
Identification of SYS1 as a Host Factor Required for Shiga Toxin-Mediated Cytotoxicity in Vero Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
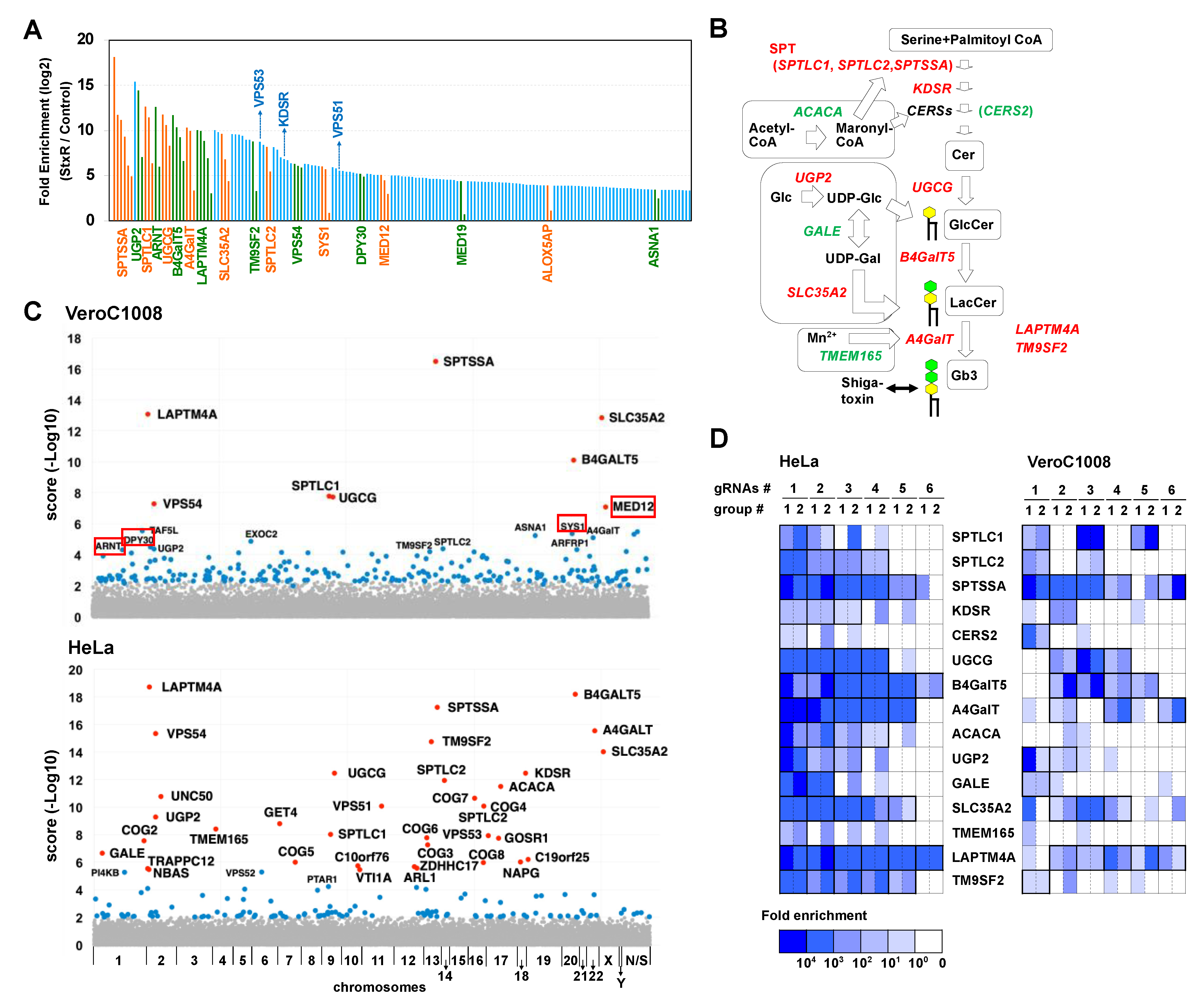
2.1. Human CRISPR KO Library Is Applicable to Vero Cell Lines
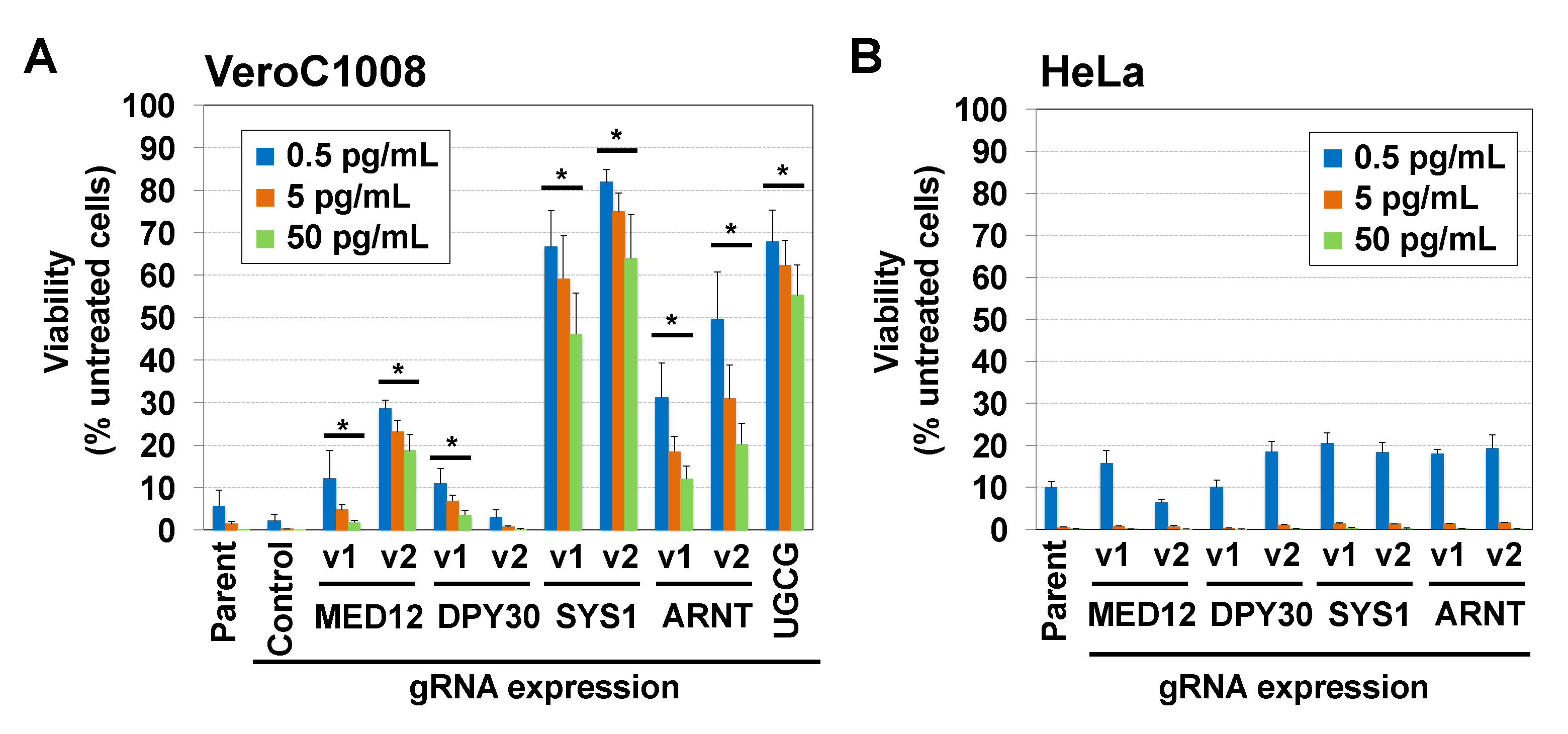
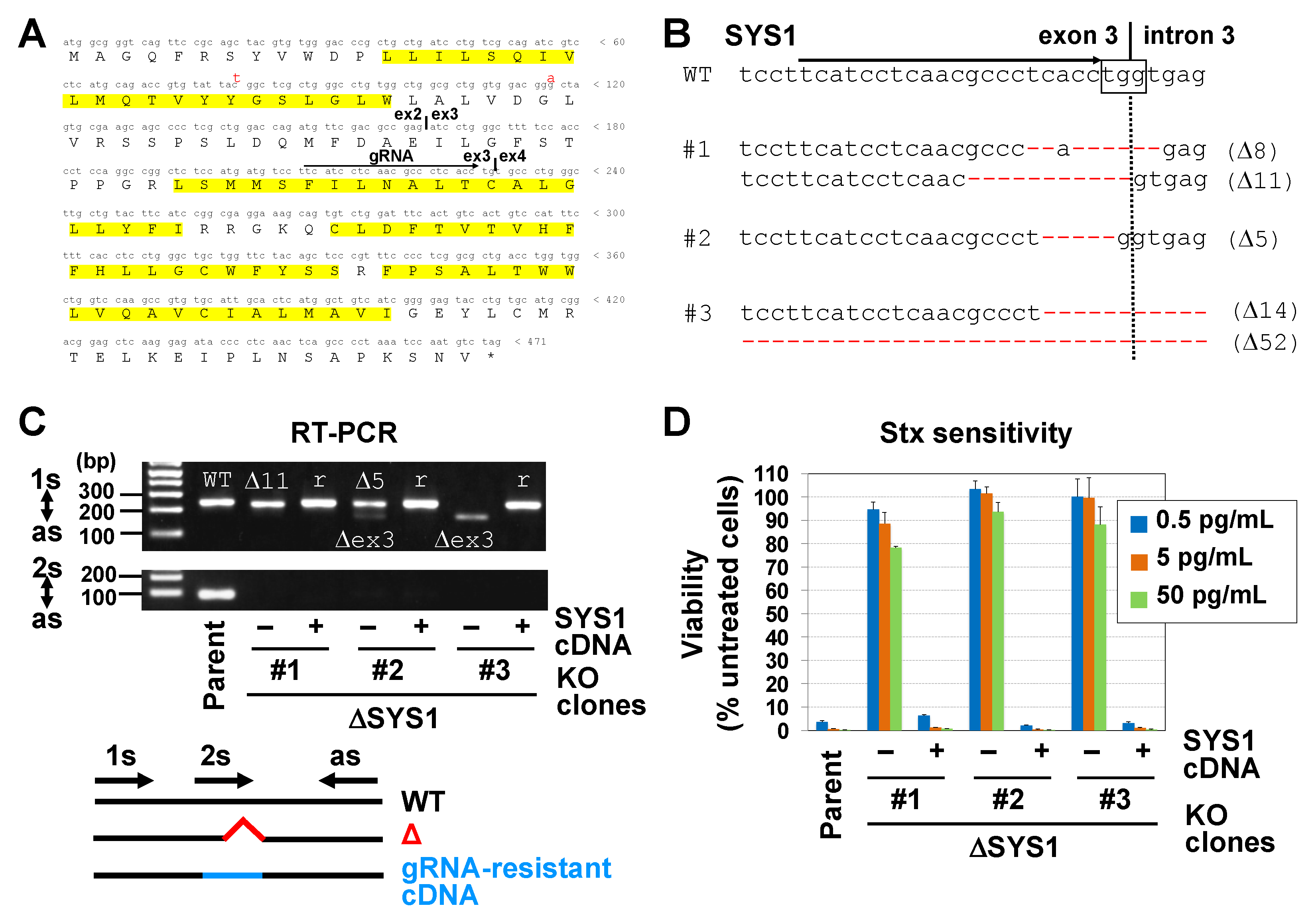
2.2. Disruption of the SYS1 Gene Confers Resistance to STx-Induced Cell Death in Vero Cells
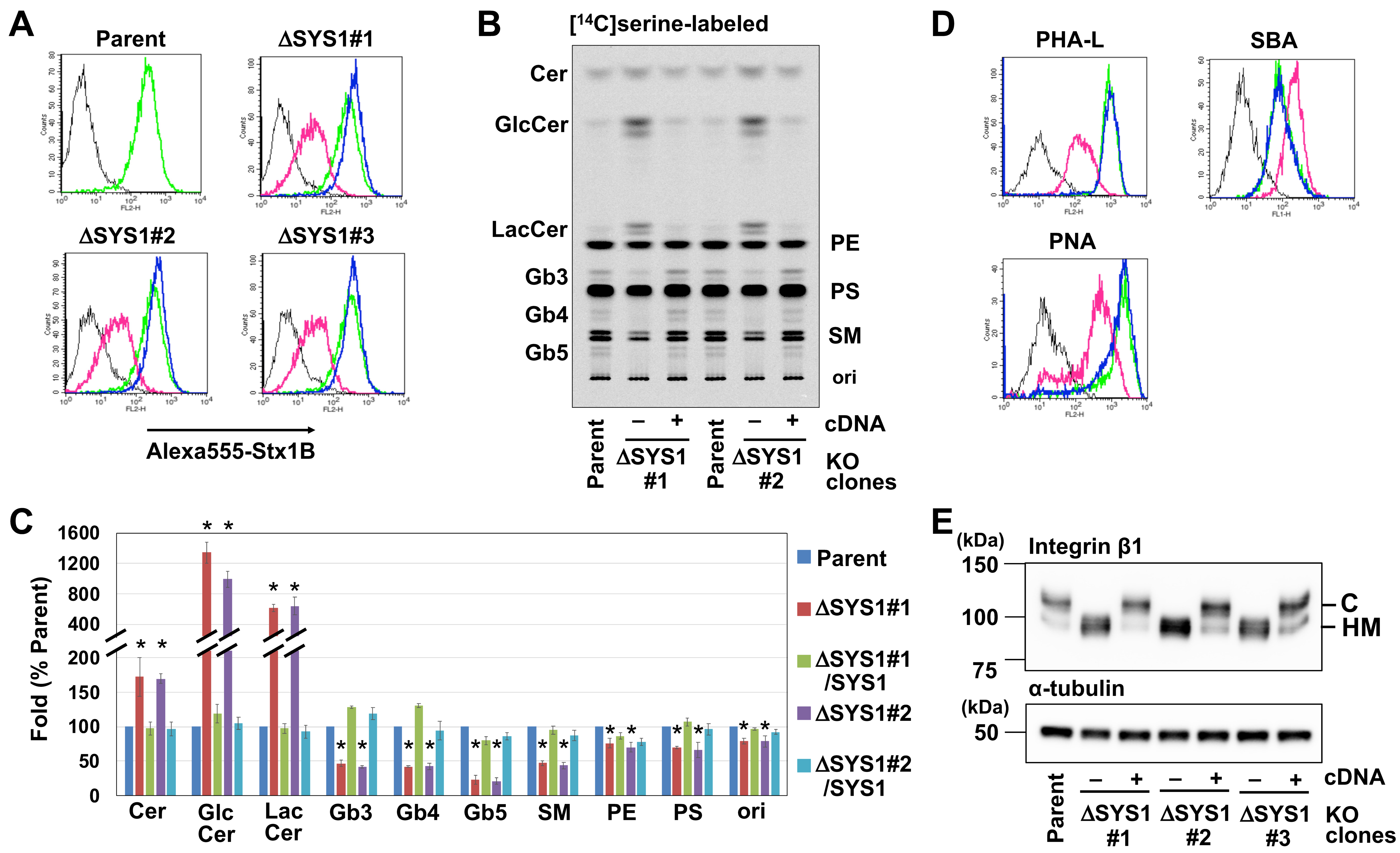
2.3. Loss of SYS1 Causes Defects in Glycan Biosynthesis
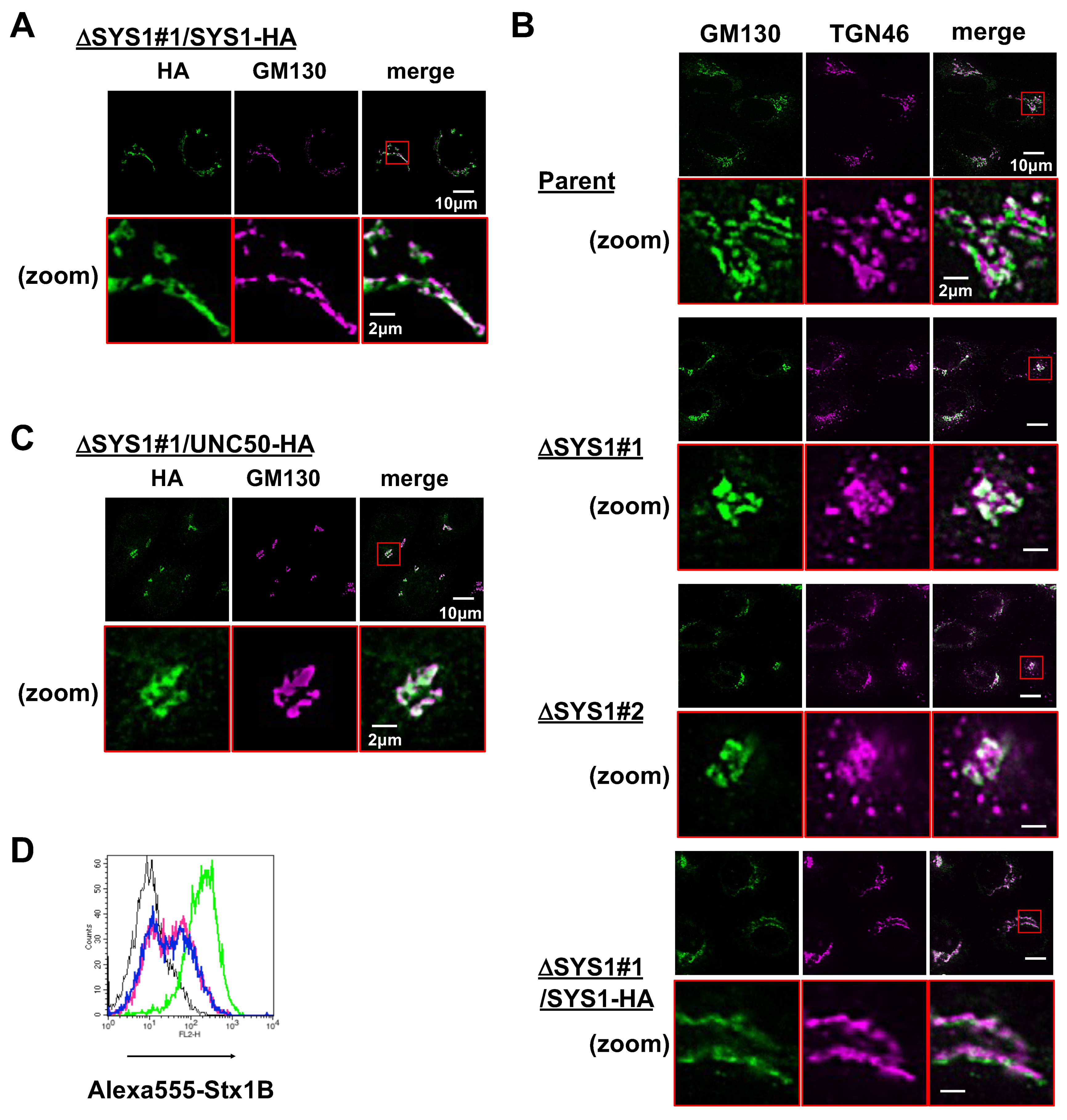
2.4. Loss of SYS1 Perturbs Golgi Morphology and TGN Distribution
2.5. SYS1 and UNC50 Are Functionally Different
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Antibodies, and Reagents
4.2. Isolation of the Cas9-Expressing Vero C1008 Cell Clone for CRISPR Screen
4.3. Preparation of sgRNA-Expressing Cell Libraries
4.4. CRISPR Screen for STx1 Treatment
4.5. Genomic DNA Sequencing
4.6. Data Processing and Analysis
4.7. Sequences of AGM Genome and Transcripts
4.8. Preparation of Lentiviruses for gRNA Expression
4.9. Construction of CRISPR KO Cell Lines
4.10. RNA Isolation, Reverse Transcription (RT)-PCR, and Sequencing
4.11. Preparation of Retroviruses for cDNA Expression and Stable Transfectants
4.12. Immunofluorescence Microscopy
4.13. Lysate Preparation and Western Blot Analysis
4.14. Metabolic Labeling of Glycolipids and TLC Analysis
4.15. FACS Analysis
4.16. STx Treatment and cell Viability Assay
4.17. Statistical Analysis
4.18. Primers Used in This Study
4.19. sgRNA Target Sequences
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Conradi, H. Uber loslishe, durch aseptische Autolyse, erhaltene Giftstoffe von Ruhr- und Typhus Bazillen. Dtsch. Med. Wochenschr. 1903, 29, 26–28. [Google Scholar] [CrossRef][Green Version]
