
Myocardial Hypertrophy and Fibrosis Are Associated with Cardiomyocyte Beta-Catenin and TRPC6/Calcineurin/NFAT Signaling in Spontaneously Hypertensive Rats with 5/6 Nephrectomy

 , and
, and
Abstract
:1. Introduction
2. Results
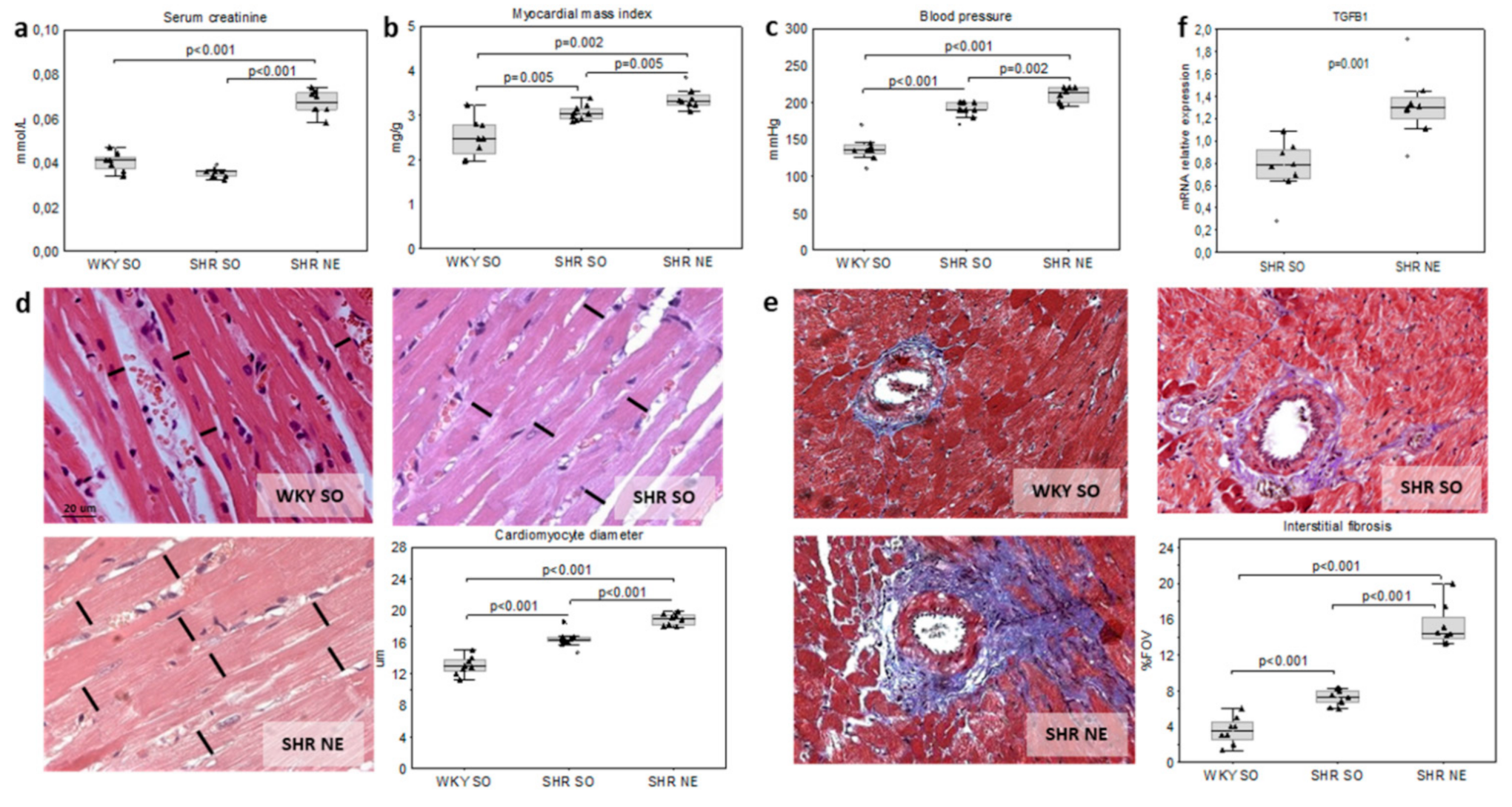
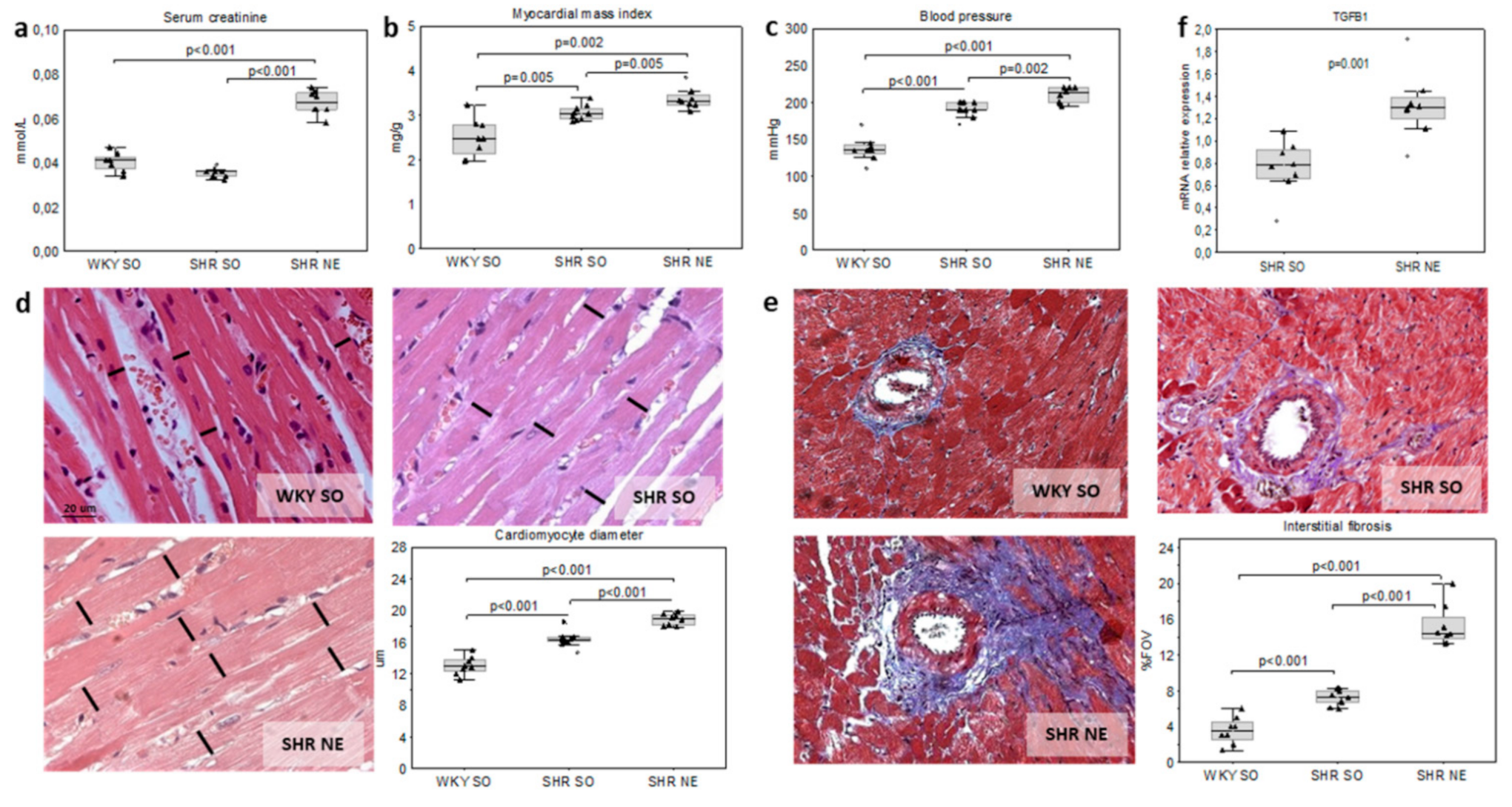
2.1. Animal Models of Hypertension and CKD
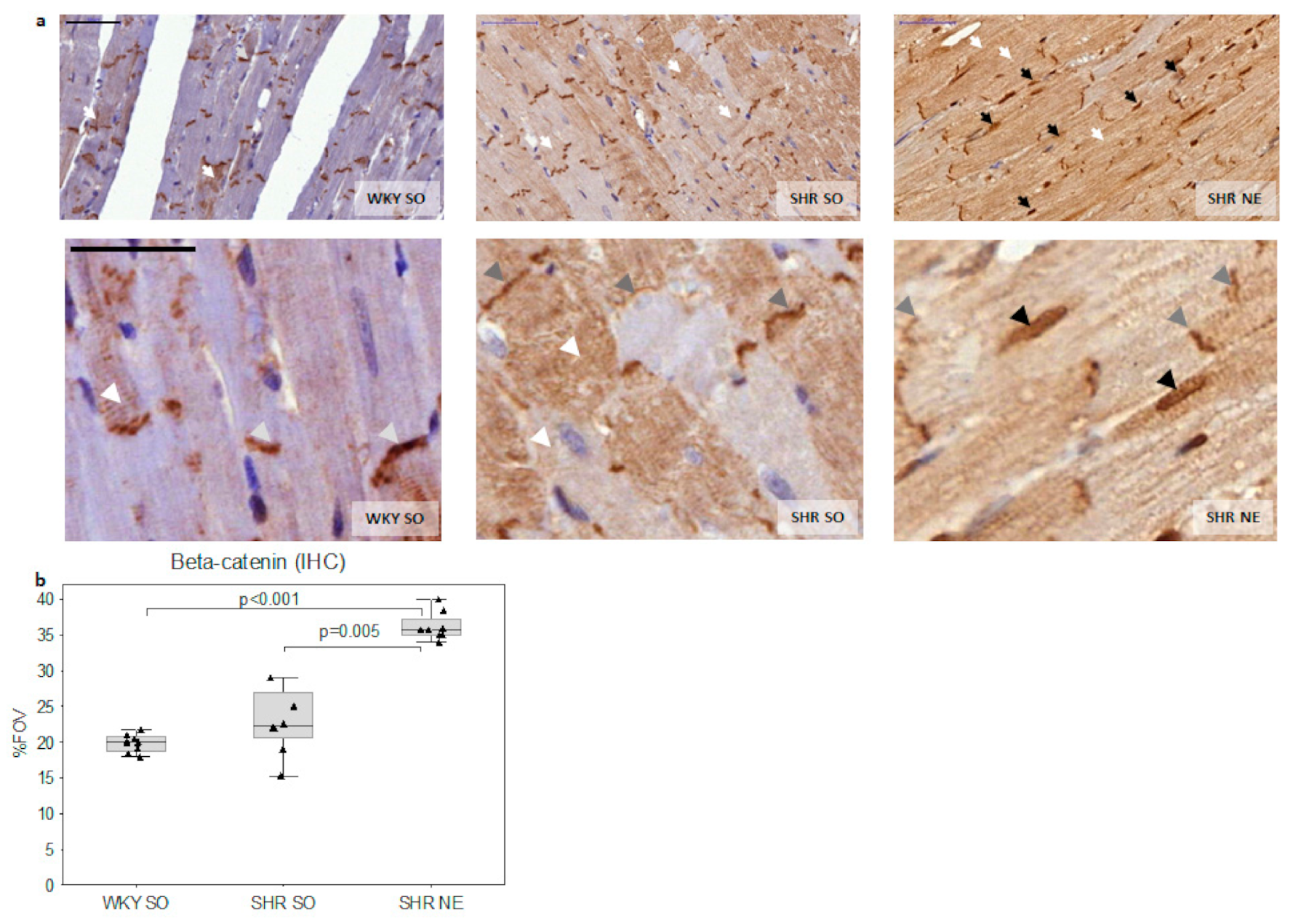
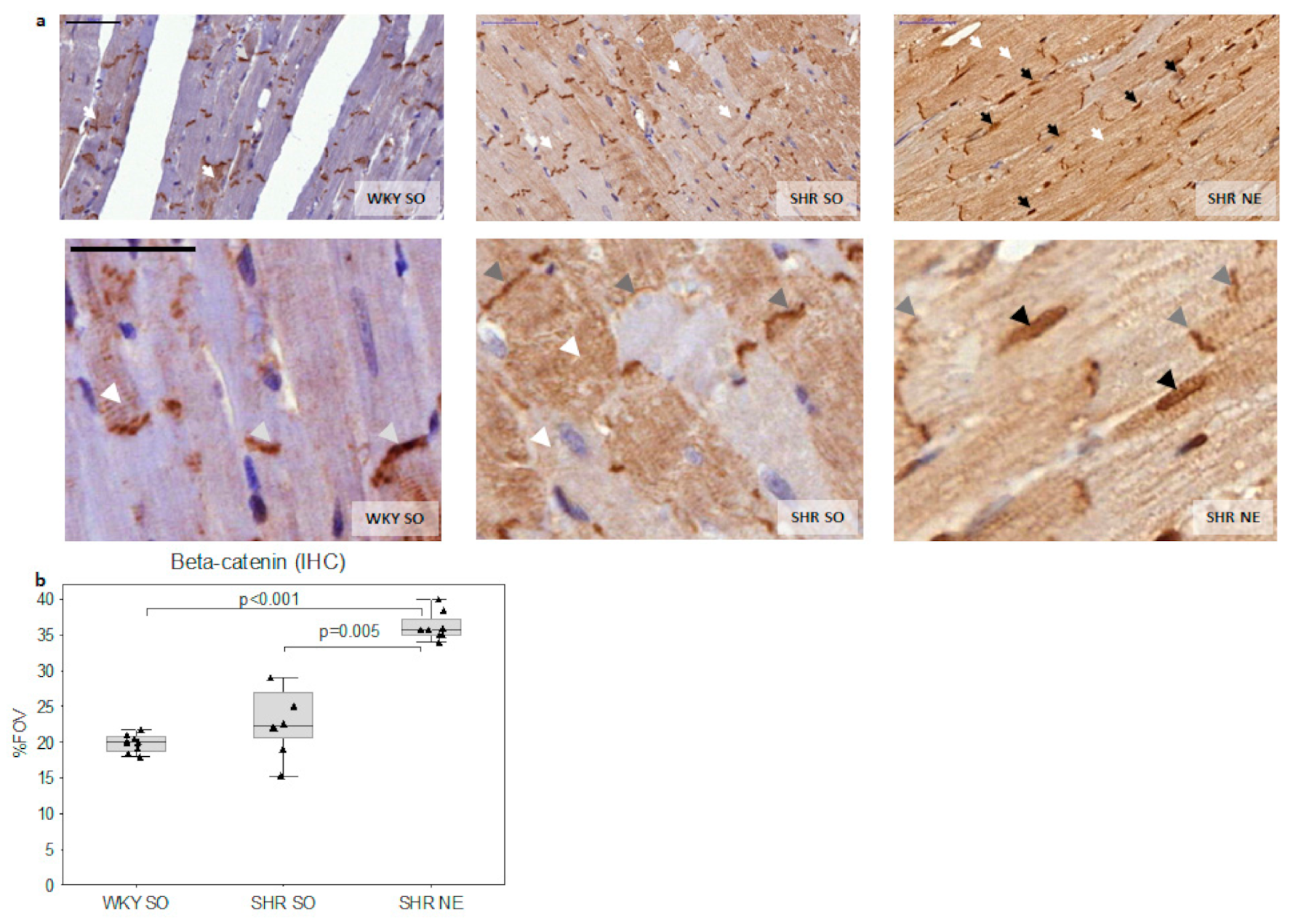
2.2. Beta-Catenin Myocardial Expression
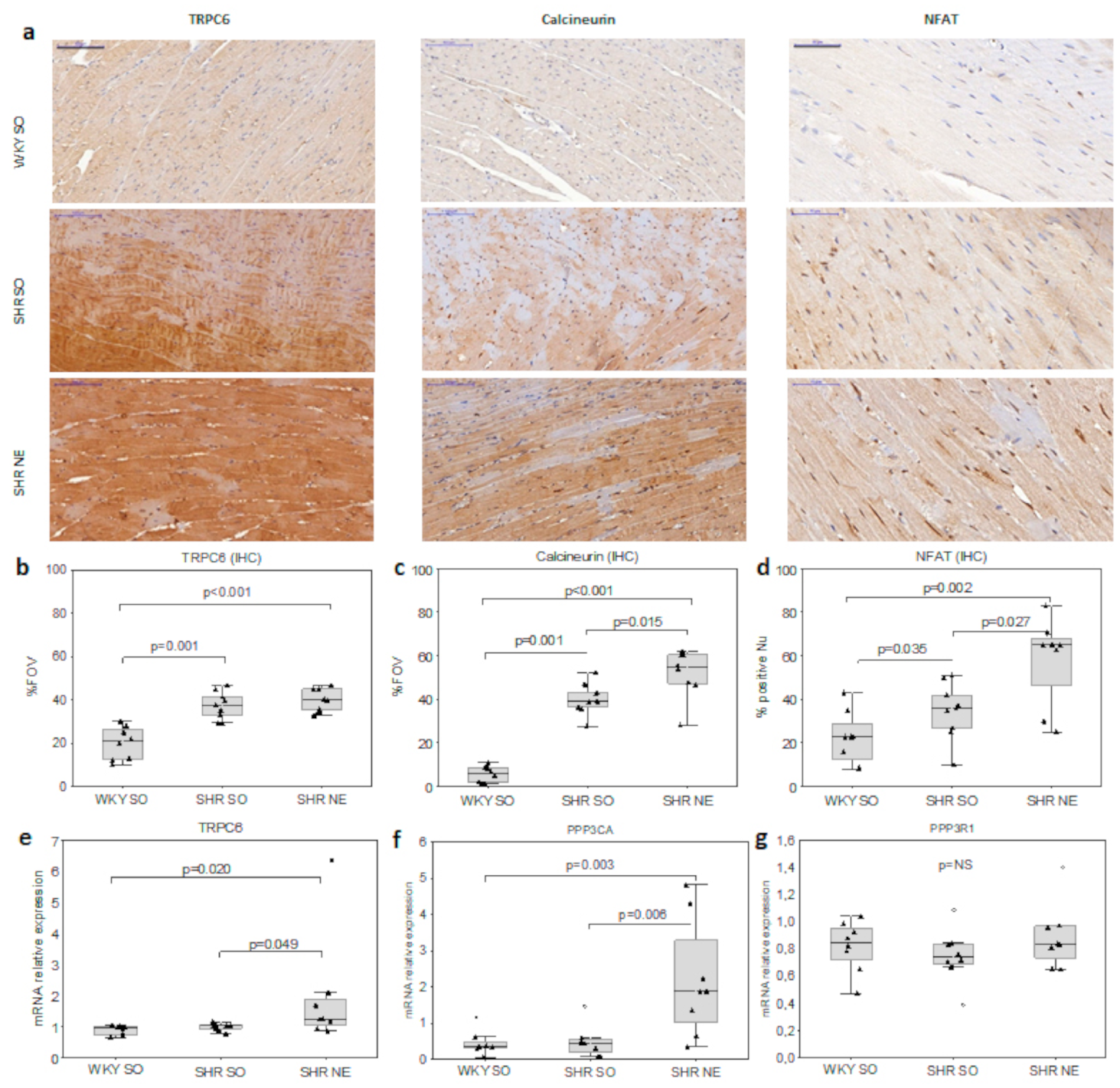
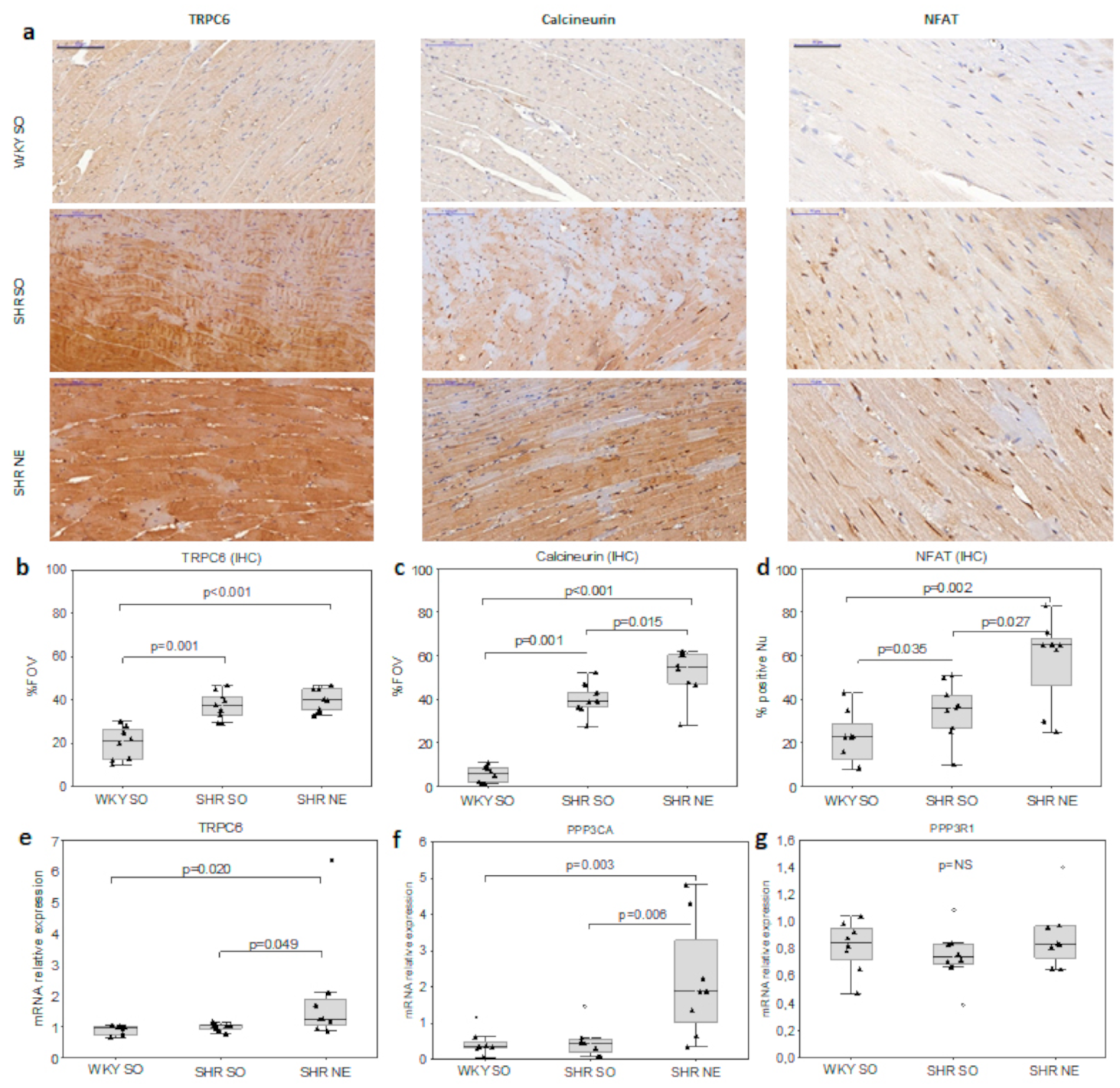
2.3. CaN/NFAT and TRPC6 Myocardial Expression
2.4. Interrelations in the Studied Parameters
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. Laboratory Measurements
5.3. Histology and IHC
5.4. Real-Time PCR
5.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Suzuki, T.; Sakamaki, Y.; Ito, S. Cardiac hypertrophy in chronic kidney disease—role of Aldosterone and FGF23. Ren. Replace. Ther. 2018, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Chen, C.; Cheng, J. The role and molecular mechanism of FoxO1 in mediating cardiac hypertrophy. ESC Heart Fail. 2020, 22, 3497–3504. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Freichel, M.; Schweig, U.; Stauffenberger, S.; Freise, D.; Schorb, W.; Flockerzi, V. Store-operated cation channels in the heart and cells of the cardiovascular system. Cell. Physiol. Biochem. 1999, 9, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Wilkin, B.J.; Bodi, I.; Molkentin, J.D. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006, 20, 1660–1670. [Google Scholar] [CrossRef] [Green Version]
- Zou, G.; Hong, H.; Lin, X.; Shi, X.; Wu, Y.; Chen, L. TRPC1, CaN and NFATC3 signaling pathway in the pathogenesis and progression of left ventricular hypertrophy in spontaneously hypertensive rats. Clin. Exp. Hypertens. 2015, 37, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Cha, S.-K.; An, S.-W.; Kuro-o, M.; Birnbaumer, L.; Huang, C.-L. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 2012, 3, 1238. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Iribe, G.; Nishida, M.; Naruse, K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: Linking physiology and pathophysiology. Prog. Biophys. Mol. Biol. 2017, 130, 264–272. [Google Scholar] [CrossRef]
