
Type I Interferon-Mediated Regulation of Antiviral Capabilities of Neutrophils

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
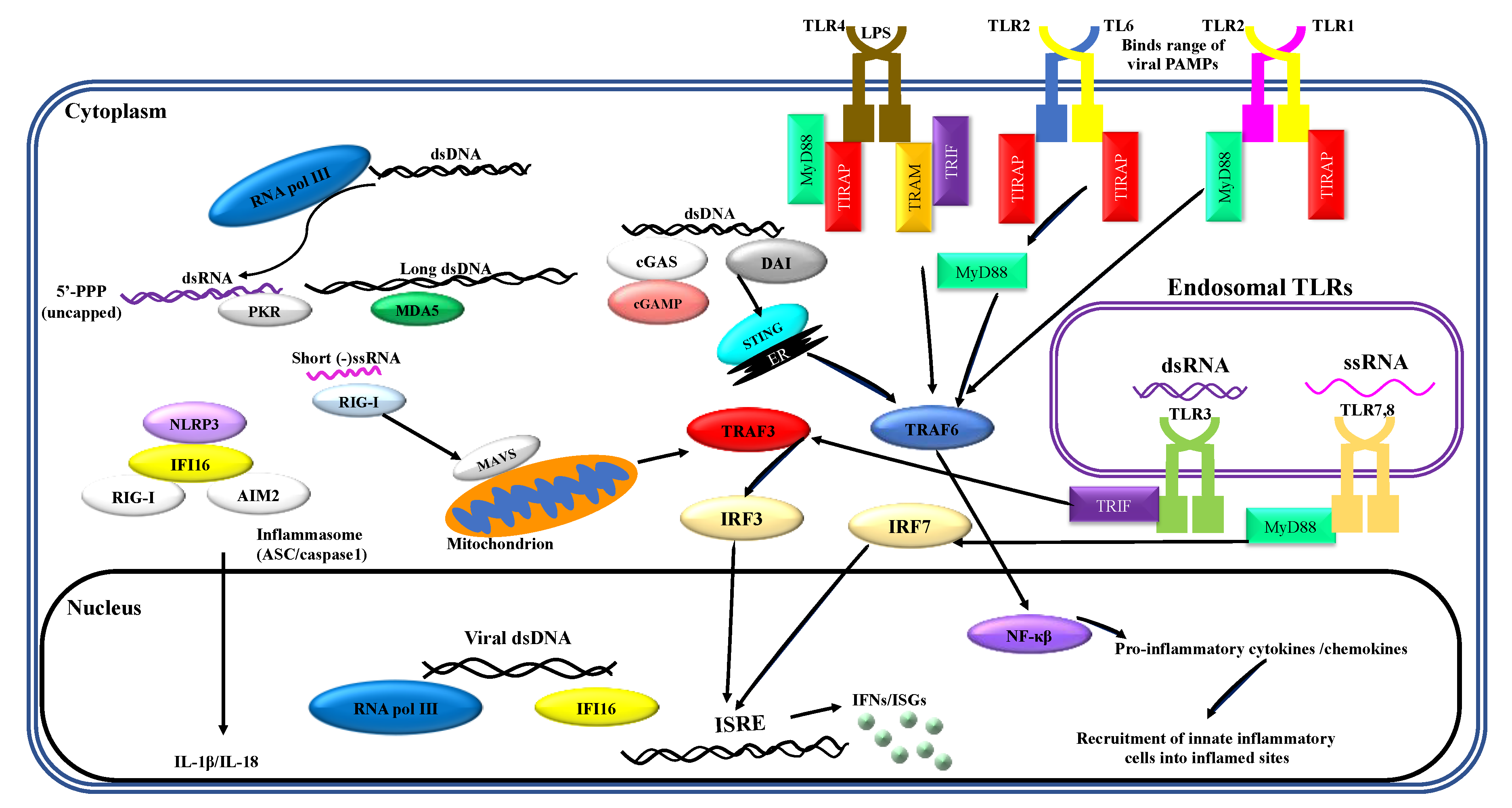
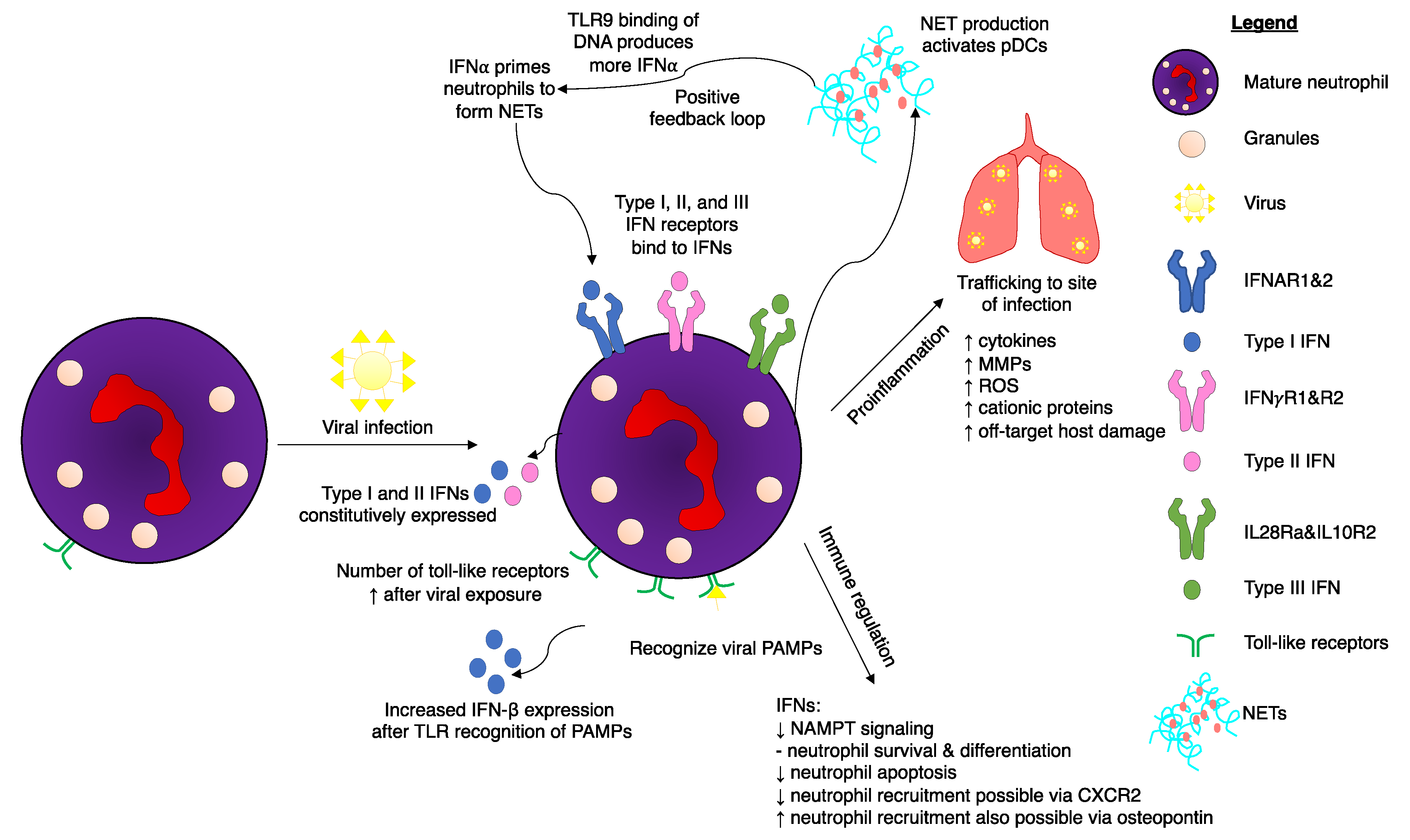
1. Recognition of Viral PAMPs by Neutrophils
2. Interferons
3. Regulation of IFN Signaling in Neutrophils
4. Walking on a Knife Edge: Host Neutrophils Associated with both Protection and Severe Adverse Events following Viral Infection
5. The importance of Neutrophil Activation/Dysregulation of Type I IFN Responses in COVID-19 Patients
6. Neutrophils Respond to Type I and Type III IFNs to Regulate Viral Infections
7. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATF2 | activating transcription factor 2 |
| cGAMP | cyclic guanosine monophosphate–adenosine monophosphate |
| cGAS | cyclic guanosine monophosphate–adenosine monophosphate (GMP–AMP) synthase |
| CHIKV | Chikungunya virus |
| COVID-19 | Coronavirus disease 2019 |
| DENV2 | Dengue virus |
| DNA | deoxyribonucleic acid |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| HMGB1 | high mobility group box protein 1 |
| IAV | influenza A virus |
| IFN | interferon |
| IFNAR | IFN-α/β receptor |
| IFNGR | interferon gamma receptor |
| IFNLR1 | interleukin 28 receptor, alpha subunit |
| IL | interleukin |
| IL10R2 | interleukin 10 receptor, beta subunit |
| iOPN | intracellular osteopontin |
| IRAK | interleukin-1 receptor-associated kinase |
| IRF | IFN regulatory factor |
| ISG | IFN-stimulated genes |
| ISGF3 | IFN-stimulated gene factor 3 |
| IRG | interferon responsive genes |
| JAK | Janus kinase |
| LPS | lipopolysaccharide |
| MAVS | mitochondrial antiviral signalingm |
| MCP-1 | monocyte chemoattractant protein 1 |
| MDA-5 | melanoma differentiation-associated protein |
| MMP | matrix metalloproteases |
| MyD88 | myeloid differentiation primary response protein 88 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NET | neutrophil extracellular traps |
| NF | nuclear factor |
| NOD | nucleotide-binding oligomerization domain |
| NOX2 | NADPH oxidase isoform 2 |
| PAMPs | pathogen-associated molecular patterns |
| pDCs | plasmacytoid dendritic cells |
| PRR | pattern recognition receptors |
| RIG-I | retinoic acid-inducible gene |
| RNA | ribonucleic acid |
| ROS | reactive oxygen species |
| RSV | respiratory syncytial virus |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SINV | Sindbis virus |
| STAT | signal transducer and activator of transcription |
| STING | stimulator of interferon genes |
| Th1 | T helper 1 |
| TIR | Toll/interleukin-1 receptor |
| TIRAP | Toll interleukin-1 receptor-associated protein |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TRAF | TNF receptor associated factor 3 |
| TRAM | TRIF-related adaptor molecule |
| TREM-1 | Triggering receptor expressed on myeloid cells-1 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| VSV | vesicular stomatitis virus |
| ZIKV | Zika virus |
References
- Pillay, J.; den Braber, I.; Vrisekoop, N.; Kwast, L.M.; de Boer, R.J.; Borghans, J.A.; Tesselaar, K.; Koenderman, L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.V.; Jonsson, C.B. A role for neutrophils in viral respiratory disease. Front. Immunol. 2017, 8, 550. [Google Scholar] [CrossRef] [PubMed]
- Stegelmeier, A.A.; van Vloten, J.P.; Mould, R.C.; Klafuric, E.M.; Minott, J.A.; Wootton, S.K.; Bridle, B.W.; Karimi, K. Myeloid cells during viral infections and inflammation. Viruses 2019, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, V.; Turk, M.; Jenne, C.N.; Kim, S.J. Neutrophils in viral infection. Cell Tissue Res. 2018, 371, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, F.; Means, T.K.; Luster, A.D. Toll-like receptors stimulate human neutrophil function. Blood 2003, 102, 2660–2669. [Google Scholar] [CrossRef]
- Hamilton, J.A. GM-CSF in inflammation. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Andonegui, G.; Goyert, S.M.; Kubes, P. Lipopolysaccharide-induced leukocyte-endothelial cell interactions: A role for CD14 versus toll-like receptor 4 within microvessels. J. Immunol. 2002, 169, 2111–2119. [Google Scholar] [CrossRef]
- Stegelmeier, A.A.; Chan, L.; Mehrani, Y.; Petrik, J.J.; Wootton, S.K.; Bridle, B.; Karimi, K. Characterization of the impact of oncolytic vesicular stomatitis virus on the trafficking, phenotype, and antigen presentation potential of neutrophils and their ability to acquire a non-structural viral protein. Int. J. Mol. Sci. 2020, 21, 6347. [Google Scholar] [CrossRef]
- van Bruggen, R.; Drewniak, A.; Tool, A.T.J.; Jansen, M.; van Houdt, M.; Geissler, J.; van den Berg, T.K.; Chapel, H.; Kuijpers, T.W. Toll-like receptor responses in IRAK-4-deficient neutrophils. J. Innate Immun. 2010, 2, 280–287. [Google Scholar] [CrossRef]
- Sturge, C.R.; Benson, A.; Raetz, M.; Wilhelm, C.L.; Mirpuri, J.; Vitetta, E.S.; Yarovinsky, F. TLR-independent neutrophil-derived IFN-gamma is important for host resistance to intracellular pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 10711–10716. [Google Scholar] [CrossRef]