- O’Brien, A.O.; Lively, T.A.; Chen, M.E.; Rothman, S.W.; Formal, S.B. Escherichia coli O157:H7 strains associated with haemorrhagic colitis in the United States produce a Shigella dysenteriae 1 (SHIGA) like cytotoxin. Lancet 1983, 1, 702. [Google Scholar] [CrossRef]
- Karmali, M.A.; Steele, B.T.; Petric, M.; Lim, C. Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet 1983, 1, 619–620. [Google Scholar] [CrossRef]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Stein, P.E.; Boodhoo, A.; Tyrrell, G.J.; Brunton, J.L.; Read, R.J. Crystal structure of the cell-binding B oligomer of Verotoxin-1 from E. coli. Nature 1992, 355, 748–750. [Google Scholar] [CrossRef]
- Fraser, M.; Chernaia, M.; Kozlov, Y.; James, M. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef]
- Jacewicz, M.; Clausen, H.; Nudelman, E.; Donohue-Rolfe, A.; Keusch, G.T. Pathogenesis of shigella diarrhea. XI. Isolation of a shigella toxin-binding glycolipid from rabbit jejunum and HeLa cells. J. Exp. Med. 1986, 163, 1391–1404. [Google Scholar] [CrossRef]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; De Grandis, S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Linstedt, A.D. Retrograde trafficking of AB5 toxins: Mechanisms to therapeutics. J. Mol. Med. 2013, 91, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Reisbig, R.; Olsnes, O.; Eiklid, K. The cytotoxic activity of Shigella toxin. Evidence for catalytic inactivation of the 60 S ribosomal subunit. J. Biol. Chem. 1981, 256, 8739–8744. [Google Scholar] [CrossRef]
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40. [Google Scholar]