- Ding, Y.; Winters, A.; Ding, M.; Graham, S.; Akopova, I.; Muallem, S.; Wang, Y.; Hong, J.H.; Gryczynski, Z.; Yang, S.-H; et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J. Biol. Chem. 2011, 286, 31799–31809. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molkentin, J.D. Calcineurin–NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Cai, W.; Sheng, J.; Yin, L. Role of SDF-1 and Wnt signaling pathway in the myocardial fibrosis of hypertensive rats. Am. J. Transl. Res. 2015, 7, 1345–1356. [Google Scholar] [PubMed]
- Zheng, Q.; Chen, P.; Xu, Z.; Yi, X.P. Expression and redistribution of β-catenin in the cardiac myocytes of left ventricle of spontaneously hypertensive rat. J. Mol. Histol. 2013, 44, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, H.-W.; Pi, L.-J.; Wang, J.; Zhan, D.-Q. Protective effect of tanshinone IIA against cardiac hypertrophy in spontaneously hypertensive rats through inhibiting the Cys-C/Wnt signaling pathway. Oncotarget 2017, 8, 10161–10170. [Google Scholar] [CrossRef] [Green Version]
- Dawson, K.; Aflaki, M.; Nattel, S. Role of the Wnt-Frizzled system in cardiac pathophysiology: A rapidly developing, poorly understood area with enormous potential. J. Physiol. 2013, 591, 1409–1432. [Google Scholar] [CrossRef]
- Qian, L.J.; Hong, J.; Zhang, J.M.; Zhu, M.L.; Wang, X.C.; Zhang, Y.J.; Chu, M.; Yao, J.; Xu, D. Downregulation of S100A4 Alleviates Cardiac Fibrosis via Wnt/β-Catenin Pathway in Mice. Cell. Physiol. Biochem. 2018, 46, 2551–2560. [Google Scholar] [CrossRef]
- Xiang, F.-L.; Fang, M.; Yutzey, K.E. Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 28, 712. [Google Scholar] [CrossRef]