- Ivan, F.X.; Tan, K.S.; Phoon, M.C.; Engelward, B.P.; Welsch, R.E.; Rajapakse, J.C.; Chow, V.T. Neutrophils infected with highly virulent influenza H3N2 virus exhibit augmented early cell death and rapid induction of type I interferon signaling pathways. Genomics 2013, 101, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.M.; White, M.R.; Hartshorn, K.L. Influenza A viruses upregulate neutrophil toll-like receptor 2 expression and function. Scand. J. Immunol. 2006, 63, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Bowen, G.N.; Padden, C.; Cerny, A.; Finberg, R.W.; Newburger, P.E.; Kurt-Jones, E.A. Toll-like receptor-mediated activation of neutrophils by influenza A virus. Blood 2008, 112, 2028–2034. [Google Scholar] [CrossRef] [PubMed]
- Tamassia, N.; Bazzoni, F.; Le Moigne, V.; Calzetti, F.; Masala, C.; Grisendi, G.; Bussmeyer, U.; Scutera, S.; De Gironcoli, M.; Costantini, C.; et al. IFN-beta expression is directly activated in human neutrophils transfected with plasmid DNA and is further increased via TLR-4-mediated signaling. J. Immunol. 2012, 189, 1500–1509. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef]
- Horng, T.; Medzhitov, R. Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 12654–12658. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Kotenko, S.V. Interferon-lambda: A new addition to an old family. J. Interferon Cytokine Res. 2010, 30, 555–564. [Google Scholar] [CrossRef]
- Walter, M.R.; Bordens, R.; Nagabhushan, T.L.; Williams, B.R.; Herberman, R.B.; Dinarello, C.A.; Borden, E.C.; Trotta, P.P.; Pestka, S.; Pfeffer, L.M. Review of recent developments in the molecular characterization of recombinant alfa interferons on the 40th anniversary of the discovery of interferon. Cancer Biother. Radiopharm. 1998, 13, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, P.J.; Williams, B.R. Fine tuning type I interferon responses. Cytokine Growth Factor Rev. 2013, 24, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Uzé, G.; Schreiber, G.; Piehler, J.; Pellegrini, S. The receptor of the type I interferon family. Curr. Top. Microbiol. Immunol. 2007, 316, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Rivera, A.; Parker, D.; Durbin, J.E. Type III IFNs: Beyond antiviral protection. Semin. Immunol. 2019, 43, 101303. [Google Scholar] [CrossRef] [PubMed]
- van Pesch, V.; Lanaya, H.; Renauld, J.C.; Michiels, T. Characterization of the murine alpha interferon gene family. J. Virol. 2004, 78, 8219–8228. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Schindler, C.; Darnell, J.E. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annu. Rev. Biochem. 1995, 64, 621–651. [Google Scholar] [CrossRef]
- Farrar, M.A.; Schreiber, R.D. The molecular cell biology of interferon-gamma and its receptor. Annu. Rev. Immunol. 1993, 11, 571–611. [Google Scholar] [CrossRef]
- Pott, J.; Mahlakõiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G.; et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.D.; Feng, N.; Sen, A.; Balan, M.; Tseng, H.C.; McElrath, C.; Smirnov, S.V.; Peng, J.; Yasukawa, L.L.; Durbin, R.K.; et al. Distinct roles of Type I and Type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog. 2016, 12, e1005600. [Google Scholar] [CrossRef]
- Mahlakõiv, T.; Hernandez, P.; Gronke, K.; Diefenbach, A.; Staeheli, P. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 2015, 11, e1004782. [Google Scholar] [CrossRef]
- Hernández, P.P.; Mahlakoiv, T.; Yang, I.; Schwierzeck, V.; Nguyen, N.; Guendel, F.; Gronke, K.; Ryffel, B.; Hoelscher, C.; Dumoutier, L.; et al. Interferon-λ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 2015, 16, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V. IFN-λs. Curr. Opin Immunol. 2011, 23, 583–590. [Google Scholar] [CrossRef]
- Witte, K.; Witte, E.; Sabat, R.; Wolk, K. IL-28A, IL-28B, and IL-29: Promising cytokines with type I interferon-like properties. Cytokine Growth Factor Rev. 2010, 21, 237–251. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Levy, D.E.; Marié, I.; Smith, E.; Prakash, A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 2002, 22, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Marié, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef]
- Wathelet, M.G.; Lin, C.H.; Parekh, B.S.; Ronco, L.V.; Howley, P.M.; Maniatis, T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1998, 1, 507–518. [Google Scholar] [CrossRef]
- Levy, D.E. Whence interferon? Variety in the production of interferon in response to viral infection. J. Exp. Med. 2002, 195, F15–F18. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Hata, N.; Asagiri, M.; Nakaya, T.; Taniguchi, T.; Tanaka, N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998, 441, 106–110. [Google Scholar] [CrossRef]
- Morin, P.; Bragança, J.; Bandu, M.T.; Lin, R.; Hiscott, J.; Doly, J.; Civas, A. Preferential binding sites for interferon regulatory factors 3 and 7 involved in interferon-A gene transcription. J. Mol. Biol. 2002, 316, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef]
