
Intrinsically Connected: Therapeutically Targeting the Cathepsin Proteases and the Bcl-2 Family of Protein Substrates as Co-regulators of Apoptosis

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
Abstract
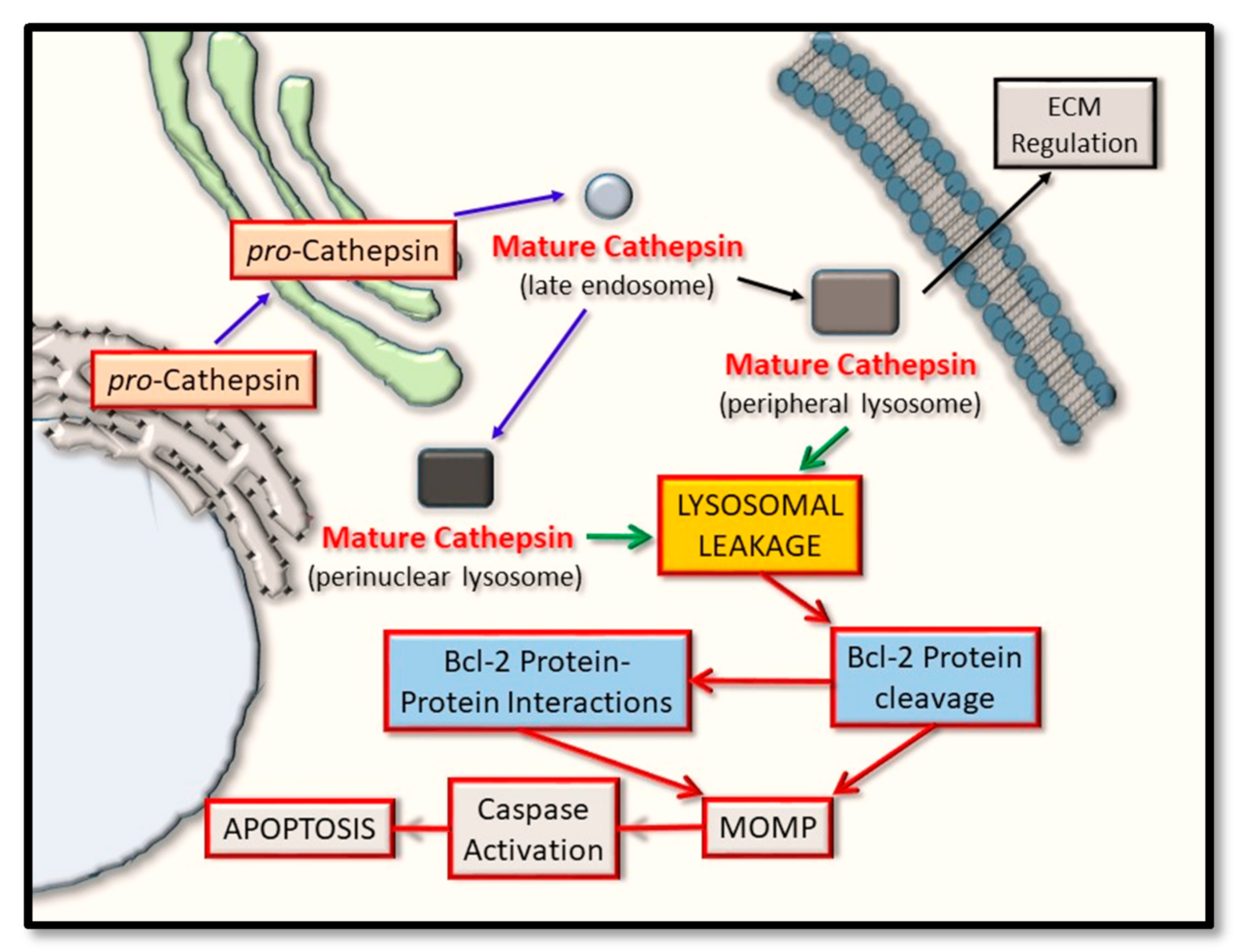
1. Introduction
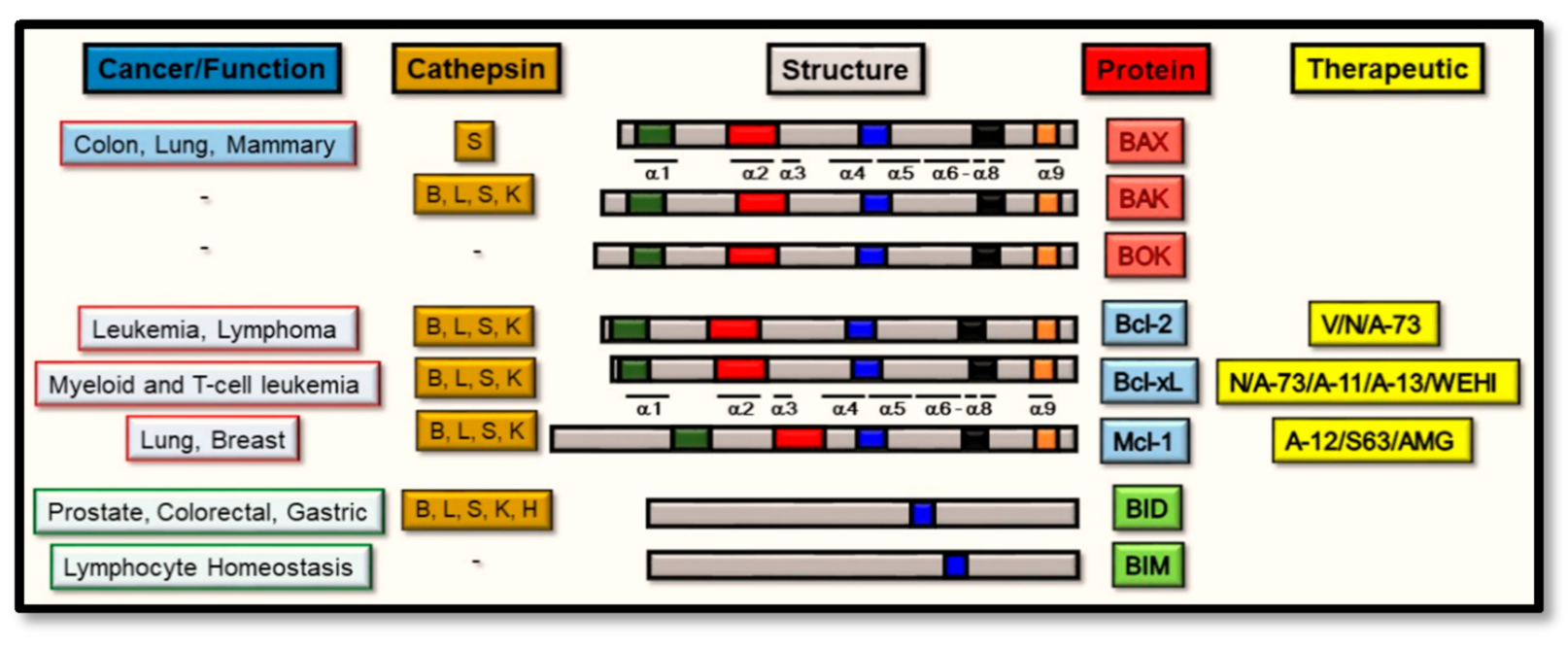
2. Pro-Apoptotic BAX and BAK as Substrate Proteins for Cathepsin Proteases
3. Anti-Apoptotic Bcl-2 Proteins as Substrates for the Cathepsin Proteases
4. BH3-Only Domain Proteins as Substrates for the Cathepsin Proteases
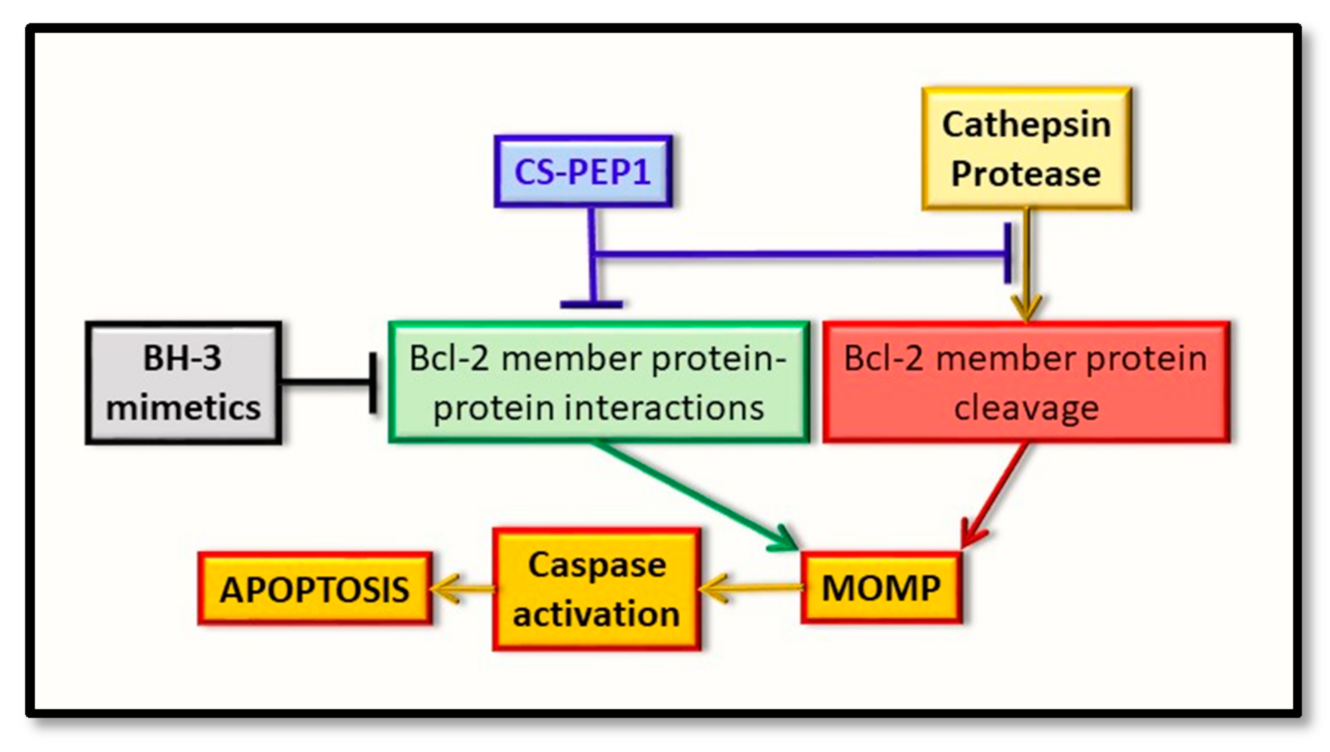
5. Cathepsin and the Bcl-2 Proteins: Targeted Therapeutic Development
6. Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Firestone, R.A.; Pisano, J.M.; Bonney, R.J. Lysosomotropic agents. 1. Synthesis and cytotoxic action of lysosomotropic detergents. J. Med. Chem. 1979, 22, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Turk, V. Lysosomes as “Suicide Bags” in Cell Death: Myth or Reality? J. Biol. Chem. 2009, 284, 21783–21787. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.N.; Salvesen, G. Handbook of Proteolytic Enzymes, 3rd ed.; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Soond, S.M.; Kozhevnikova, M.V.; Zamyatnin, A.A., Jr. ‘Patchiness’ and basic cancer research: Unravelling the proteases. Cell Cycle 2019, 18, 1687–1701. [Google Scholar] [CrossRef]
- Jordans, S.; Jenko-Kokalj, S.; Kühl, N.M.; Tedelind, S.; Sendt, W.; Brömme, D.; Turk, D.; Brix, K. Monitoring compartment-specific substrate cleavage by cathepsins B, K, L, and S at physiological pH and redox conditions. BMC Biochem. 2009, 10, 23. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Soond, S.M.; Kozhevnikova, M.V.; Townsend, P.A.; Zamyatnin, J.A.A. Cysteine Cathepsin Protease Inhibition: An update on its Diagnostic, Prognostic and Therapeutic Potential in Cancer. Pharmaceuticals 2019, 12, 87. [Google Scholar] [CrossRef]
- Soond, S.M.; Kozhevnikova, M.V.; Frolova, A.S.; Savvateeva, L.V.; Plotnikov, E.Y.; Townsend, P.A.; Han, Y.-P.; Zamyatnin, A.A. Lost or Forgotten: The nuclear cathepsin protein isoforms in cancer. Cancer Lett. 2019, 462, 43–50. [Google Scholar] [CrossRef]
- Soond, S.M.; Savvateeva, L.V.; Makarov, V.A.; Gorokhovets, N.V.; Townsend, P.A.; Zamyatnin, J.A.A. Making Connections: p53 and the Cathepsin Proteases as Co-Regulators of Cancer and Apoptosis. Cancers 2020, 12, 3476. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Worth, A.; Thrasher, A.J.; Gaspar, H.B. Autoimmune lymphoproliferative syndrome: Molecular basis of disease and clinical phenotype. Br. J. Haematol. 2006, 133, 124–140. [Google Scholar] [CrossRef]
- Ethell, D.W.; Buhler, L.A. Fas ligand-mediated apoptosis in degenerative disorders of the brain. J. Clin. Immunol. 2003, 23, 439–446. [Google Scholar] [CrossRef]