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Bogdanova, E.O.; Beresneva, O.N.; Zubina, I.M.; Ivanova, G.T.; Parastaeva, M.M.; Galkina, O.V.; Dobronravov, V.A. Canonical WNT signaling and myocardial remodeling in arterial hypertension and chronic kidney dysfunction. Nephrol. (Saint-Petersburg) J. 2020, 24, 85–92. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun. 2012, 13, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.; Hsu, C.Y.; Li, Y.; Mishra, R.K.; Keane, M.; Rosas, S.E.; Dries, D.; Xie, D.; Chen, J.; He, J.; et al. Associations between kidney function and subclinical cardiac abnormalities in CKD. J. Am. Soc. Nephrol. 2012, 23, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farshid, A.; Pathak, R.; Shadbolt, B.; Arnolda, L.; Talaulikar, G. Diastolic function is a strong predictor of mortality in patients with chronic kidney disease. BMC Nephrol. 2013, 14, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, K.K.; Levy, D.; Kannel, W.B.; Pinsky, J.L. The epidemiology of heart failure: The Framingham study. J. Am. Coll. Cardiol. 1993, 22, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Zannad, F.; Rossignol, P. Cardiorenal syndrome revisited. Circulation 2018, 138, 929–944. [Google Scholar] [CrossRef]
- Mall, G.; Huther, W.; Schneider, J.; Lundin, P.; Ritz, E. Diffuse intermyocardiocytic fibrosis in uraemic patients. Nephrol. Dial. Transplant. 1990, 5, 39–44. [Google Scholar] [CrossRef]
- Dobronravov, V.A.; Bogdanova, E.O.; Semenova, N.Y.; Beresneva, O.N.; Parastaeva, M.M.; Galkina, O.V.; Zubina, I.M.; Zueva, E.E.; Ivanova, G.T.; Kaukov, I.G.; et al. Renal expression of αKlotho protein, fibroblast growth factor 23 and parathyroid hormone in experimental modeling of early stages of chronic kidney damage. Nephrol. (Saint-Petersburg) J. 2014, 18, 72–78. [Google Scholar]
- Yang, K.; Wang, C.; Nie, L.; Zhao, X.; Gu, J.; Guan, X.; Wang, S.; Xiao, T.; Xu, X.; He, T.; et al. Klotho protects against indoxyl sulphate-induced myocardial hypertrophy. J. Am. Soc. Nephrol. 2015, 26, 2434–2446. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Kong, J.; Chen, S.; Cao, L.P.; Qiao, G.; Zheng, W.; Liu, W.; Li, X.; Gardner, D.G.; Li, Y.C. Cardiac hypertrophy in vitamin D receptor knockout mice: Role of the systemic and cardiac renin-angiotensin systems. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 125–132. [Google Scholar] [CrossRef]