- Stirnweiss, A.; Ksienzyk, A.; Klages, K.; Rand, U.; Grashoff, M.; Hauser, H.; Kröger, A. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J. Immunol. 2010, 184, 5179–5185. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Hata, N.; Sato, M.; Takaoka, A.; Asagiri, M.; Tanaka, N.; Taniguchi, T. Constitutive IFN-alpha/beta signal for efficient IFN-alpha/beta gene induction by virus. Biochem. Biophys. Res. Commun. 2001, 285, 518–525. [Google Scholar] [CrossRef]
- Tanaka, N.; Sato, M.; Lamphier, M.S.; Nozawa, H.; Oda, E.; Noguchi, S.; Schreiber, R.D.; Tsujimoto, Y.; Taniguchi, T. Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells 1998, 3, 29–37. [Google Scholar] [CrossRef]
- Liu, B.; Liao, J.; Rao, X.; Kushner, S.A.; Chung, C.D.; Chang, D.D.; Shuai, K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. USA 1998, 95, 10626–10631. [Google Scholar] [CrossRef]
- van Boxel-Dezaire, A.H.; Rani, M.R.; Stark, G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006, 25, 361–372. [Google Scholar] [CrossRef]
- David, M. Signal transduction by type I interferons. Biotechniques 2002, 33 (Suppl. 4), 58–65. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Huynh, L.; Wang, L.; Shi, C.; Park-Min, K.H.; Ivashkiv, L.B. ITAM-coupled receptors inhibit IFNAR signaling and alter macrophage responses to TLR4 and Listeria monocytogenes. J. Immunol. 2012, 188, 3447–3457. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Shen, Y.; Yang, W.; Mecklenbrauker, I.; Neel, B.G.; Ivashkiv, L.B. Inhibition of IFN-alpha signaling by a PKC- and protein tyrosine phosphatase SHP-2-dependent pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 10267–10272. [Google Scholar] [CrossRef]
- Davidson, S.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic potential of interferon αβ in acute influenza infection. Nat. Commun. 2014, 5, 3864. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Weber, F. Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev. 2008, 19, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Rocha, B.C.; Marques, P.E.; Leoratti, F.M.S.; Junqueira, C.; Pereira, D.B.; Antonelli, L.R.D.V.; Menezes, G.B.; Golenbock, D.T.; Gazzinelli, R.T. Type I interferon transcriptional signature in neutrophils and low-density granulocytes are associated with tissue damage in malaria. Cell Rep. 2015, 13, 2829–2841. [Google Scholar] [CrossRef]
- Berry, M.P.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef]
- López de Padilla, C.M.; Niewold, T.B. The type I interferons: Basic concepts and clinical relevance in immune-mediated inflammatory diseases. Gene 2016, 576, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.U.; Kwon, H.J.; Ko, H.J.; Byun, Y.H.; Seong, B.L.; Uematsu, S.; Akira, S.; Kweon, M.N. Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog. 2011, 7, e1001304. [Google Scholar] [CrossRef]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef]
- Herold, S.; Steinmueller, M.; von Wulffen, W.; Cakarova, L.; Pinto, R.; Pleschka, S.; Mack, M.; Kuziel, W.A.; Corazza, N.; Brunner, T.; et al. Lung epithelial apoptosis in influenza virus pneumonia: The role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008, 205, 3065–3077. [Google Scholar] [CrossRef]
- Noyce, R.S.; Taylor, K.; Ciechonska, M.; Collins, S.E.; Duncan, R.; Mossman, K.L. Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J. Virol. 2011, 85, 10926–10931. [Google Scholar] [CrossRef]
- Noyce, R.S.; Collins, S.E.; Mossman, K.L. Identification of a novel pathway essential for the immediate-early, interferon-independent antiviral response to enveloped virions. J. Virol. 2006, 80, 226–235. [Google Scholar] [CrossRef]
- Iversen, M.B.; Reinert, L.S.; Thomsen, M.K.; Bagdonaite, I.; Nandakumar, R.; Cheshenko, N.; Prabakaran, T.; Vakhrushev, S.Y.; Krzyzowska, M.; Kratholm, S.K.; et al. An innate antiviral pathway acting before interferons at epithelial surfaces. Nat. Immunol. 2016, 17, 150–158. [Google Scholar] [CrossRef]
- Denney, L.; Ho, L.P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018, 41, 218–233. [Google Scholar] [CrossRef]
- Prescott, J.B.; Hall, P.R.; Bondu-Hawkins, V.S.; Ye, C.; Hjelle, B. Early innate immune responses to Sin Nombre hantavirus occur independently of IFN regulatory factor 3, characterized pattern recognition receptors, and viral entry. J. Immunol. 2007, 179, 1796–1802. [Google Scholar] [CrossRef]
- Klinkhammer, J.; Schnepf, D.; Ye, L.; Schwaderlapp, M.; Gad, H.H.; Hartmann, R.; Garcin, D.; Mahlakoiv, T.; Staeheli, P. IFN-lambda prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. Elife 2018, 7, e33354. [Google Scholar] [CrossRef]
- Fung, K.Y.; Mangan, N.E.; Cumming, H.; Horvat, J.C.; Mayall, J.R.; Stifter, S.A.; De Weerd, N.; Roisman, L.C.; Rossjohn, J.; Robertson, S.A.; et al. Interferon-ε protects the female reproductive tract from viral and bacterial infection. Science 2013, 339, 1088–1092. [Google Scholar] [CrossRef]
- Stavrou, S.; Blouch, K.; Kotla, S.; Bass, A.; Ross, S.R. Nucleic acid recognition orchestrates the anti-viral response to retroviruses. Cell Host Microbe 2015, 17, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 2017, 46, 875–890.e876. [Google Scholar] [CrossRef]
