) to Determine MBP-CC1 Structural Domains Responsible for Adhesion
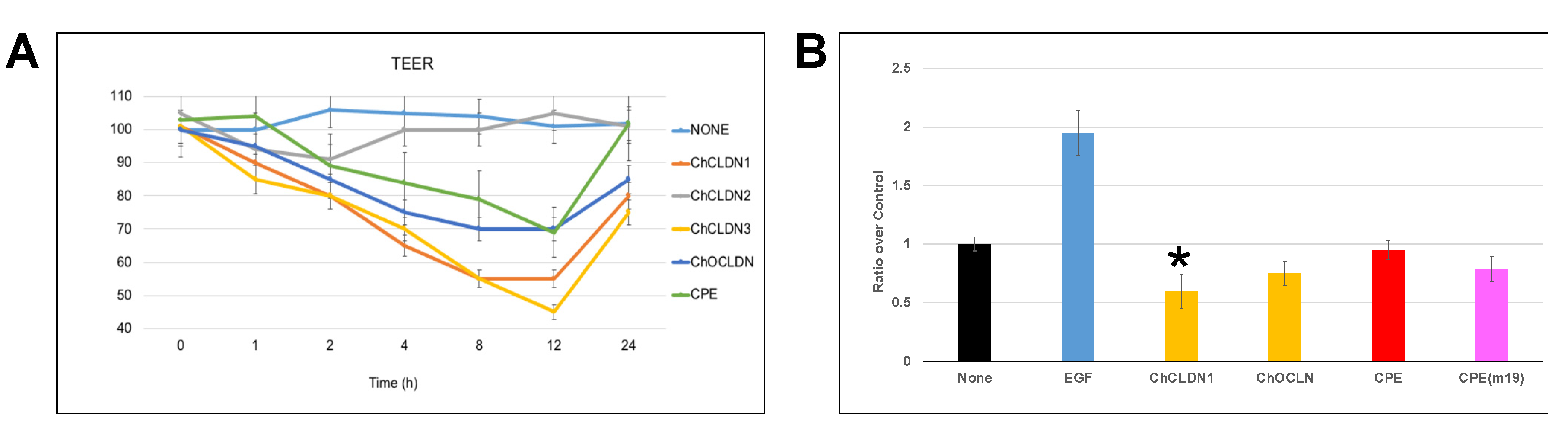
2.4. MBP-CC1 In Vitro Experiments
2.5. Zebrafish MBP-CC11A Effects In Vivo
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Protein Expression and Purification
4.3. Protein Models
4.4. Small-Angle X-ray Scattering for Biomolecules (bioSAXS)
4.5. Surface Plasmon Resonance (SPR)
4.6. Tissue Culture
4.7. Trans Epithelial Electrical Resistance (TEER)
4.8. Proliferation Assay
4.9. Zebrafish
4.9.1. Transgenic Line
4.9.2. Zebrafish Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Otani, T.; Furuse, M. Tight junction structure and function revisited. Trends Cell Biol. 2020, 30, 805–817. [Google Scholar] [CrossRef]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Jayagopal, A.; Yang, J.L.; Haselton, F.R.; Chang, M.S. Tight junction-associated signaling pathways modulate cell proliferation in uveal melanoma. Invest. Ophthalmol. Vis. Sci. 2011, 52, 588–593. [Google Scholar] [CrossRef]
- Diaz-Coranguez, M.; Liu, X.; Antonetti, D.A. Tight junctions in cell proliferation. Int. J. Mol. Sci. 2019, 20, 5972. [Google Scholar] [CrossRef] [PubMed]
- Irudayanathan, F.J.; Wang, N.; Wang, X.; Nangia, S. Architecture of the paracellular channels formed by claudins of the blood-brain barrier tight junctions. Ann. N. Y. Acad. Sci. 2017, 1405, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, U.; Schuetz, A. Structural features of tight-junction proteins. Int. J. Mol. Sci. 2019, 20, 6020. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Fromm, M. Claudins and other tight junction proteins. Compr. Physiol. 2012, 2, 1819–1852. [Google Scholar]
- Li, Y.; Fanning, A.S.; Anderson, J.M.; Lavie, A. Structure of the conserved cytoplasmic C-terminal domain of occludin: Identification of the ZO-1 binding surface. J. Mol. Biol. 2005, 352, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Tani, K.; Fujiyoshi, Y. Crystal structures of claudins: Insights into their intermolecular interactions. Ann. N. Y. Acad. Sci. 2017, 1397, 25–34. [Google Scholar] [CrossRef]
- Nomme, J.; Antanasijevic, A.; Caffrey, M.; Van Itallie, C.M.; Anderson, J.M.; Fanning, A.S.; Lavie, A. Structural basis of a key factor regulating the affinity between the zonula occludens first PDZ domain and claudins. J. Biol. Chem. 2015, 290, 16595–16606. [Google Scholar] [CrossRef] [PubMed]
- Argueso, P.; Gipson, I.K. Assessing mucin expression and function in human ocular surface epithelia In Vivo and In Vitro. Methods Mol. Biol. 2012, 842, 313–325. [Google Scholar]
- Furuse, M.; Sasaki, H.; Fujimoto, K.; Tsukita, S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 1998, 143, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Steed, E.; Rodrigues, N.T.; Balda, M.S.; Matter, K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009, 10, 95. [Google Scholar] [CrossRef]