- Ichikawa, S.; Sakiyama, H.; Suzuki, G.; Hidari, K.I.; Hirabayashi, Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 4638–4643. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.L.; Dominguez, M.; Wu, K.; Pagano, R.E. Identification of active site residues in glucosylceramide synthase. A nucleotide-binding catalytic motif conserved with processive beta-glycosyltransferases. J. Biol. Chem. 2001, 276, 26492–26498. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Sato, T.; Natsuka, S.; Kobayashi, Y.; Zhou, D.; Shinkai, T.; Hayakawa, S.; Furukawa, K. Involvement of murine β-1,4-galactosyltransferase V in lactosylceramide biosynthesis. Glycoconj. J. 2010, 27, 685–695. [Google Scholar] [CrossRef]
- Nishie, T.; Hikimochi, Y.; Zama, K.; Fukusumi, Y.; Ito, M.; Yokoyama, H.; Naruse, C.; Ito, M.; Asano, M. Beta4-galactosyltransferase-5 is a lactosylceramide synthase essential for mouse extra-embryonic development. Glycobiology 2010, 20, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Fukumoto, S.; Furukawa, K.; Okajima, T.; Wiels, J.; Yokoyama, K.; Suzuki, Y.; Urano, T.; Ohta, M.; Furukawa, K. Molecular cloning of globotriaosylceramide/CD77 synthase, a glycosyltransferase that initiates the synthesis of globo series glycosphingolipids. J. Biol. Chem. 2000, 275, 15152–15156. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, T.; Nishikawa, K.; Hanada, K. Transmembrane BAX inhibitor motif containing (TMBIM) family proteins perturbs a trans-Golgi network enzyme, Gb3 synthase, and reduces Gb3 biosynthesis. J. Biol. Chem. 2010, 285, 35505–35518. [Google Scholar] [CrossRef]
- D’Angelo, G.; Uemura, T.; Chuang, C.C.; Polishchuk, E.; Santoro, M.; Ohvo-Rekilä, H.; Sato, T.; Di Tullio, G.; Varriale, A.; D’Auria, S.; et al. Vesicular and non-vesicular transport feed distinct glycosylation pathways in the Golgi. Nature 2013, 501, 116–120. [Google Scholar] [CrossRef]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Yamaji, T.; Hanada, K. Sphingolipid metabolism and interorganellar transport: Localization of sphingolipid enzymes and lipid transfer proteins. Traffic 2015, 16, 101–122. [Google Scholar] [CrossRef]