- Almeida, S.; Sarmento-Ribeiro, A.B.; Januário, C.; Rego, A.C.; Oliveira, C.R. Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2008, 374, 599–603. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Duprez, L.; Wirawan, E.; Berghe, T.V.; Vandenabeele, P. Major cell death pathways at a glance. Microbes Infect. 2009, 11, 1050–1062. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med. 2010, 31, 1–20. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Brenner, D.; Mak, T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009, 21, 871–877. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 2009, 122, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kass, G.E.N.; Szegezdi, E.; Joseph, B. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner Caspase-3, -6, and -7 Perform Distinct, Non-redundant Roles during the Demolition Phase of Apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Chimini, G. The phagocytosis of apoptotic cells. Semin. Immunol. 2001, 13, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Nawaz, M.; Ahsan, H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell. Biochem. 2011, 351, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Saez, A.J. The secrets of the Bcl-2 family. Cell Death Differ. 2012, 19, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Kvansakul, M.; Yang, H.; Fairlie, W.D.; Czabotar, P.E.; Fischer, S.F.; Perugini, M.A.; Huang, D.C.S.; Colman, P.M. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 2008, 15, 1564–1571. [Google Scholar] [CrossRef]
- Huang, D.C.; Adams, J.M.; Cory, S. The conserved N-terminal BH4 domain of Bcl-2 homologues is essential for inhibition of apoptosis and interaction with CED-4. EMBO J. 1998, 17, 1029–1039. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; Garcia-Saez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef]
- Huang, D.C.; Strasser, A. BH3-Only Proteins—Essential Initiators of Apoptotic Cell Death. Cell 2000, 103, 839–842. [Google Scholar] [CrossRef]
- Petros, A.M.; Olejniczak, E.T.; Fesik, S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 2004, 1644, 83–94. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef]
- Kim, H.; Tu, H.-C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Stepwise Activation of BAX and BAK by tBID, BIM, and PUMA Initiates Mitochondrial Apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef]
- Wang, K.; Gross, A.; Waksman, G.; Korsmeyer, S.J. Mutagenesis of the BH3 Domain of BAX Identifies Residues Critical for Dimerization and Killing. Mol. Cell. Biol. 1998, 18, 6083–6089. [Google Scholar] [CrossRef]
- Hinds, M.G.; Lackmann, M.; Skea, G.L.; Harrison, P.J.; Huang, D.C.S.; Day, C.L. The structure of Bcl-w reveals a role for the C-terminal residues in modulating biological activity. EMBO J. 2003, 22, 1497–1507. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak Peptide Complex: Recognition Between Regulators of Apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Walensky, L.D. Targeting BAX to drug death directly. Nat. Chem. Biol. 2019, 15, 657–665. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Cosentino, K.; García-Sáez, A.J. Bax and Bak Pores: Are We Closing the Circle? Trends Cell Biol. 2017, 27, 266–275. [Google Scholar] [CrossRef]