- Otani, T.; Nguyen, T.P.; Tokuda, S.; Sugihara, K.; Sugawara, T.; Furuse, K.; Miura, T.; Ebnet, K.; Furuse, M. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J. Cell Biol. 2019, 218, 3372–3396. [Google Scholar] [CrossRef]
- Baumholtz, A.I.; Gupta, I.R.; Ryan, A.K. Claudins in morphogenesis: Forming an epithelial tube. Tissue Barriers 2017, 5, e1361899. [Google Scholar] [CrossRef]
- Baumholtz, A.I.; Simard, A.; Nikolopoulou, E.; Oosenbrug, M.; Collins, M.M.; Piontek, A.; Krause, G.; Piontek, J.; Greene, N.D.; Ryan, A.K. Claudins are essential for cell shape changes and convergent extension movements during neural tube closure. Dev. Biol. 2017, 428, 25–38. [Google Scholar] [CrossRef]
- Gupta, I.R.; Ryan, A.K. Claudins: Unlocking the code to tight junction function during embryogenesis and in disease. Clin. Genet. 2010, 77, 314–325. [Google Scholar] [CrossRef]
- Siddiqui, M.; Sheikh, H.; Tran, C.; Bruce, A.E. The tight junction component Claudin E is required for zebrafish epiboly. Dev. Dyn. 2010, 239, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Tsukita, S.; Furuse, M. Tight junctions containing claudin 4 and 6 are essential for blastocyst formation in preimplantation mouse embryos. Dev. Biol. 2007, 312, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Piontek, J.; Wolburg, H.; Piehl, C.; Liss, M.; Otten, C.; Christ, A.; Willnow, T.E.; Blasig, I.E.; Abdelilah-Seyfried, S. Establishment of a neuroepithelial barrier by Claudin5a is essential for zebrafish brain ventricular lumen expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 1425–1430. [Google Scholar] [CrossRef]
- El Andalousi, J.; Khairallah, H.; Zhuang, Y.; Ryan, A.K.; Gupta, I.R. Role of claudins in renal branching morphogenesis. Physiol. Rep. 2020, 8, e14492. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Bissell, M.J. The organization of tight junctions in epithelia: Implications for mammary gland biology and breast tumorigenesis. J. Mammary Gland Biol. Neoplasia 2003, 8, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Campbell, J.A.; Schelling, P.; Forrest, J.C.; Watson, M.J.; Peters, T.R.; Aurrand-Lions, M.; Imhof, B.A.; Dermody, T.S.; Stehle, T. Crystal structure of human junctional adhesion molecule 1: Implications for reovirus binding. Proc. Natl. Acad. Sci. USA 2003, 100, 5366–5371. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, A.; Radusheva, V.; Krug, S.M.; Heinemann, U. Crystal structure of the tricellulin C-terminal coiled-coil domain reveals a unique mode of dimerization. Ann. N. Y. Acad. Sci. 2017, 1405, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Fuladi, S.; Jannat, R.W.; Shen, L.; Weber, C.R.; Khalili-Araghi, F. Computational modeling of claudin structure and function. Int. J. Mol. Sci. 2020, 21, 742. [Google Scholar] [CrossRef]
- Andersen, J.L.; Gourdon, P.; Moller, J.V.; Morth, J.P.; Nissen, P. Crystallization and preliminary structural analysis of the Listeria monocytogenes Ca(2+)-ATPase LMCA1. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 718–722. [Google Scholar] [CrossRef]