- Davidson, S.; McCabe, T.M.; Crotta, S.; Gad, H.H.; Hessel, E.M.; Beinke, S.; Hartmann, R.; Wack, A. IFNλ is a potent anti-influenza therapeutic without the inflammatory side effects of IFNα treatment. EMBO Mol. Med. 2016, 8, 1099–1112. [Google Scholar] [CrossRef]
- Kanazawa, N.; Kurosaki, M.; Sakamoto, N.; Enomoto, N.; Itsui, Y.; Yamashiro, T.; Tanabe, Y.; Maekawa, S.; Nakagawa, M.; Chen, C.H.; et al. Regulation of hepatitis C virus replication by interferon regulatory factor 1. J. Virol. 2004, 78, 9713–9720. [Google Scholar] [CrossRef] [PubMed]
- Kolb, J.P.; Casella, C.R.; SenGupta, S.; Chilton, P.M.; Mitchell, T.C. Type I interferon signaling contributes to the bias that Toll-like receptor 4 exhibits for signaling mediated by the adaptor protein TRIF. Sci. Signal. 2014, 7, ra108. [Google Scholar] [CrossRef] [PubMed]
- Tamassia, N.; Moigne, V.L.; Rossato, M.; Donini, M.; McCartney, S.; Calzetti, F.; Colonna, M.; Bazzoni, F.; Cassatella, M.A. Activation of an immunoregulatory and antiviral gene expression program in poly(I:C)-transfected human neutrophils. J. Immunol. 2008, 181, 6563–6573. [Google Scholar] [CrossRef]
- Martinelli, S.; Urosevic, M.; Daryadel, A.; Oberholzer, P.A.; Baumann, C.; Fey, M.F.; Dummer, R.; Simon, H.U.; Yousefi, S. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J. Biol. Chem. 2004, 279, 44123–44132. [Google Scholar] [CrossRef] [PubMed]
- Esser, M.A.G.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nat. Lett. 2009, 458, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Trans. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef]
- Pylaeva, E.; Bordbari, S.; Spyra, I.; Decker, A.S.; Haussler, S.; Vybornov, V.; Lang, S.; Jablonska, J. Detrimental effect of type I IFNs during acute lung infection with Pseudomonas aeruginosa is mediated through the stimulation of neutrophil NETosis. Front. Immunol. 2019, 10, 2190. [Google Scholar] [CrossRef] [PubMed]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef]
- Meunier, I.; von Messling, V. NS1-mediated delay of type I interferon induction contributes to influenza A virulence in ferrets. J. Gen. Virol. 2011, 92, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Svitek, N.; Rudd, P.A.; Obojes, K.; Pillet, S.; von Messling, V. Severe seasonal influenza in ferrets correlates with reduced interferon and increased IL-6 induction. Virology 2008, 376, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; Garcia-Sastre, A. Influenza A virus NS1 protein prevents activation of NF-κB and induction of alpha:beta interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar] [CrossRef]
- Hufford, M.M.; Richardson, G.; Zhou, H.; Manicassamy, B.; García-Sastre, A.; Enelow, R.I.; Braciale, T.J. Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PLoS ONE 2012, 7, e46581. [Google Scholar] [CrossRef]
- Tate, M.D.; Brooks, A.G.; Reading, P.C.; Mintern, J.D. Neutrophils sustain effective CD8(+) T-cell responses in the respiratory tract following influenza infection. Immunol. Cell Biol. 2012, 90, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Siakaeva, E.; Pylaeva, E.; Spyra, I.; Bordbari, S.; Hoing, B.; Kurten, C.; Lang, S.; Jablonska, J. Neutrophil maturation and survival is controlled by IFN-dependent regulation of NAMPT signaling. Int. J. Mol. Sci. 2019, 20, 5584. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, E.; Hato, F.; Kato, T.; Sakamoto, C.; Akahori, M.; Hino, M.; Kitagawa, S. Type I and type II interferons delay human neutrophil apoptosis via activation of STAT3 and up-regulation of cellular inhibitor of apoptosis. J. Leukoc. Biol. 2005, 78, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Pylaeva, E.; Lang, S.; Jablonska, J. The essential role of type I interferons in differentiation and activation of tumor-associated neutrophils. Front. Immunol. 2016, 7, 629. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Wu, C.F.; Lienenklaus, S.; Kroger, A.; Weiss, S.; Jablonska, J. Delayed apoptosis of tumor associated neutrophils in the absence of endogenous IFN-beta. Int. J. Cancer 2015, 136, 572–583. [Google Scholar] [CrossRef] [PubMed]
- de Wit, H.; Dokter, W.H.; Esselink, M.T.; Halie, M.R.; Vellenga, E. Interferon-gamma enhances the LPS-induced G-CSF gene expression in human adherent monocytes, which is regulated at transcriptional and posttranscriptional levels. Exp. Hematol. 1993, 21, 785–790. [Google Scholar]
- Nguyen-Jackson, H.; Panopoulos, A.D.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 controls the neutrophil migratory response to CXCR2 ligands by direct activation of G-CSF–induced CXCR2 expression and via modulation of CXCR2 signal transduction. Blood 2010, 115, 3354–3363. [Google Scholar] [CrossRef]
- Scheel-Toellner, D.; Wang, K.; Henriquez, N.V.; Webb, P.R.; Craddock, R.; Pilling, D.; Akbar, A.N.; Salmon, M.; Lord, J.M. Cytokine-mediated inhibition of apoptosis in non-transformed T cells and neutrophils can be dissociated from protein kinase B activation. Eur. J. Immunol. 2002, 32, 486–493. [Google Scholar] [CrossRef]
- Li, L.; Huang, L.; Sung, S.S.; Lobo, P.I.; Brown, M.G.; Gregg, R.K.; Engelhard, V.H.; Okusa, M.D. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J. Immunol. 2007, 178, 5899–5911. [Google Scholar] [CrossRef]