- Dobronravov, V.A. Phosphate, kidneys, bones and cardiovascular system. Nephrol. (Saint-Petersburg) J. 2016, 20, 10–24. [Google Scholar]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [Green Version]
- Leifheit-Nestler, M.; große Siemer, R.; Flasbart, K.; Richter, B.; Kirchhoff, F.; Ziegler, W.H.; Klintschar, M.; Becker, J.U.; Erbersdobler, A.; Aufricht, C.; et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2016, 31, 1088. [Google Scholar] [CrossRef] [Green Version]
- Law, J.P.; Price, A.M.; Pickup, L.; Radhakrishnan, A.; Weston, C.; Jones, A.M.; McGettrick, H.M.; Chua, W.; Steeds, R.P.; Fabritz, L.; et al. Clinical Potential of Targeting Fibroblast Growth Factor-23 and αKlotho in the Treatment of Uremic Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e016041. [Google Scholar] [CrossRef] [PubMed]
- Suassuna, P.G.A.; Cherem, P.M.; de Castro, B.B.; Maquigussa, E.; Cenedeze, M.A.; Lovisi, J.C.M.; Custódio, M.R.; Sanders-Pinheiro, H.; de Paula, R.B. αKlotho attenuates cardiac hypertrophy and increases myocardial fibroblast growth factor 21 expression in uremic rats. Exp. Biol. Med. (Maywood) 2020, 245, 66–78. [Google Scholar] [CrossRef]
- Czaya, B.; Seeherunvong, W.; Singh, S.; Yanucil, C.; Ruiz, P.; Quiroz, Y.; Grabner, A.; Katsoufis, C.; Swaminathan, S.; Abitbol, C.; et al. Cardioprotective Effects of Paricalcitol Alone and in Combination With FGF23 Receptor Inhibition in Chronic Renal Failure: Experimental and Clinical Studies. Am. J. Hypertens. 2019, 32, 34–44. [Google Scholar] [CrossRef]
- Cerasola, G.; Nardi, E.; Palermo, A.; Mulè, G.; Cottone, S. Epidemiology and pathophysiology of left ventricular abnormalities in chronic kidney disease: A review. J. Nephrol. 2011, 24, 1–10. [Google Scholar] [CrossRef]
- Bogdanova, E.O.; Semenova, N.Y.; Beresneva, O.N.; Parastaeva, M.M.; Ivanova, G.T.; Galkina, O.V.; Zubina, I.M.; Kayukov, I.G.; Kovalenko, T.L.; Kotenko, L.V.; et al. Renal αKlotho expression is associated with myocardial hypertrophy (experimental study). Arter. Hypertens. (Russian Federation) 2014, 20, 522–530. [Google Scholar] [CrossRef]