- Guo, Y. Be Cautious with Crystal Structures of Membrane Proteins or Complexes Prepared in Detergents. Crystals 2020, 10, 86. [Google Scholar] [CrossRef]
- Hardy, D.; Bill, R.M.; Jawhari, A.; Rothnie, A.J. Overcoming bottlenecks in the membrane protein structural biology pipeline. Biochem. Soc. Trans. 2016, 44, 838–844. [Google Scholar] [CrossRef]
- Raynal, B.; Brule, S.; Uebel, S.; Knauer, S.H. Assessing and improving protein sample quality. Methods Mol. Biol. 2021, 2263, 3–46. [Google Scholar]
- Smyth, D.R.; Mrozkiewicz, M.K.; McGrath, W.J.; Listwan, P.; Kobe, B. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 2003, 12, 1313–1322. [Google Scholar] [CrossRef]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, Q.; Tong, H.; Xu, D.; Wang, W.; Ran, T. Crystal structure of MBP-PigG fusion protein and the essential function of PigG in the prodigiosin biosynthetic pathway in Serratia marcescens FS14. Int. J. Biol. Macromol. 2017, 99, 394–400. [Google Scholar] [CrossRef]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, K.; Liu, R.; Zhao, F.; Gupta, S.; Zhang, N.; Prud’homme, G.J. Novel GLP-1 fusion chimera as potent long acting GLP-1 receptor agonist. PLoS ONE 2010, 5, e12734. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Nishizawa, T.; Tani, K.; Yamazaki, Y.; Tamura, A.; Ishitani, R.; Dohmae, N.; Tsukita, S.; Nureki, O.; Fujiyoshi, Y. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 2014, 344, 304–307. [Google Scholar] [CrossRef]
- Vecchio, A.J.; Stroud, R.M. Claudin-9 structures reveal mechanism for toxin-induced gut barrier breakdown. Proc. Natl. Acad. Sci. USA 2019, 116, 17817–17824. [Google Scholar] [CrossRef]
- Sonoda, N.; Furuse, M.; Sasaki, H.; Yonemura, S.; Katahira, J.; Horiguchi, Y.; Tsukita, S. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 1999, 147, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Kermani, A.A. A guide to membrane protein X-ray crystallography. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.J.; Afrose, F.; Koeppe II, R.E.; Greathouse, D.V. Helix formation and stability in membranes. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, N.; Irudayanathan, F.J.; Nangia, S. Computational nanoscopy of tight junctions at the blood-brain barrier interface. Int. J. Mol. Sci. 2019, 20, 5583. [Google Scholar] [CrossRef]
- Krause, G.; Protze, J.; Piontek, J. Assembly and function of claudins: Structure-function relationships based on homology models and crystal structures. Semin. Cell Dev. Biol. 2015, 42, 3–12. [Google Scholar] [CrossRef]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Cording, J.; Arslan, B.; Staat, C.; Dithmer, S.; Krug, S.M.; Krüger, A.; Berndt, P.; Günther, R.; Winkler, L.; Blasig, I.E.; et al. Trictide, a tricellulin-derived peptide to overcome cellular barriers. Ann. N. Y. Acad. Sci. 2017, 1405, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zwanziger, D.; Hackel, D.; Staat, C.; Böcker, A.; Brack, A.; Beyermann, M.; Rittner, H.; Blasig, I.E. A peptidomimetic tight junction modulator to improve regional analgesia. Mol. Pharm. 