- Shalaby, R.; Flores-Romero, H.; García-Sáez, A.J. The Mysteries around the BCL-2 Family Member BOK. Biomolecules 2020, 10, 1638. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Emschermann, F.; Lauterwasser, J.; Kaiser, A.; Ichim, G.; Tait, S.W.G.; Frank, R.; Langer, H.F.; et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015, 34, 67–80. [Google Scholar] [CrossRef]
- Setoguchi, K.; Otera, H.; Mihara, K. Cytosolic factor- and TOM-independent import of C-tail-anchored mitochondrial outer membrane proteins. EMBO J. 2006, 25, 5635–5647. [Google Scholar] [CrossRef] [PubMed]
- Echeverry, N.; Bachmann, D.C.G.; Ke, F.; Strasser, A.; Simon, H.-U.; Kaufmann, T. Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 2013, 20, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Schulman, J.J.; Wright, F.A.; Han, X.; Zluhan, E.J.; Szczesniak, L.M.; Wojcikiewicz, R.J.H. The Stability and Expression Level of Bok Are Governed by Binding to Inositol 1,4,5-Trisphosphate Receptors. J. Biol. Chem. 2016, 291, 11820–11828. [Google Scholar] [CrossRef] [PubMed]
- Schulman, J.J.; Wright, F.A.; Kaufmann, T.; Wojcikiewicz, R.J.H. The Bcl-2 Protein Family Member Bok Binds to the Coupling Domain of Inositol 1,4,5-Trisphosphate Receptors and Protects Them from Proteolytic Cleavage. J. Biol. Chem. 2013, 288, 25340–25349. [Google Scholar] [CrossRef]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic Frameshift Mutations in theBAXGene in Colon Cancers of the Microsatellite Mutator Phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef]
- Zhang, M.; Zheng, J.; Nussinov, R.; Ma, B. Oncogenic Mutations Differentially Affect Bax Monomer, Dimer, and Oligomeric Pore Formation in the Membrane. Sci. Rep. 2016, 6, 33340. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, W.; Lapolla, S.M.; Miao, Y.; Shao, Y.; Falcone, M.; Boreham, D.; McFarlane, N.; Ding, J.; Johnson, A.E.; et al. Bax Forms an Oligomer via Separate, Yet Interdependent, Surfaces. J. Biol. Chem. 2010, 285, 17614–17627. [Google Scholar] [CrossRef]
- Shibata, M.-A.; Liu, M.-L.; Knudson, M.C.; Shibata, E.; Yoshidome, K.; Bandey, T.; Korsmeyer, S.J.; Green, J.E. Haploid loss of bax leads to accelerated mammary tumor development in C3(1)/SV40-TAg transgenic mice: Reduction in protective apoptotic response at the preneoplastic stage. EMBO J. 1999, 18, 2692–2701. [Google Scholar] [CrossRef]
- Knudson, C.M.; Tung, K.S.K.; Tourtellotte, W.G.; Brown, G.A.J.; Korsmeyer, S.J. Bax-Deficient Mice with Lymphoid Hyperplasia and Male Germ Cell Death. Science 1995, 270, 96–99. [Google Scholar] [CrossRef]
- Lindsten, T.; Ross, A.J.; King, A.; Zong, W.-X.; Rathmell, J.C.; Shiels, H.A.; Ulrich, E.; Waymire, K.G.; Mahar, P.; Frauwirth, K.; et al. The Combined Functions of Proapoptotic Bcl-2 Family Members Bak and Bax Are Essential for Normal Development of Multiple Tissues. Mol. Cell 2000, 6, 1389–1399. [Google Scholar] [CrossRef]