- Bogdanova, E.O.; Galkina, O.V.; Zubina, I.M.; Dobronravov, V.A. Klotho, fibroblast growth factor 23 and inorganic phosphate in early stages of chronic kidney disease. Nephrol. (Saint-Petersburg) J. 2016, 20, 61–67. [Google Scholar]
- Hu, M.C.; Kuro-o, M.; Orson, W. Secreted klotho and chronic kidney disease. Adv. Exp. Med. Biol. 2012, 728, 126–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.J.; Yang, H.C.; Fogo, A.B. Animal models of regression/progression of kidney disease. Drug Discov. Today Dis. Models 2014, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Hou, F.F.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling mediates both heart and kidney injury in type 2 cardiorenal syndrome. Kidney Int. 2019, 95, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Vite, A.; Radice, G. N-cadherin/catenin complex as a master regulator of intercalated disc function. Cell Commun. Adhes. 2014, 21, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, X.; Zhang, H.; Wang, Z.; Nan, G.; Li, Y.; Zhang, F.; Mohammed, M.K.; Haydon, R.C.; Luu, H.H.; et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: Implications in targeted cancer therapies. Lab. Investig. 2016, 96, 116–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, C.C.; Robbins, D.J.; Ahmed, Y.; Lee, E. Nuclear Regulation of Wnt/β-Catenin Signaling: It’s a Complex Situation. Genes 2020, 11, 886. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Wang, C.; Hong, X. An essential role for Wnt/β-catenin signaling in mediating hypertensive heart disease. Sci. Rep. 8996, 8, 899. [Google Scholar] [CrossRef]
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.M.; Benitah, J.P.; Perros, F.; Humbert, M.; Antigny, F. Ca2+ handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Ohba, T.; Watanabe, H.; Murakami, M.; Takahashi, Y.; Iino, K.; Kuromitsu, S.; Mori, Y.; Ono, K.; Iijima, T.; Ito, H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Xiao, J.H.; Wang, Y.; Xu, B.M.; Gao, L.; Wang, J.L. Upregulation of TRPC1 contributes to contractile function in isoproterenol-induced hypertrophic myocardium of rat. Cell. Physiol. Biochem. 2013, 32, 951–959. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsnefes, M.M.; Betoko, A.; Schneider, M.F.; Salusky, I.B.; Wolf, M.S.; Jüppner, H.; Warady, B.A.; Furth, S.L.; Portale, A.A. FGF23 and Left Ventricular Hypertrophy in Children with CKD. Clin. J. Am. Soc. Nephrol. 2018, 6, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; deFilippi, C.; Isakova, T.; Gutiérrez, O.M.; Laliberte, K.; Seliger, S.; Kelley, W.; Duh, S.H.; Hise, M.; Christenson, R.; et al. Fibroblast growth factor 23, high-sensitivity cardiac troponin, and left ventricular hypertrophy in CKD. Am. J. Kidney Dis. 2013, 61, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Andrukhova, O.; Slavic, S.; Odorfer, K.I.; Erben, R.G. Experimental myocardial infarction upregulates circulating fibroblast growth factor-23. J. Bone Miner. Res. 2015, 30, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Cai, C.; Xiao, Z.; Quarles, L.D. FGF23 induced left ventricular hypertrophy mediated by FGFR4 signaling in the myocardium is attenuated by soluble Klotho in mice. J. Mol. Cell. Cardiol. 2020, 138, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Touchberry, C.D.; Green, T.M.; Tchikrizov, V.; Mannix, J.E.; Mao, T.F.; Carney, B.W.; Girgis, M.; Vincent, R.J.; Wetmore, L.A.; Dawn, B.; et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E863–E873. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Li, X.; Li, Q.; Lin, H.; Chen, Z.; Xie, J.; Xuan, W.; Liao, W.; Bin, J.; Huang, X.; et al. FGF23 promotes myocardial fibrosis in mice through activation of beta-catenin. Oncotarget 2016, 7, 64649–64664. [Google Scholar] [CrossRef] [Green Version]
- Dalton, G.D.; Xie, J.; An, S.W.; Huang, C.L. New Insights into the Mechanism of Action of Soluble Klotho. Front. Endocrinol. 2017, 8, 323. [Google Scholar] [CrossRef] [Green Version]
- Dalton, G.; An, S.W.; Al-Juboori, S.I.; Nischan, N.; Yoon, J.; Dobrinskikh, E.; Hilgemann, D.W.; Xie, J.; Luby-Phelps, K.; Kohler, J.J.; et al. Soluble klotho binds monosialoganglioside to regulate membrane microdomains and growth factor signaling. Proc. Natl. Acad. Sci. USA 2017, 114, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [Green Version]