2012, 9, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Staat, C.; Coisne, C.; Dabrowski, S.; Stamatovic, S.M.; Andjelkovic, A.V.; Wolburg, H.; Engelhardt, B.; Blasig, I.E. Mode of action of claudin peptidomimetics in the transient opening of cellular tight junction barriers. Biomaterials 2015, 54, 9–20. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray crystallography and drug discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef]
- Irudayanathan, F.J.; Trasatti, J.P.; Karande, P.; Nangia, S. Molecular architecture of the blood brain barrier tight junction proteins--A synergistic computational and In Vitro approach. J. Phys. Chem. B 2016, 120, 77–88. [Google Scholar] [CrossRef]
- Irudayanathan, F.J.; Wang, X.; Wang, N.; Willsey, S.R.; Seddon, I.A.; Nangia, S. Self-Assembly simulations of classic claudins-insights into the pore structure, selectivity, and higher order complexes. J. Phys. Chem. B 2018, 122, 7463–7474. [Google Scholar] [CrossRef]
- Go, A.; Kim, S.; Baum, J.; Hecht, M.H. Structure and dynamics of de novo proteins from a designed superfamily of 4-helix bundles. Protein Sci. 2008, 17, 821–832. [Google Scholar] [CrossRef]
- Bradley, L.H.; Thumfort, P.P.; Hecht, M.H. De novo proteins from binary-patterned combinatorial libraries. Methods Mol. Biol. 2006, 340, 53–69. [Google Scholar] [PubMed]
- Waugh, D.S. Crystal structures of MBP fusion proteins. Protein Sci. 2016, 25, 559–571. [Google Scholar] [CrossRef]
- Hong, P.; Koza, S.; Bouvier, E.S. Size-Exclusion chromatography for the analysis of protein biotherapeutics and their aggregates. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2923–2950. [Google Scholar] [CrossRef] [PubMed]
- Kachala, M.; Valentini, E.; Svergun, D.I. Application of SAXS for the structural characterization of IDPs. Adv. Exp. Med. Biol. 2015, 870, 261–289. [Google Scholar]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef]
- Grawert, T.W.; Svergun, D.I. Structural modeling using solution small-angle x-ray scattering (SAXS). J. Mol. Biol. 2020, 432, 3078–3092. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Mitic, L.L.; Anderson, J.M. Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J. Biol. Chem. 2011, 286, 3442–3450. [Google Scholar] [CrossRef]
- Zhao, J.; Krystofiak, E.S.; Ballesteros, A.; Cui, R.; Van Itallie, C.M.; Anderson, J.M.; Fenollar-Ferrer, C.; Kachar, B. Multiple claudin-claudin cis interfaces are required for tight junction strand formation and inherent flexibility. Commun. Biol. 2018, 1, 50. [Google Scholar] [CrossRef] [PubMed]
- Joh, N.H.; Grigoryan, G.; Wu, Y.; DeGrado, W.F. Design of self-assembling transmembrane helical bundles to elucidate principles required for membrane protein folding and ion transport. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160214. [Google Scholar] [CrossRef]