- Yasunami, Y.; Kojo, S.; Kitamura, H.; Toyofuku, A.; Satoh, M.; Nakano, M.; Nabeyama, K.; Nakamura, Y.; Matsuoka, N.; Ikeda, S.; et al. Valpha14 NK T cell-triggered IFN-gamma production by Gr-1+CD11b+ cells mediates early graft loss of syngeneic transplanted islets. J. Exp. Med. 2005, 202, 913–918. [Google Scholar] [CrossRef]
- Gomez, J.C.; Yamada, M.; Martin, J.R.; Dang, H.; Brickey, W.J.; Bergmeier, W.; Dinauer, M.C.; Doerschuk, C.M. Mechanisms of interferon-gamma production by neutrophils and its function during Streptococcus pneumoniae pneumonia. Am. J. Respir. Cell Mol. Biol. 2015, 52, 349–364. [Google Scholar] [CrossRef]
- Yamada, M.; Gomez, J.C.; Chugh, P.E.; Lowell, C.A.; Dinauer, M.C.; Dittmer, D.P.; Doerschuk, C.M. Interferon-gamma production by neutrophils during bacterial pneumonia in mice. Am. J. Respir. Crit. Care Med. 2011, 183, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.N.; Beaman, B.L. Interferon-gamma activation of polymorphonuclear neutrophil function. Immunology 2004, 112, 2–12. [Google Scholar] [CrossRef]
- Pine, R. Constitutive expression of an ISGF2/IRF1 transgene leads to interferon-independent activation of interferon-inducible genes and resistance to virus infection. J. Virol. 1992, 66, 4470–4478. [Google Scholar] [CrossRef] [PubMed]
- Paladino, P.; Cummings, D.T.; Noyce, R.S.; Mossman, K.L. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J. Immunol. 2006, 177, 8008–8016. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Gjinaj, E.; Bachu, M.; Squire, E.; Novatt, H.; Ozato, K.; Rabin, R.L. IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front. Immunol. 2019, 10, 1019. [Google Scholar] [CrossRef]
- Paludan, S.R. Innate antiviral defenses independent of inducible IFNα/β production. Trends Immunol. 2016, 37, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, W.; Peppelenbosch, M.P.; Pan, Q. Noncanonical antiviral mechanisms of ISGs: Dispensability of inducible interferons. Trends Immunol. 2017, 38, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, Y.; Xu, L.; Su, J.; Huang, F.; Wang, Y.; Boor, P.P.C.; Chen, K.; Cao, W.; Zhou, X.; et al. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci. Signal. 2017, 10, eaah4248. [Google Scholar] [CrossRef]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil diversity in health and disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Peteranderl, C.; Morales-Nebreda, L.; Selvakumar, B.; Lecuona, E.; Vadász, I.; Morty, R.E.; Schmoldt, C.; Bespalowa, J.; Wolff, T.; Pleschka, S.; et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J. Clin. Investig. 2016, 126, 1566–1580. [Google Scholar] [CrossRef]
- Lim, K.; Hyun, Y.M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8⁺ T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [PubMed]
- Nicolás-Ávila, J.; Adrover, J.M.; Hidalgo, A. Neutrophils in homeostasis, immunity, and cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Goritzka, M.; Makris, S.; Kausar, F.; Durant, L.R.; Pereira, C.; Kumagai, Y.; Culley, F.J.; Mack, M.; Akira, S.; Johansson, C. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J. Exp. Med. 2015, 212, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Hibbs, M.L.; Chen, W. The contributions of lung macrophage and monocyte heterogeneity to influenza pathogenesis. Immunol. Cell Biol. 2017, 95, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef]
- Alazawi, W.; Knolle, P.A. Interfering with Kupffer cell replenishment: New insights into liver injury. J. Hepatol. 2018, 68, 635–637. [Google Scholar] [CrossRef]
- Borst, K.; Frenz, T.; Spanier, J.; Tegtmeyer, P.K.; Chhatbar, C.; Skerra, J.; Ghita, L.; Namineni, S.; Lienenklaus, S.; Köster, M.; et al. Type I interferon receptor signaling delays Kupffer cell replenishment during acute fulminant viral hepatitis. J. Hepatol. 2017, 68, 682–690. [Google Scholar] [CrossRef]
- Lang, P.A.; Recher, M.; Honke, N.; Scheu, S.; Borkens, S.; Gailus, N.; Krings, C.; Meryk, A.; Kulawik, A.; Cervantes-Barragan, L.; et al. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology 2010, 52, 25–32. [Google Scholar] [CrossRef]
- Movita, D.; van de Garde, M.D.; Biesta, P.; Kreefft, K.; Haagmans, B.; Zuniga, E.; Herschke, F.; De Jonghe, S.; Janssen, H.L.; Gama, L.; et al. Inflammatory monocytes recruited to the liver within 24 hours after virus-induced inflammation resemble Kupffer cells but are functionally distinct. J. Virol. 2015, 89, 4809–4817. [Google Scholar] [CrossRef] [PubMed]
- Polakos, N.K.; Cornejo, J.C.; Murray, D.A.; Wright, K.O.; Treanor, J.J.; Crispe, I.N.; Topham, D.J.; Pierce, R.H. Kupffer cell-dependent hepatitis occurs during influenza infection. Am. J. Pathol. 2006, 168, 1169–1178, quiz 1404–1165. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Walsh, K.B.; Rice, S.; Rosen, H.; Oldstone, M.B. Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3799–3804. [Google Scholar] [CrossRef]