- Fannjiang, Y.; Kim, C.-H.; Huganir, R.L.; Zou, S.; Lindsten, T.; Thompson, C.B.; Mito, T.; Traystman, R.J.; Larsen, T.; Griffin, D.E.; et al. BAK Alters Neuronal Excitability and Can Switch from Anti- to Pro-Death Function during Postnatal Development. Dev. Cell 2003, 4, 575–585. [Google Scholar] [CrossRef]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Youle, R.J.; Edlich, F. The C-terminal helix of Bcl-xL mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. 2012, 20, 333–342. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic bax and bak: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Lovell, J.F.; Billen, L.P.; Bindner, S.; Shamas-Din, A.; Fradin, C.; Leber, B.; Andrews, D.W. Membrane Binding by tBid Initiates an Ordered Series of Events Culminating in Membrane Permeabilization by Bax. Cell 2008, 135, 1074–1084. [Google Scholar] [CrossRef]
- Hsu, Y.-T.; Wolter, K.G.; Youle, R.J. Cytosol-to-membrane redistribution of Bax and Bcl-XL during apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef]
- Llambi, F.; Wang, Y.-M.; Victor, B.; Yang, M.; Schneider, D.M.; Gingras, S.; Parsons, M.J.; Zheng, J.H.; Brown, S.A.; Pelletier, S.; et al. BOK Is a Non-canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell 2016, 165, 421–433. [Google Scholar] [CrossRef]
- Einsele-Scholz, S.; Malmsheimer, S.; Bertram, K.; Stehle, D.; Johänning, J.; Manz, M.; Daniel, P.T.; Gillissen, B.F.; Schulze-Osthoff, K.; Essmann, F. Bok is a genuine multi-BH-domain protein that triggers apoptosis in the absence of Bax and Bak. J. Cell Sci. 2016, 129, 2213–2223. [Google Scholar] [CrossRef]
- Fernández-Marrero, Y.; Bleicken, S.; Das, K.K.; Bachmann, D.; Kaufmann, T.; Garcia-Saez, A.J. The membrane activity of BOK involves formation of large, stable toroidal pores and is promoted by cBID. FEBS J. 2017, 284, 711–724. [Google Scholar] [CrossRef]
- Ke, F.; Voss, A.; Kerr, J.B.; O’Reilly, L.A.; Tai, L.; Echeverry, N.; Bouillet, P.; Strasser, A.; Kaufmann, T. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. 2012, 19, 915–925. [Google Scholar] [CrossRef]
- Suominen, J.S.; Yan, W.; Toppari, J.; Kaipia, A. The expression and regulation of Bcl-2-related ovarian killer (Bok) mRNA in the developing and adult rat testis. Eur. J. Endocrinol. 2001, 145, 771–778. [Google Scholar] [CrossRef]
- Jääskeläinen, M.; Nieminen, A.; Pökkylä, R.-M.; Kauppinen, M.; Liakka, A.; Heikinheimo, M.; Vaskivuo, T.E.; Klefström, J.; Tapanainen, J.S. Regulation of cell death in human fetal and adult ovaries—Role of Bok and Bcl-XL. Mol. Cell. Endocrinol. 2010, 330, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Galleano, M.; Cederbaum, A.I. Cytotoxicity and Apoptosis Produced by Arachidonic Acid in Hep G2 Cells Overexpressing Human Cytochrome P4502E1. J. Biol. Chem. 1997, 272, 14532–14541. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Iskandar, K.B.; Hirpara, J.L.; Clement, M.-V.; Pervaiz, S. Hydrogen Peroxide-Mediated Cytosolic Acidification Is a Signal for Mitochondrial Translocation of Bax during Drug-Induced Apoptosis of Tumor Cells. Cancer Res. 2004, 64, 7867–7878. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Tian, C.; Zhao, L.; Petit, P.X.; Mehrpour, M.; Chen, Q. Cysteine 62 of Bax Is Critical for Its Conformational Activation and Its Proapoptotic Activity in Response to H2O2-induced Apoptosis. J. Biol. Chem. 2008, 283, 15359–15369. [Google Scholar] [CrossRef]