- Badilescu, S.; Raju, D.; Bathini, S.; Packirisamy, M. Gold Nano-Island platforms for localized surface plasmon resonance sensing: A short review. Molecules 2020, 25, 4661. [Google Scholar] [CrossRef] [PubMed]
- Vendome, J.; Felsovalyi, K.; Song, H.; Yang, Z.; Jin, X.; Brasch, J.; Harrison, O.J.; Ahlsen, G.; Bahna, F.; Kaczynska, A.; et al. Structural and energetic determinants of adhesive binding specificity in type I cadherins. Proc. Natl. Acad. Sci. USA 2014, 111, E4175–E4184. [Google Scholar] [CrossRef]
- Parisini, E.; Higgins, J.M.; Liu, J.H.; Brenner, M.B.; Wang, J.H. The crystal structure of human E-cadherin domains 1 and 2, and comparison with other cadherins in the context of adhesion mechanism. J. Mol. Biol. 2007, 373, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Brynestad, S.; Sarker, M.R.; McClane, B.A.; Granum, P.E.; Rood, J.I. Enterotoxin plasmid from Clostridium perfringens is conjugative. Infect. Immun. 2001, 69, 3483–3487. [Google Scholar] [CrossRef]
- McClane, B.A. The complex interactions between Clostridium perfringens enterotoxin and epithelial tight junctions. Toxicon 2001, 39, 1781–1791. [Google Scholar] [CrossRef]
- Takahashi, A.; Saito, Y.; Kondoh, M.; Matsushita, K.; Krug, S.M.; Suzuki, H.; Tsujino, H.; Li, X.; Aoyama, H.; Matsuhisa, K.; et al. Creation and biochemical analysis of a broad-specific claudin binder. Biomaterials 2012, 33, 3464–3474. [Google Scholar] [CrossRef]
- Li, J.; Angelow, S.; Linge, A.; Zhuo, M.; Yu, A.S. Claudin-2 pore function requires an intramolecular disulfide bond between two conserved extracellular cysteines. Am. J. Physiol. Cell Physiol. 2013, 305, C190–C196. [Google Scholar] [CrossRef] [PubMed]
- Angelow, S.; Yu, A.S. Cysteine mutagenesis to study the structure of claudin-2 paracellular pores. Ann. N. Y. Acad. Sci. 2009, 1165, 143–147. [Google Scholar] [CrossRef]
- Angelow, S.; Yu, A.S. Structure-function studies of claudin extracellular domains by cysteine-scanning mutagenesis. J. Biol. Chem. 2009, 284, 29205–29217. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Van Itallie, C.; Rahner, C.; Anderson, J.M. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am. J. Physiol. Cell Physiol. 2003, 284, C1346–C1354. [Google Scholar] [CrossRef] [PubMed]
- Mrsny, R.J.; Brown, G.T.; Gerner-Smidt, K.; Buret, A.G.; Meddings, J.B.; Quan, C.; Koval, M.; Nusrat, A. A key claudin extracellular loop domain is critical for epithelial barrier integrity. Am. J. Pathol. 2008, 172, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Lu, Z.; Lu, Q.; Chen, Y.H. The claudin family of proteins in human malignancy: A clinical perspective. Cancer Manag. Res. 2013, 5, 367–375. [Google Scholar]