- Blackburn, J.B.; Lupashin, V.V. Creating Knockouts of Conserved Oligomeric Golgi Complex Subunits Using CRISPR-Mediated Gene Editing Paired with a Selection Strategy Based on Glycosylation Defects Associated with Impaired COG Complex Function. Methods Mol. Biol. 2016, 1496, 145–161. [Google Scholar] [PubMed]
- Wu, X.; Steet, R.A.; Bohorov, O.; Bakker, J.; Newell, J.; Krieger, M.; Spaapen, L.; Kornfeld, S.; Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004, 10, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, R.; Foulquier, F.; Jaeken, J.; Matthijs, G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Mol. Genet. Metab. 2008, 93, 15–21. [Google Scholar] [CrossRef]
- Yamaji, T.; Sekizuka, T.; Tachida, Y.; Sakuma, C.; Morimoto, K.; Kuroda, M.; Hanada, K. A CRISPR Screen Identifies LAPTM4A and TM9SF Proteins as Glycolipid-Regulating Factors. iScience. 2019, 11, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Muneeruddin, K.; Choi, M.Y.; Tao, L.; Bhuiyan, R.H.; Ohmi, Y.; Furukawa, K.; Furukawa, K.; Boland, S.; Shaffer, S.A.; et al. Genome-wide CRISPR screens for Shiga toxins and ricin reveal Golgi proteins critical for glycosylation. PLoS Biol. 2018, 16, e2006951. [Google Scholar] [CrossRef]
- Majumder, S.; Kono, M.; Lee, Y.T.; Byrnes, C.; Li, C.; Tuymetova, G.; Proia, R.L. A genome-wide CRISPR/Cas9 screen reveals that the aryl hydrocarbon receptor stimulates sphingolipid levels. J. Biol. Chem. 2020, 295, 4341–4349. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.R.; Lazarus, J.E.; Sit, B.; Schmieder, S.; Lencer, W.I.; Blondel, C.J.; Doench, J.G.; Davis, B.M.; Waldor, M.K. CRISPR Screen Reveals that EHEC’s T3SS and Shiga Toxin Rely on Shared Host Factors for Infection. mBio 2018, 9, e01003-18. [Google Scholar] [CrossRef]