- Short, K.R.; Kroeze, E.J.B.V.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Jamieson, A.M.; Pasman, L.; Yu, S.; Gamradt, P.; Homer, R.J.; Decker, T.; Medzhitov, R. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science 2013, 340, 1230–1234. [Google Scholar] [CrossRef]
- Read, A.F.; Graham, A.L.; Råberg, L. Animal defenses against infectious agents: Is damage control more important than pathogen control. PLoS Biol. 2008, 6, e4. [Google Scholar] [CrossRef]
- Brandes, M.; Klauschen, F.; Kuchen, S.; Germain, R.N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 2013, 154, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Oshansky, C.M.; Gartland, A.J.; Wong, S.S.; Jeevan, T.; Wang, D.; Roddam, P.L.; Caniza, M.A.; Hertz, T.; Devincenzo, J.P.; Webby, R.J.; et al. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care Med. 2014, 189, 449–462. [Google Scholar] [CrossRef]
- de Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Poon, L.L.; Lau, A.S.; Luk, W.; Lau, Y.L.; Shortridge, K.F.; Gordon, S.; Guan, Y.; Peiris, J.S. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: A mechanism for the unusual severity of human disease? Lancet 2002, 360, 1831–1837. [Google Scholar] [CrossRef]
- Talmi-Frank, D.; Altboum, Z.; Solomonov, I.; Udi, Y.; Jaitin, D.A.; Klepfish, M.; David, E.; Zhuravlev, A.; Keren-Shaul, H.; Winter, D.R.; et al. Extracellular matrix proteolysis by MT1-MMP contributes to influenza-related tissue damage and mortality. Cell Host Microbe 2016, 20, 458–470. [Google Scholar] [CrossRef]
- Medeiros, N.I.; Fares, R.C.; Franco, E.P.; Sousa, G.R.; Mattos, R.T.; Chaves, A.T.; Nunes, M.D.; Dutra, W.O.; Correa-Oliveira, R.; Rocha, M.O.; et al. Differential expression of matrix metalloproteinases 2, 9 and cytokines by neutrophils and monocytes in the clinical forms of Chagas disease. PLoS Negl. Trop. Dis. 2017, 11, e0005284. [Google Scholar] [CrossRef]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef]
- Gao, R.; Bhatnagar, J.; Blau, D.M.; Greer, P.; Rollin, D.C.; Denison, A.M.; Deleon-Carnes, M.; Shieh, W.J.; Sambhara, S.; Tumpey, T.M.; et al. Cytokine and chemokine profiles in lung tissues from fatal cases of 2009 pandemic influenza A (H1N1): Role of the host immune response in pathogenesis. Am. J. Pathol. 2013, 183, 1258–1268. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, A.; Wan, Y.; Liu, X.; Qiu, C.; Xi, X.; Ren, Y.; Wang, J.; Dong, Y.; Bao, M.; et al. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 769–774. [Google Scholar] [CrossRef]
- Klein, S.L.; Hodgson, A.; Robinson, D.P. Mechanisms of sex disparities in influenza pathogenesis. J. Leukoc. Biol. 2012, 92, 67–73. [Google Scholar] [CrossRef]
- Bradley, L.M.; Douglass, M.F.; Chatterjee, D.; Akira, S.; Baaten, B.J. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 2012, 8, e1002641. [Google Scholar] [CrossRef]
- Keck, T.; Balcom, J.H.; Fernández-del Castillo, C.; Antoniu, B.A.; Warshaw, A.L. Matrix metalloproteinase-9 promotes neutrophil migration and alveolar capillary leakage in pancreatitis-associated lung injury in the rat. Gastroenterology 2002, 122, 188–201. [Google Scholar] [CrossRef]
- Simonsen, L.; Clarke, M.J.; Schonberger, L.B.; Arden, N.H.; Cox, N.J.; Fukuda, K. Pandemic versus epidemic influenza mortality: A pattern of changing age distribution. J. Infect. Dis. 1998, 178, 53–60. [Google Scholar] [CrossRef]
- Morris, D.E.; Cleary, D.W.; Clarke, S.C. Secondary bacterial infections associated with influenza pandemics. Front. Microbiol. 2017, 8, 1041. [Google Scholar] [CrossRef]
- Berthiaume, Y.; Lesur, O.; Dagenais, A. Treatment of adult respiratory distress syndrome: Plea for rescue therapy of the alveolar epithelium. Thorax 1999, 54, 150–160. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Fensterl, V.; Chattopadhyay, S.; Sen, G.C. No love lost between viruses and interferons. Annu. Rev. Virol. 2015, 2, 549–572. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, K.E.; Lewerenz, M.; Reboul, J.; Lutfalla, G.; Uzé, G. The type I interferon receptor: Structure, function, and evolution of a family business. J. Interferon Cytokine Res. 1999, 19, 1069–1098. [Google Scholar] [CrossRef]
- González-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Conrad, E.; Resch, T.K.; Gogesch, P.; Kalinke, U.; Bechmann, I.; Bogdan, C.; Waibler, Z. Protection against RNA-induced liver damage by myeloid cells requires type I interferon and IL-1 receptor antagonist in mice. Hepatology 2014, 59, 1555–1563. [Google Scholar] [CrossRef]
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011, 140, 697–708.e694. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.J.; Bermejo-Martin, J.F.; Danesh, A.; Muller, M.P.; Kelvin, D.J. Human immunopathogenesis of severe acute respiratory syndrome (SARS). Virus Res. 2008, 133, 13–19. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef]
- García-Sastre, A. Ten strategies of interferon evasion by viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef]
- Fujisawa, H. Inhibitory role of neutrophils on influenza virus multiplication in the lungs of mice. Microbiol. Immunol. 2001, 45, 679–688. [Google Scholar] [CrossRef]