- Chen, S.; Einspanier, R.; Schoen, J. Transepithelial electrical resistance (TEER): A functional parameter to monitor the quality of oviduct epithelial cells cultured on filter supports. Histochem. Cell Biol. 2015, 144, 509–515. [Google Scholar] [CrossRef]
- Gioanni, J.; Fischel, J.L.; Lambert, J.C.; Demard, F.; Mazeau, C.; Zanghellini, E.; Ettore, F.; Formento, P.; Chauvel, P.; Lalanne, C.M.; et al. Two new human tumor cell lines derived from squamous cell carcinomas of the tongue: Establishment, characterization and response to cytotoxic treatment. Eur. J. Cancer Clin. Oncol. 1988, 24, 1445–1455. [Google Scholar] [CrossRef]
- Cuellar, P.; Hernández-Nava, E.; García-Rivera, G.; Chávez-Munguía, B.; Schnoor, M.; Betanzos, A.; Orozco, E. Entamoeba histolytica EhCP112 dislocates and degrades claudin-1 and claudin-2 at tight junctions of the intestinal epithelium. Front. Cell Infect. Microbiol. 2017, 7, 372. [Google Scholar] [CrossRef]
- Nakamura, S.; Irie, K.; Tanaka, H.; Nishikawa, K.; Suzuki, H.; Saitoh, Y.; Tamura, A.; Tsukita, S.; Fujiyoshi, Y. Morphologic determinant of tight junctions revealed by claudin-3 structures. Nat. Commun. 2019, 10, 816. [Google Scholar] [CrossRef]
- Shen, Z.; Song, W.; Qian, L.; Zhu, J.; Li, Y.; Li, M.; Zhang, T.; Zhao, W.; Zhou, Y.; Yang, X. Effect of claudin 1 on cell proliferation, migration and apoptosis in human cervical squamous cell carcinoma. Oncol. Rep. 2020, 45, 606–618. [Google Scholar] [CrossRef]
- Tsukita, S.; Yamazaki, Y.; Katsuno, T.; Tamura, A.; Tsukita, S. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene 2008, 27, 6930–6938. [Google Scholar] [CrossRef]
- Bhat, A.A.; Pope, J.L.; Smith, J.J.; Ahmad, R.; Chen, X.; Washington, M.K.; Beauchamp, R.D.; Singh, A.B.; Dhawan, P. Claudin-7 expression induces mesenchymal to epithelial transformation (MET) to inhibit colon tumorigenesis. Oncogene 2015, 34, 4570–4580. [Google Scholar] [CrossRef]
- Eckert, J.J.; Fleming, T.P. Tight junction biogenesis during early development. Biochim. Biophys. Acta 2008, 1778, 717–728. [Google Scholar] [CrossRef]
- Kolosov, D.; Bui, P.; Chasiotis, H.; Kelly, S.P. Claudins in teleost fishes. Tissue Barriers 2013, 1, e25391. [Google Scholar] [CrossRef] [PubMed]
- Matsunuma, R.; Chan, D.W.; Kim, B.J.; Singh, P.; Han, A.; Saltzman, A.B.; Cheng, C.; Lei, J.T.; Wang, J.; da Silva, L.R.; et al. DPYSL3 modulates mitosis, migration, and epithelial-to-mesenchymal transition in claudin-low breast cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E11978–E11987. [Google Scholar] [CrossRef]
- Tada, M.; Heisenberg, C.P. Convergent extension: Using collective cell migration and cell intercalation to shape embryos. Development 2012, 139, 3897–3904. [Google Scholar] [CrossRef] [PubMed]
- Schwayer, C.; Shamipour, S.; Pranjic-Ferscha, K.; Schauer, A.; Balda, M.; Tada, M.; Matter, K.; Heisenberg, C.P. Mechanosensation of tight junctions depends on ZO-1 phase separation and flow. Cell 2019, 179, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Gordon, H.B.; Lusk, S.; Carney, K.R.; Wirick, E.O.; Murray, B.F.; Kwan, K.M. Hedgehog signaling regulates cell motility and optic fissure and stalk formation during vertebrate eye morphogenesis. Development 2018, 145. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPI Evaluated | Ka (1/(M*s)) | Kd (1/s) | KD (M) |