- Yasumura, Y.; Kawakita, Y. Studies on SV40 in tissue culture: Preliminary step for cancer reserach in vitro. Nihon Rinsho 1963, 21, 1201–1215. (In Japanese) [Google Scholar]
- Osada, N.; Kohara, A.; Yamaji, T.; Hirayama, N.; Kasai, F.; Sekizuka, T.; Kuroda, M.; Hanada, K. The genome landscape of the african green monkey kidney-derived vero cell line. DNA Res. 2014, 21, 673–683. [Google Scholar] [CrossRef]
- Konowalchuk, J.; Speirs, J.I.; Stavric, S. Vero response to a cytotoxin of Escherichia coli. Infect. Immun. 1977, 18, 775–779. [Google Scholar] [CrossRef]
- Sakuma, C.; Sekizuka, T.; Kuroda, M.; Kasai, F.; Saito, K.; Ikeda, M.; Yamaji, T.; Osada, N.; Hanada, K. Novel endogenous simian retroviral integrations in Vero cells: Implications for quality control of a human vaccine cell substrate. Sci Rep. 2018, 8, 644. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef]
- Tsukada, M.; Gallwitz, D. Isolation and characterization of SYS genes from yeast, multicopy suppressors of the functional loss of the transport GTPase Ypt6p. J. Cell Sci. 1996, 109, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Isaji, T.; Sato, Y.; Fukuda, T.; Gu, J. N-glycosylation of the I-like domain of beta1 integrin is essential for beta1 integrin expression and biological function: Identification of the minimal N-glycosylation requirement for alpha5beta1. J. Biol. Chem. 2009, 284, 12207–12216. [Google Scholar] [CrossRef] [PubMed]
- Behnia, R.; Panic, B.; Whyte, J.R.; Munro, S. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 2004, 6, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Bonifacino, J.S. ARFRP1 functions upstream of ARL1 and ARL5 to coordinate recruitment of distinct tethering factors to the trans-Golgi network. J. Cell Biol. 2019, 218, 3681–3696. [Google Scholar] [CrossRef]