- Hernández, A R.; Klein, A.M.; Kirschner, M.W. Kinetic responses of β-catenin specify the sites of Wnt control. Science 2012, 338, 1337–1340. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Parameters | Group 1 WKY-SO | Group 2 SHR-SO | Group 3 SHR-NE |
|---|---|---|---|
| Strain | Wistar Kyoto rats | Spontaneously hypertensive rats | |
| Model | control | hypertension | hypertension and CKD |
| Surgery | sham-operated | sham-operated | 5/6 nephrectomy |
| Observation period, mo | 2 | 2 | 2 |
| Rats number, n | 8 | 8 | 8 |
| HR, bpm | 401 (393–417) | 400 (374–416) | 412 (397–436) |
| PTH, ng/mL | 96.9 (53.3–137.3) | 37.2 (21.5–105.3) | 95.3 (80.3–123.0) |
| FGF23, ng/mL | 293.4 (243.0–361.2) | 627.6 (383.7–688.9) a | 767.5 (701.1–889.9) a b |
| rKlotho, % FOV | 34.2 (29.5–36.0) | 25.3 (16.2–28.0) a | 13.1 (12.5–14.8) c |
| sKlotho, ng/mL | 2698.3 (2413.3–2830.9) | 800.2 (606.6–890.4) a | 1310.4 (1115.7–1541.9) d |
| mKl/Gapdh, Hprt1 | 0.39 (0.31–0.41) | 0.35 (0.27–0.40) | 0.36 (0.25–0.43) |
| Parameters | TRPC6, %FOV | CaN, %FOV | NFAT, %FOV | b-catenin, %FOV |
|---|---|---|---|---|
| sKlotho, pg/mL | −0.47 * | −0.37 | −0.21 | −0.28 |
| rKlotho, %FOV | −0.69 * | −0.82 * | −0.67 * | −0.61 * |
| FGF23, ng/mL | 0.58 * | 0.68 * | 0.46 * | 0.57 * |
| PTH, ng/mL | −0.27 | −0.14 | −0.05 | 0.19 |
| sCr, mmol/L | 0.11 | 0.29 | 0.22 | 0.48 |
| MMI, mg/g | 0.61 * | 0.75 * | 0.68 * | 0.45 |
| dCM, um | 0.74 * | 0.84 * | 0.74 * | 0.74 * |
| IF, %FOV | 0.71 * | 0.88 * | 0.80 * | 0.69 * |
| Primer Name | Sequence (from 5′ to 3′) | Accession Number | Gene Product |
|---|---|---|---|
| Ppp3ca | NM_017041.2 | Protein phosphatase 3 catalytic subunit alpha | |
| Forward | CAGTAACTTTCGAGCCAGCC | ||
| Reverse | GACTTGGCGGAAATGGAACG | ||
| Ppp3r1 | NM_017309.2 | Protein phosphatase 3 regulatory subunit B, alpha | |
| Forward | AGCTTGACTTGGACAACTCT | ||
| Reverse | ATATCTAGGCCACCTACAAC | ||
| Trpc6 | NM_053559.1 | Transient receptor potential cation channel, subfamily C, member 6 | |
| Forward | GTGAACGAAGGGGAGCTGAA | ||
| Reverse | GCGGCTTTCCTCTTGTTTCG | ||
| Tgfb1 | NM_021578.2 | Transforming growth factor, beta 1 | |