- Cao, X.; Deng, X.; May, W.S. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood 2003, 102, 2605–2614. [Google Scholar] [CrossRef]
- Jürgensmeier, J.M.; Xie, Z.; Deveraux, Q.; Ellerby, L.; Bredesen, D.; Reed, J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 4997–5002. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojič, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine Cathepsins Trigger Caspase-dependent Cell Death through Cleavage of Bid and Antiapoptotic Bcl-2 Homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef]
- Soond, S.; Savvateeva, L.; Makarov, V.; Gorokhovets, N.; Townsend, P.; Zamyatnin, A. Cathepsin S Cleaves BAX as a Novel and Therapeutically Important Regulatory Mechanism for Apoptosis. Pharmaceutics 2021, 13, 339. [Google Scholar] [CrossRef]
- Bidère, N.; Lorenzo, H.K.; Carmona, S.; Laforge, M.; Harper, F.; Dumont, C.; Senik, A. Cathepsin D Triggers Bax Activation, Resulting in Selective Apoptosis-inducing Factor (AIF) Relocation in T Lymphocytes Entering the Early Commitment Phase to Apoptosis. J. Biol. Chem. 2003, 278, 31401–31411. [Google Scholar] [CrossRef]
- Shahar, N.; Larisch, S. Inhibiting the inhibitors: Targeting anti-apoptotic proteins in cancer and therapy resistance. Drug Resist. Updat. 2020, 52, 100712. [Google Scholar] [CrossRef]
- Jullien, M.; Gomez-Bougie, P.; Chiron, D.; Touzeau, C. Restoring Apoptosis with BH3 Mimetics in Mature B-Cell Malignancies. Cells 2020, 9, 717. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Bruncko, M.; Oost, T.K.; Belli, B.A.; Ding, H.; Joseph, M.K.; Kunzer, A.; Martineau, D.; McClellan, W.J.; Mitten, M.; Ng, S.-C.; et al. Studies Leading to Potent, Dual Inhibitors of Bcl-2 and Bcl-xL. J. Med. Chem. 2007, 50, 641–662. [Google Scholar] [CrossRef]
- Gassian, N.; Frontczak, A.; El Kaddissi, A.; Calcagno, F.; Almotlak, H.; Barkatz, J.; Mouillet, G.; Maurina, T.; Stein, U.; Hon, T.N.T.; et al. Traitement systémique du cancer du pénis localement avancé ou métastatique. Bull Cancer 2020, 107, S17–S23. [Google Scholar] [CrossRef]
- Kaiser, U.; Schilli, M.; Haag, U.; Neumann, K.; Kreipe, H.; Kogan, E.; Havemann, K. Expression of bcl-2—Protein in small cell lung cancer. Lung Cancer 1996, 15, 31–40. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef]
- Veis, D.J.; Sorenson, C.M.; Shutter, J.R.; Korsmeyer, S.J. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993, 75, 229–240. [Google Scholar] [CrossRef]
- Motoyama, N.; Wang, F.; Roth, K.A.; Sawa, H.; Nakayama, K.; Negishi, I.; Senju, S.; Zhang, Q.; Fujii, S.; Et, A. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 1995, 267, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T. Obligate Role of Anti-Apoptotic MCL-1 in the Survival of Hematopoietic Stem Cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Letai, A.; Beard, C.; Sorcinelli, M.D.; Ong, C.C.; Korsmeyer, S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003, 426, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Lessene, G.; Czabotar, P.E.; Colman, P.M. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.D.; Shen, W.; Kunzer, A.; McClellan, W.J.; Bruncko, M.; Oost, T.K.; Ding, H.; Joseph, M.K.; Zhang, H.; Nimmer, P.M.; et al. Discovery and Structure−Activity Relationship of Antagonists of B-Cell Lymphoma 2 Family Proteins with Chemopotentiation Activity in Vitro and in Vivo. J. Med. Chem. 2006, 49, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.-L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients With Relapsed or Refractory Disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; De Oliveira, M.R.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II Study of Single-Agent Navitoclax (ABT-263) and Biomarker Correlates in Patients with Relapsed Small Cell Lung Cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef]
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 2826–2833. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed Anuclear Cell Death Delimits Platelet Life Span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Debrincat, M.A.; Pleines, I.; Lebois, M.; Lane, R.M.; Holmes, M.L.; Corbin, J.; Vandenberg, C.J.; Alexander, W.S.; Ng, A.P.; Strasser, A.; et al. BCL-2 is dispensable for thrombopoiesis and platelet survival. Cell Death Dis. 2015, 6, e1721. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.-M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.A.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Cerella, C.; Dicato, M.; Diederich, M. BH3 Mimetics in AML Therapy: Death and Beyond? Trends Pharmacol. Sci. 2020, 41, 793–814. [Google Scholar] [CrossRef]
- Tahir, S.K.; Smith, M.L.; Hessler, P.; Rapp, L.R.; Idler, K.B.; Park, C.H.; Leverson, J.D.; Lam, L.T. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017, 17, 1–10. [Google Scholar] [CrossRef]
- Lessene, G.; Czabotar, P.E.; Sleebs, B.E.; Zobel, K.; Lowes, K.N.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective BCL-XL inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef]
- Koehler, M.F.T.; Bergeron, P.; Choo, E.F.; Lau, K.; Ndubaku, C.; Dudley, D.; Gibbons, P.; Sleebs, B.E.; Rye, C.S.; Nikolakopoulos, G.; et al. Structure-Guided Rescaffolding of Selective Antagonists of BCL-XL. ACS Med. Chem. Lett. 2014, 5, 662–667. [Google Scholar] [CrossRef]
- Tao, Z.-F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef]
- Wei, G.; Margolin, A.A.; Haery, L.; Brown, E.; Cucolo, L.; Julian, B.; Shehata, S.; Kung, A.L.; Beroukhim, R.; Golub, T.R. Chemical Genomics Identifies Small-Molecule MCL1 Repressors and BCL-xL as a Predictor of MCL1 Dependency. Cancer Cell 2012, 21, 547–562. [Google Scholar] [CrossRef]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Lee, E.F.; Czabotar, P.E.; Van Delft, M.F.; Michalak, E.M.; Boyle, M.J.; Willis, S.N.; Puthalakath, H.; Bouillet, P.; Colman, P.M.; Huang, D.C.; et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J. Cell Biol. 2008, 180, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.F.; Czabotar, P.E.; Yang, H.; Sleebs, B.E.; Lessene, G.; Colman, P.M.; Smith, B.J.; Fairlie, W. Conformational Changes in Bcl-2 Pro-survival Proteins Determine Their Capacity to Bind Ligands. J. Biol. Chem. 2009, 284, 30508–30517. [Google Scholar] [CrossRef] [PubMed]