- Selyunin, A.S.; Iles, L.R.; Bartholomeusz, G.; Mukhopadhyay, S. Genome-wide siRNA screen identifies UNC50 as a regulator of Shiga toxin 2 trafficking. J. Cell Biol. 2017, 216, 3249–3262. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Osakada, H.; Kojidani, T.; Waga, M.; Suda, Y.; Asakawa, H.; Haraguchi, T.; Nakano, A. Visualization of secretory cargo transport within the Golgi apparatus. J. Cell Biol. 2019, 218, 1602–1618. [Google Scholar] [CrossRef] [PubMed]
- Virreira Winter, S.; Zychlinsky, A.; Bardoel, B.W. Genome-wide CRISPR screen reveals novel host factors required for Staphylococcus aureus α-hemolysin-mediated toxicity. Sci. Rep. 2016, 6, 24242. [Google Scholar] [CrossRef]
- Setty, S.R.; Strochlic, T.I.; Tong, A.H.; Boone, C.; Burd, C.G. Golgi targeting of ARF-like GTPase Arl3p requires its Nalpha-acetylation and the integral membrane protein Sys1p. Nat. Cell Biol. 2004, 6, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Spessott, W.; Uliana, A.; Maccioni, H.J. Defective GM3 synthesis in Cog2 null mutant CHO cells associates to mislocalization of lactosylceramide sialyltransferase in the Golgi complex. Neurochem. Res. 2010, 35, 2161–2167. [Google Scholar] [CrossRef] [PubMed]
- Young, W.W., Jr.; Lutz, M.S.; Mills, S.E.; Lechler-Osborn, S. Use of brefeldin A to define sites of glycosphingolipid synthesis: GA2/GM2/GD2 synthase is trans to the brefeldin A block. Proc. Natl. Acad. Sci. USA 1990, 87, 6838–6842. [Google Scholar] [CrossRef] [PubMed]
- Kjellberg, M.A.; Mattjus, P. Glycolipid transfer protein expression is affected by glycosphingolipid synthesis. PLoS ONE 2013, 8, e70283. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, T.; Horie, A.; Tachida, Y.; Sakuma, C.; Suzuki, Y.; Kushi, Y.; Hanada, K. Role of Intracellular Lipid Logistics in the Preferential Usage of Very Long Chain-Ceramides in Glucosylceramide. Int. J. Mol. Sci. 2016, 17, 1761. [Google Scholar] [CrossRef] [PubMed]