- Tate, M.D.; Deng, Y.M.; Jones, J.E.; Anderson, G.P.; Brooks, A.G.; Reading, P.C. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J. Immunol. 2009, 183, 7441–7450. [Google Scholar] [CrossRef] [PubMed]
- Srikiatkhachorn, A.; Mathew, A.; Rothman, A.L. Immune-mediated cytokine storm and its role in severe dengue. Semin. Immunopathol 2017, 39, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Furuya, Y.; Steiner, D.; Metzger, D.W. Does type I interferon limit protective neutrophil responses during pulmonary Francisella tularensis infection? Front. Immunol. 2014, 5, 355. [Google Scholar] [CrossRef]
- Tate, M.D.; Brooks, A.G.; Reading, P.C. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 2008, 9, 57. [Google Scholar] [CrossRef]
- Tang, B.M.; Shojaei, M.; Teoh, S.; Meyers, A.; Ho, J.; Ball, T.B.; Keynan, Y.; Pisipati, A.; Kumar, A.; Eisen, D.P.; et al. Neutrophils-related host factors associated with severe disease and fatality in patients with influenza infection. Nat. Commun. 2019, 10, 3422. [Google Scholar] [CrossRef]
- Xin, L.; Vargas-Inchaustegui, D.A.; Raimer, S.S.; Kelly, B.C.; Hu, J.; Zhu, L.; Sun, J.; Soong, L. Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J. Immunol. 2010, 184, 7047–7056. [Google Scholar] [CrossRef]
- Besteman, S.B.; Callaghan, A.; Langedijk, A.C.; Hennus, M.P.; Meyaard, L.; Mokry, M.; Bont, L.J.; Calis, J.J.A. Transcriptome of airway neutrophils reveals an interferon response in life-threatening respiratory syncytial virus infection. Clin. Immunol. 2020, 220, 108593. [Google Scholar] [CrossRef]
- de Steenhuijsen Piters, W.A.; Heinonen, S.; Hasrat, R.; Bunsow, E.; Smith, B.; Suarez-Arrabal, M.C.; Chaussabel, D.; Cohen, D.M.; Sanders, E.A.; Ramilo, O.; et al. Nasopharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am. J. Respir. Crit. Care Med. 2016, 194, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Jong, V.L.; Ahout, I.M.; van den Ham, H.J.; Jans, J.; Zaaraoui-Boutahar, F.; Zomer, A.; Simonetti, E.; Bijl, M.A.; Brand, H.K.; van, I.W.F.; et al. Transcriptome assists prognosis of disease severity in respiratory syncytial virus infected infants. Sci. Rep. 2016, 6, 36603. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.C.; Anderson, D.; Galbraith, S.; Fantino, E.; Gutierrez Cardenas, D.; Read, J.F.; Serralha, M.; Holt, B.J.; Strickland, D.H.; Sly, P.D.; et al. Personalized transcriptomics reveals heterogeneous immunophenotypes in children with viral bronchiolitis. Am. J. Respir. Crit. Care Med. 2019, 199, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, R.S.; Coates, M.; Ito, K.; Ghazaly, M.; Feather, C.; Abdulla, F.; Tunstall, T.; Jain, P.; Cass, L.; Rapeport, G.; et al. Reduced nasal viral load and IFN responses in infants with respiratory syncytial virus bronchiolitis and respiratory failure. Am. J. Respir. Crit. Care Med. 2018, 198, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.E.; Locke, M.C.; Young, A.R.; Monte, K.; Hedberg, M.L.; Shimak, R.M.; Sheehan, K.C.F.; Veis, D.J.; Diamond, M.S.; Lenschow, D.J. Distinct roles of interferon alpha and beta in controlling Chikungunya virus replication and modulating neutrophil-mediated inflammation. J. Virol. 2019, 94, e00841-19. [Google Scholar] [CrossRef]
- Palha, N.; Guivel-Benhassine, F.; Briolat, V.; Lutfalla, G.; Sourisseau, M.; Ellett, F.; Wang, C.H.; Lieschke, G.J.; Herbomel, P.; Schwartz, O.; et al. Real-time whole-body visualization of Chikungunya Virus infection and host interferon response in zebrafish. PLoS Pathog. 2013, 9, e1003619. [Google Scholar] [CrossRef]
- Hiroki, C.H.; Toller-Kawahisa, J.E.; Fumagalli, M.J.; Colon, D.F.; Figueiredo, L.T.M.; Fonseca, B.; Franca, R.F.O.; Cunha, F.Q. Neutrophil extracellular traps effectively control acute Chikungunya virus infection. Front. Immunol. 2019, 10, 3108. [Google Scholar] [CrossRef]
- Stock, A.T.; Smith, J.M.; Carbone, F.R. Type I IFN suppresses CXCR2 driven neutrophil recruitment into the sensory ganglia during viral infection. J. Exp. Med. 2014, 211, 751–759. [Google Scholar] [CrossRef]
- Zukor, K.; Wang, H.; Siddharthan, V.; Julander, J.G.; Morrey, J.D. Zika virus-induced acute myelitis and motor deficits in adult interferon alphabeta/gamma receptor knockout mice. J. Neurovirol. 2018, 24, 273–290. [Google Scholar] [CrossRef]
- Boucontet, L.; Passoni, G.; Thiry, V.; Maggi, L.; Herbomel, P.; Levraud, J.P.; Colucci-Guyon, E. A model of superinfection of virus-infected zebrafish larvae: Increased susceptibility to bacteria associated with neutrophil death. Front. Immunol. 2018, 9, 1084. [Google Scholar] [CrossRef]
- Alvarez-Torres, D.; Gomez-Abellan, V.; Arizcun, M.; Garcia-Rosado, E.; Bejar, J.; Sepulcre, M.P. Identification of an interferon-stimulated gene, isg15, involved in host immune defense against viral infections in gilthead seabream (Sparus aurata L.). Fish. Shellfish Immunol. 2018, 73, 220–227. [Google Scholar] [CrossRef]
- Morales, D.J.; Lenschow, D.J. The antiviral activities of ISG15. J. Mol. Biol. 2013, 425, 4995–5008. [Google Scholar] [CrossRef]