|---|---|---|---|
| MBP-eCAD vs. MBP-eCAD * | 4.65 × 103 ± 1.61 × 102 | 6.96 × 10−4 ± 7.87 × 10−5 | 1.97 × 10−7 ± 2.25 × 10−8 |
| MBP-CC1 vs. MBP-CC1 | 8.88 × 104 ± 4.11 × 103 | 2.42 × 10−5 ± 1.26 × 10−6 | 2.70 × 10−10 ± 2.45 × 10−11 |
| MBP-CC1 vs. MBP-CC1 (50%ΔC) | 1.47 × 103 ± 1.05 × 101 | 5.30 × 10−5 ± 1.78 × 10−6 | 3.60 × 10−8 ± 3.76 × 10−9 |
| MBP-CC1 vs. MBP-CC1 (40%) | 3.92 × 103 ± 2.02 × 101 | 8.90 × 10−5 ± 5.71 × 10−7 | 2.27 × 10−8 ± 2.63 × 10−10 |
| MBP-CC1 vs. MBP-CC1 (30%) | 6.60 × 103 ± 0.29 × 101 | 4.98 × 10−3 ± 8.89 × 10−8 | 7.54 × 10−7 ± 3.36 × 10−8 |
| MBP-CC1 vs. MBP-CC1 (30%ΔEL1) | 5.35 × 102 ± 7.43 × 101 | 2.13 × 10−3 ± 8.17 × 10−6 | 3.98 × 10−6 ± 5.79 × 10−7 |
| MBP-CC1 vs. MBP-CC1 (30%ΔEL2) | 1.32 × 103 ± 2.31 × 101 | 3.80 × 10−4 ± 1.95 × 10−6 | 2.87 × 10−7 ± 6.49 × 10−9 |
| MBP-CC1 vs. MBP-CPE | 2.06 × 103 ± 1.71 × 102 | 8.71 × 10−4 ± 2.09 × 10−5 | 4.22 × 10−7 ± 4.53 × 10−8 |
| MBP-CC1 vs. MBP-CPE (m19) | 9.64 × 103 ± 2.41 × 102 | 1.88 × 10−3 ± 4.86 × 10−6 | 1.95 × 10−7 ± 5.39 × 10−9 |
| MBP-CC1 vs. MBP−2jua | 1.31 × 102 ± 2.12 × 101 | 1.80 × 10−1 ± 6.84 × 10−2 | 1.38 × 10−4 ± 2.34 × 10−5 |
| MBP-COC vs. MBP-COC | 3.67 × 103 ± 4.17 × 101 | 6.47 × 10−5 ± 3.39 × 10−6 | 1.76 × 10−8 ± 1.13 × 10−9 |
| MBP-CC1 vs. MBP-COC | 2.09 × 103 ± 2.61 × 101 | 2.48 × 10−4 ± 1.11 × 10−5 | 1.18 × 10−7 ± 6.27 × 10−9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, A.; Warner, M.; Mendoza, C.; Memmott, C.; LeCheminant, T.; Bailey, S.; Christensen, C.; Keller, J.; Suli, A.; Mizrachi, D. Chimeric Claudins: A New Tool to Study Tight Junction Structure and Function. Int. J. Mol. Sci. 2021, 22, 4947. https://doi.org/10.3390/ijms22094947
Taylor A, Warner M, Mendoza C, Memmott C, LeCheminant T, Bailey S, Christensen C, Keller J, Suli A, Mizrachi D. Chimeric Claudins: A New Tool to Study Tight Junction Structure and Function. International Journal of Molecular Sciences. 2021; 22(9):4947. https://doi.org/10.3390/ijms22094947
Chicago/Turabian StyleTaylor, Abigail, Mark Warner, Christopher Mendoza, Calvin Memmott, Tom LeCheminant, Sara Bailey, Colter Christensen, Julie Keller, Arminda Suli, and Dario Mizrachi. 2021. "Chimeric Claudins: A New Tool to Study Tight Junction Structure and Function" International Journal of Molecular Sciences 22, no. 9: 4947. https://doi.org/10.3390/ijms22094947
APA StyleTaylor, A., Warner, M., Mendoza, C., Memmott, C., LeCheminant, T., Bailey, S., Christensen, C., Keller, J., Suli, A., & Mizrachi, D. (2021). Chimeric Claudins: A New Tool to Study Tight Junction Structure and Function. International Journal of Molecular Sciences, 22(9), 4947. https://doi.org/10.3390/ijms22094947