- Cirman, T.; Orešić, K.; Mazovec, G.D.; Turk, V.; Reed, J.C.; Myers, R.M.; Salvesen, G.S.; Turk, B. Selective Disruption of Lysosomes in HeLa Cells Triggers Apoptosis Mediated by Cleavage of Bid by Multiple Papain-like Lysosomal Cathepsins. J. Biol. Chem. 2004, 279, 3578–3587. [Google Scholar] [CrossRef]
- Appelqvist, H.; Johansson, A.C.; Linderoth, E.; Johansson, U.; Antonsson, B.; Steinfeld, R.; Kagedal, K.; Ollinger, K. Lysosome-mediated apoptosis is associated with cathepsin d-specific processing of bid at phe24, trp48, and phe183. Ann. Clin. Lab. Sci. 2012, 42, 231–242. [Google Scholar]
- Strasser, A. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 189–200. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Zong, W.-X.; Lindsten, T.; Ross, A.J.; MacGregor, G.R.; Thompson, C.B. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001, 15, 1481–1486. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.-J.; Yuan, J. Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 Interacting Protein, Mediates Cytochrome c Release from Mitochondria in Response to Activation of Cell Surface Death Receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Gross, A.; Yin, X.-M.; Wang, K.; Wei, M.C.; Jockel, J.; Milliman, C.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Caspase Cleaved BID Targets Mitochondria and Is Required for Cytochrome c Release, while BCL-XL Prevents This Release but Not Tumor Necrosis Factor-R1/Fas Death. J. Biol. Chem. 1999, 274, 1156–1163. [Google Scholar] [CrossRef]
- Slee, E.A.; Keogh, S.A.; Martin, S.J. Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: A potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ. 2000, 7, 556–565. [Google Scholar] [CrossRef]
- Eskes, R.; Desagher, S.; Antonsson, B.; Martinou, J.-C. Bid Induces the Oligomerization and Insertion of Bax into the Outer Mitochondrial Membrane. Mol. Cell. Biol. 2000, 20, 929–935. [Google Scholar] [CrossRef]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. Tbid, a membrane-targeted death ligand, oligomerizes bak to release cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar]
- Stoka, V.; Turk, B.; Schendel, S.L.; Kim, T.-H.; Cirman, T.; Snipas, S.J.; Ellerby, L.M.; Bredesen, D.; Freeze, H.; Abrahamson, M.; et al. Lysosomal Protease Pathways to Apoptosis. J. Biol. Chem. 2001, 276, 3149–3157. [Google Scholar] [CrossRef]
- Reiners, J.J., Jr.; Caruso, J.A.; Mathieu, P.; Chelladurai, B.; Yin, X.-M.; Kessel, D. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves bid cleavage. Cell Death Differ. 2002, 9, 934–944. [Google Scholar] [CrossRef]
- Blomgran, R.; Zheng, L.; Stendahl, O. Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J. Leukoc. Biol. 2007, 81, 1213–1223. [Google Scholar] [CrossRef]
- Egle, A.; Harris, A.W.; Bouillet, P.; Cory, S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 6164–6169. [Google Scholar] [CrossRef]