- Takatsuki, A.; Tamura, G. Brefeldin A, a Specific Inhibitor of Intracellular Translocation of Vesicular Stomatitis Virus G Protein: Intracellular Accumulation of High-mannose Type G Protein and Inhibition of Its Cell Surface Expression. Agric. Biol. Chem. 1985, 49, 899–902. [Google Scholar]
- Lippincott-Schwartz, J.; Yuan, L.C.; Bonifacino, J.S.; Klausner, R.D. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: Evidence for membrane cycling from Golgi to ER. Cell 1989, 56, 801–813. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Finazzi, D.; Klausner, R.D. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 1992, 360, 350–352. [Google Scholar] [CrossRef]
- Billmann, M.; Horn, T.; Fischer, B.; Sandmann, T.; Huber, W.; Boutros, M. A genetic interaction map of cell cycle regulators. Mol. Biol. Cell 2016, 27, 1397–1407. [Google Scholar] [CrossRef]
- Yamaji, T.; Hanamatsu, H.; Sekizuka, T.; Kuroda, M.; Iwasaki, N.; Ohnishi, M.; Furukawa, J.I.; Yahiro, K.; Hanada, K. A CRISPR Screen Using Subtilase Cytotoxin Identifies SLC39A9 as a Glycan-Regulating Factor. iScience 2019, 15, 407–420. [Google Scholar] [CrossRef]
- Stewart, S.E.; Menzies, S.A.; Popa, S.J.; Savinykh, N.; Petrunkina Harrison, A.; Lehner, P.J.; Moreau, K. A genome-wide CRISPR screen reconciles the role of N-linked glycosylation in galectin-3 transport to the cell surface. J. Cell Sci. 2017, 130, 3234–3247. [Google Scholar] [CrossRef]
- Park, J.S.; Helble, J.D.; Lazarus, J.E.; Yang, G.; Blondel, C.J.; Doench, J.G.; Starnbach, M.N.; Waldor, M.K. A FACS-Based Genome-wide CRISPR Screen Reveals a Requirement for COPI in Chlamydia trachomatis Invasion. iScience 2019, 11, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Kojima, T.; Kitamura, T. Plat-E: An efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000, 7, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Matsuoka, K.; Kita, E.; Igai, K.; Higashi, N.; Miyagawa, A.; Watanabe, T.; Yanoshita, R.; Samejima, Y.; Terunuma, D.; et al. Oral therapeutic agents with highly clustered globotriose for treatment of Shiga toxigenic Escherichia coli infections. J. Infect. Dis. 2004, 189, 360–368. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Yamaji, T.; Hanada, K. Establishment of HeLa cell mutants deficient in sphingolipid-related genes using TALENs. PLoS ONE 2014, 9, e88124. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Holm, S. A simple sequentially rejective multiple test procedrure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakuma, C.; Sekizuka, T.; Kuroda, M.; Hanada, K.; Yamaji, T. Identification of SYS1 as a Host Factor Required for Shiga Toxin-Mediated Cytotoxicity in Vero Cells. Int. J. Mol. Sci. 2021, 22, 4936. https://doi.org/10.3390/ijms22094936
Sakuma C, Sekizuka T, Kuroda M, Hanada K, Yamaji T. Identification of SYS1 as a Host Factor Required for Shiga Toxin-Mediated Cytotoxicity in Vero Cells. International Journal of Molecular Sciences. 2021; 22(9):4936. https://doi.org/10.3390/ijms22094936
Chicago/Turabian StyleSakuma, Chisato, Tsuyoshi Sekizuka, Makoto Kuroda, Kentaro Hanada, and Toshiyuki Yamaji. 2021. "Identification of SYS1 as a Host Factor Required for Shiga Toxin-Mediated Cytotoxicity in Vero Cells" International Journal of Molecular Sciences 22, no. 9: 4936. https://doi.org/10.3390/ijms22094936
APA StyleSakuma, C., Sekizuka, T., Kuroda, M., Hanada, K., & Yamaji, T. (2021). Identification of SYS1 as a Host Factor Required for Shiga Toxin-Mediated Cytotoxicity in Vero Cells. International Journal of Molecular Sciences, 22(9), 4936. https://doi.org/10.3390/ijms22094936