- D’Cunha, J.; Knight, E., Jr.; Haas, A.L.; Truitt, R.L.; Borden, E.C. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc. Natl. Acad. Sci. USA 1996, 93, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.M.; Acharya, D.; Duty, L.; Thompson, E.A.; Le, L.; Stokic, D.S.; Leis, A.A.; Bai, F. Osteopontin facilitates West Nile virus neuroinvasion via neutrophil "Trojan horse" transport. Sci. Rep. 2017, 7, 4722. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhang, M.; Zhang, L.; Wang, P.; Song, G.; Liu, B.; Wu, H.; Yin, Z.; Gao, C. Intracellular osteopontin stabilizes TRAF3 to positively regulate innate antiviral response. Sci Rep. 2016, 6, 23771. [Google Scholar] [CrossRef] [PubMed]
- Rojas Marquez, J.D.; Li, T.; McCluggage, A.R.R.; Tan, J.M.J.; Muise, A.; Higgins, D.E.; Brumell, J.H. Cutting edge: NOX2 NADPH oxidase controls infection by an intracellular bacterial pathogen through limiting the type 1 IFN response. J. Immunol. 2021, 206, 323–328. [Google Scholar] [CrossRef]
- To, E.E.; Luong, R.; Diao, J.; JJ, O.L.; Brooks, D.A.; Vlahos, R.; Selemidis, S. Novel endosomal NOX2 oxidase inhibitor ameliorates pandemic influenza A virus-induced lung inflammation in mice. Respirology 2019, 24, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.; Andreakos, E. Neutrophils in viral infections: Current concepts and caveats. J. Leukoc. Biol. 2015, 98, 557–564. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell Infect. Microbiol. 2017, 7, 373. [Google Scholar]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg 2020, 76, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, S.M.; Geller, A.E.; Hu, X.; Tieri, D.; Cooke, E.A.; Ding, C.; Woeste, M.; Zhange, H.-G.; Cavallazi, R.; Clifford, S.P.; et al. Emergence of low-density inflammatory neutrophils correlates with hypercoagulable state and disease severity in COVID-19 patients. bioRxiv 2020, 6724. [Google Scholar] [CrossRef]
- Rosa, B.A.; Ahmed, M.; Singh, D.K.; Choreno-Parra, J.A.; Cole, J.; Jimenez-Alvarez, L.A.; Rodriguez-Reyna, T.S.; Singh, B.; Golzalez, O.; Carrion, R.; et al. IFN signaling and neutrophil degranulation transcriptional signatures are induced during SARS-CoV-2 infection. bioRxiv 2020, 4, 290. [Google Scholar] [CrossRef]
- Didangelos, A. COVID-19 hyperinflammation- what about neutrophils? mSphere 2020, 5, e00367-20. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Cicco, S.; Cicco, G.; Racanelli, V.; Vacca, A. Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): Two potential targets for COVID-19 treatment. Mediators Inflamm. 2020, 2020, 7527953. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Petrosillo, N.; Viceconte, G.; Ergonul, O.; Ippolito, G.; Petersen, E. COVID-19, SARS and MERS: Are they closely related? Clin. Microbiol. Infect. 2020, 26, 729–734. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B., Jr.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-H.; Wang, Y.-N.; Chang, Q.-Y.; Ma, P.; Hu, Y.; Cao, X. Type III interferons in viral infection and antiviral immunity. Cell Physiol. Biochem. 2018, 51, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, V.; Dutta, O.; McElrath, C.; Du, P.; Chang, Y.J.; Cicciarelli, B.; Pitler, A.; Whitehead, I.; Obar, J.J.; Durbin, J.E.; et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol. 2017, 2, eaan5357. [Google Scholar] [CrossRef] [PubMed]
- Blazek, K.; Eames, H.L.; Weiss, M.; Byrne, A.J.; Perocheau, D.; Pease, J.E.; Doyle, S.; McCann, F.; Williams, R.O.; Udalova, I.A. IFN-lambda resolves inflammation via suppression of neutrophil infiltration and IL-1beta production. J. Exp. Med. 2015, 212, 845–853. [Google Scholar] [CrossRef]
- Santer, D.M.; Minty, G.E.S.; Golec, D.P.; Lu, J.; May, J.; Namdar, A.; Shah, J.; Elahi, S.; Proud, D.; Joyce, M.; et al. Differential expression of interferon-lambda receptor 1 splice variants determines the magnitude of the antiviral response induced by interferon-lambda 3 in human immune cells. PLoS Pathog. 2020, 16, e1008515. [Google Scholar] [CrossRef]
- Nice, T.J.; Bladridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stegelmeier, A.A.; Darzianiazizi, M.; Hanada, K.; Sharif, S.; Wootton, S.K.; Bridle, B.W.; Karimi, K. Type I Interferon-Mediated Regulation of Antiviral Capabilities of Neutrophils. Int. J. Mol. Sci. 2021, 22, 4726. https://doi.org/10.3390/ijms22094726
Stegelmeier AA, Darzianiazizi M, Hanada K, Sharif S, Wootton SK, Bridle BW, Karimi K. Type I Interferon-Mediated Regulation of Antiviral Capabilities of Neutrophils. International Journal of Molecular Sciences. 2021; 22(9):4726. https://doi.org/10.3390/ijms22094726
Chicago/Turabian StyleStegelmeier, Ashley A., Maedeh Darzianiazizi, Kiersten Hanada, Shayan Sharif, Sarah K. Wootton, Byram W. Bridle, and Khalil Karimi. 2021. "Type I Interferon-Mediated Regulation of Antiviral Capabilities of Neutrophils" International Journal of Molecular Sciences 22, no. 9: 4726. https://doi.org/10.3390/ijms22094726
APA StyleStegelmeier, A. A., Darzianiazizi, M., Hanada, K., Sharif, S., Wootton, S. K., Bridle, B. W., & Karimi, K. (2021). Type I Interferon-Mediated Regulation of Antiviral Capabilities of Neutrophils. International Journal of Molecular Sciences, 22(9), 4726. https://doi.org/10.3390/ijms22094726