- Tan, T.-T.; Degenhardt, K.; Nelson, D.A.; Beaudoin, B.; Nieves-Neira, W.; Bouillet, P.; Villunger, A.; Adams, J.M.; White, E. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 2005, 7, 227–238. [Google Scholar] [CrossRef]
- Sunters, A.; de Mattos, S.F.; Stahl, M.; Brosens, J.J.; Zoumpoulidou, G.; Saunders, C.A.; Coffer, P.J.; Medema, R.H.; Coombes, R.C.; Lam, E.W.-F. FoxO3a Transcriptional Regulation of Bim Controls Apoptosis in Paclitaxel-treated Breast Cancer Cell Lines. J. Biol. Chem. 2003, 278, 49795–49805. [Google Scholar] [CrossRef]
- Li, R.; Moudgil, T.; Ross, H.J.; Hu, H.-M. Apoptosis of non-small-cell lung cancer cell lines after paclitaxel treatment involves the BH3-only proapoptotic protein Bim. Cell Death Differ. 2005, 12, 292–303. [Google Scholar] [CrossRef]
- Kutuk, O.; Letai, A. Displacement of Bim by Bmf and Puma rather than increase in Bim level mediates paclitaxel-induced apoptosis in breast cancer cells. Cell Death Differ. 2010, 17, 1624–1635. [Google Scholar] [CrossRef]
- Czernick, M.; Rieger, A.; Goping, I.S. Bim is reversibly phosphorylated but plays a limited role in paclitaxel cytotoxicity of breast cancer cell lines. Biochem. Biophys. Res. Commun. 2009, 379, 145–150. [Google Scholar] [CrossRef]
- Shukla, S.; Saxena, S.; Singh, B.K.; Kakkar, P. BH3-only protein BIM: An emerging target in chemotherapy. Eur. J. Cell Biol. 2017, 96, 728–738. [Google Scholar] [CrossRef]
- Opydo-Chanek, M.; Gonzalo, O.; Marzo, I. Multifaceted anticancer activity of BH3 mimetics: Current evidence and future prospects. Biochem. Pharmacol. 2017, 136, 12–23. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soond, S.M.; Kozhevnikova, M.V.; Savvateeva, L.V.; Townsend, P.A.; Zamyatnin, A.A., Jr. Intrinsically Connected: Therapeutically Targeting the Cathepsin Proteases and the Bcl-2 Family of Protein Substrates as Co-regulators of Apoptosis. Int. J. Mol. Sci. 2021, 22, 4669. https://doi.org/10.3390/ijms22094669
Soond SM, Kozhevnikova MV, Savvateeva LV, Townsend PA, Zamyatnin AA Jr. Intrinsically Connected: Therapeutically Targeting the Cathepsin Proteases and the Bcl-2 Family of Protein Substrates as Co-regulators of Apoptosis. International Journal of Molecular Sciences. 2021; 22(9):4669. https://doi.org/10.3390/ijms22094669
Chicago/Turabian StyleSoond, Surinder M., Maria V. Kozhevnikova, Lyudmila V. Savvateeva, Paul A. Townsend, and Andrey A. Zamyatnin, Jr. 2021. "Intrinsically Connected: Therapeutically Targeting the Cathepsin Proteases and the Bcl-2 Family of Protein Substrates as Co-regulators of Apoptosis" International Journal of Molecular Sciences 22, no. 9: 4669. https://doi.org/10.3390/ijms22094669
APA StyleSoond, S. M., Kozhevnikova, M. V., Savvateeva, L. V., Townsend, P. A., & Zamyatnin, A. A., Jr. (2021). Intrinsically Connected: Therapeutically Targeting the Cathepsin Proteases and the Bcl-2 Family of Protein Substrates as Co-regulators of Apoptosis. International Journal of Molecular Sciences, 22(9), 4669. https://doi.org/10.3390/ijms22094669