| Forward | TGGCGTTACCTTGGTAACC | ||
| Reverse | GGTGTTGAGCCCTTTCCAG | ||
| Kl | NM_031336.2 | Klotho (Kl) | |
| Forward | ACTTCGGTGGTCAGGTCAAG | ||
| Reverse | CTCTTTGGGTAGTCGCCATC | ||
| Gapdh | NM_017008.4 | Glyceraldehyde-3-phosphate dehydrogenase | |
| Forward | AGATGGTGAAGGTCGGTGTG | ||
| Reverse | GATCTCGCTCCTGGAAGATG | ||
| Hprt1 | NM_012583.2 | Hypoxanthine phosphoribosyltransferase 1 | |
| Forward | GTTGGATACAGGCCAGACTT | ||
| Reverse | GCCACATCAACAGGACTCTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogdanova, E.; Beresneva, O.; Galkina, O.; Zubina, I.; Ivanova, G.; Parastaeva, M.; Semenova, N.; Dobronravov, V. Myocardial Hypertrophy and Fibrosis Are Associated with Cardiomyocyte Beta-Catenin and TRPC6/Calcineurin/NFAT Signaling in Spontaneously Hypertensive Rats with 5/6 Nephrectomy. Int. J. Mol. Sci. 2021, 22, 4645. https://doi.org/10.3390/ijms22094645
Bogdanova E, Beresneva O, Galkina O, Zubina I, Ivanova G, Parastaeva M, Semenova N, Dobronravov V. Myocardial Hypertrophy and Fibrosis Are Associated with Cardiomyocyte Beta-Catenin and TRPC6/Calcineurin/NFAT Signaling in Spontaneously Hypertensive Rats with 5/6 Nephrectomy. International Journal of Molecular Sciences. 2021; 22(9):4645. https://doi.org/10.3390/ijms22094645
Chicago/Turabian StyleBogdanova, Evdokia, Olga Beresneva, Olga Galkina, Irina Zubina, Galina Ivanova, Marina Parastaeva, Natalia Semenova, and Vladimir Dobronravov. 2021. "Myocardial Hypertrophy and Fibrosis Are Associated with Cardiomyocyte Beta-Catenin and TRPC6/Calcineurin/NFAT Signaling in Spontaneously Hypertensive Rats with 5/6 Nephrectomy" International Journal of Molecular Sciences 22, no. 9: 4645. https://doi.org/10.3390/ijms22094645
APA StyleBogdanova, E., Beresneva, O., Galkina, O., Zubina, I., Ivanova, G., Parastaeva, M., Semenova, N., & Dobronravov, V. (2021). Myocardial Hypertrophy and Fibrosis Are Associated with Cardiomyocyte Beta-Catenin and TRPC6/Calcineurin/NFAT Signaling in Spontaneously Hypertensive Rats with 5/6 Nephrectomy. International Journal of Molecular Sciences, 22(9), 4645. https://doi.org/10.3390/ijms22094645