
The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies

 ,
,  and
and 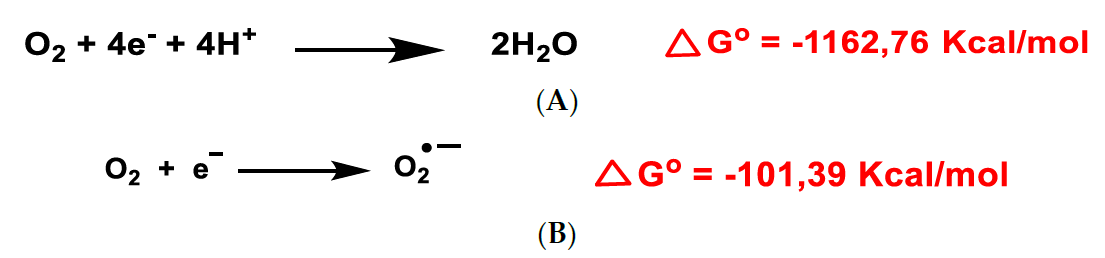{kind=link}
{kind=link}
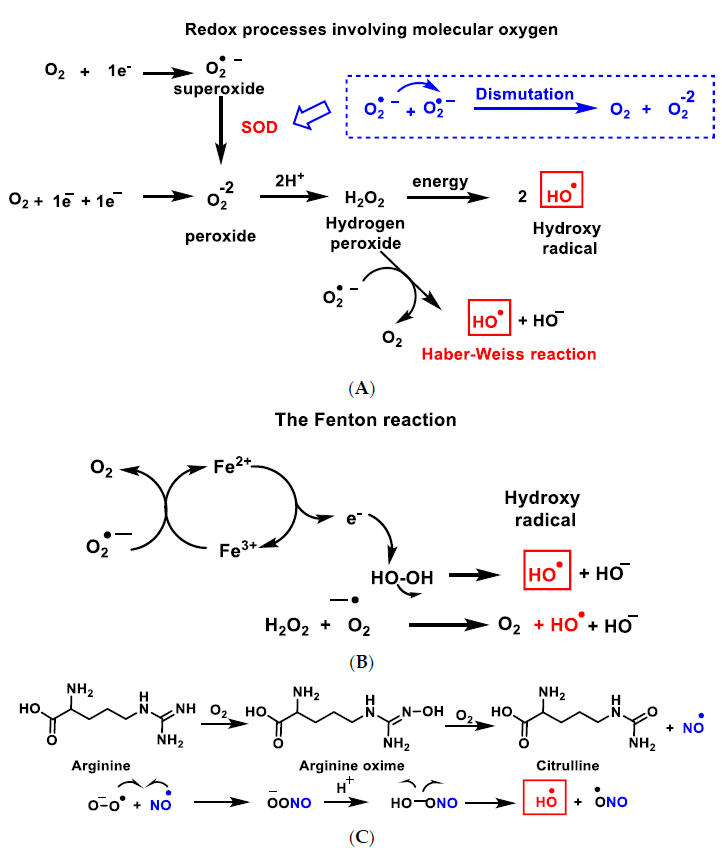{kind=link}
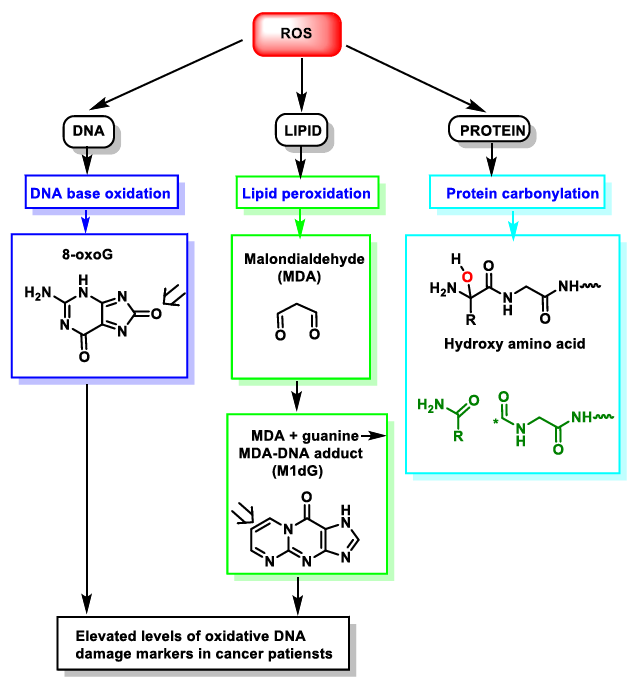{kind=link}
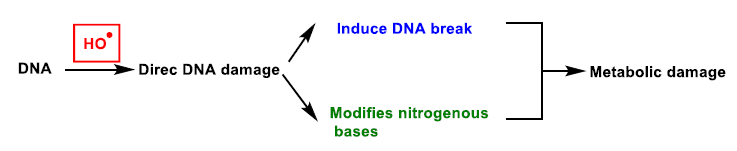{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
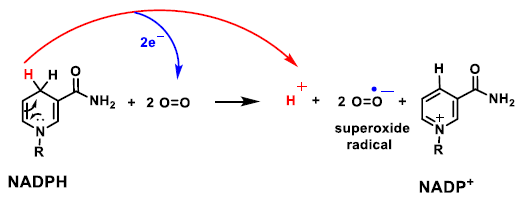
2. Genesis and Effects of ROS
3. Dual Role of ROS: Physiological and Pathological
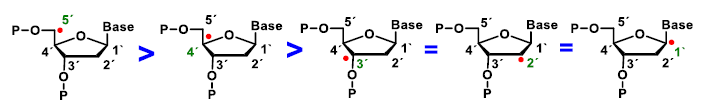
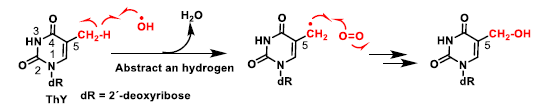
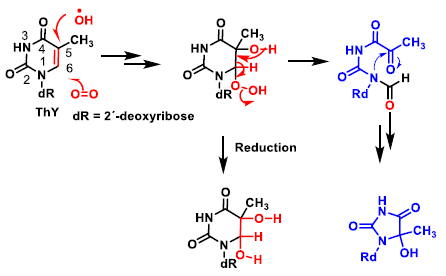
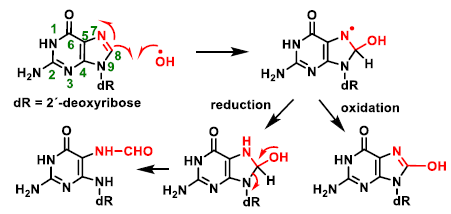
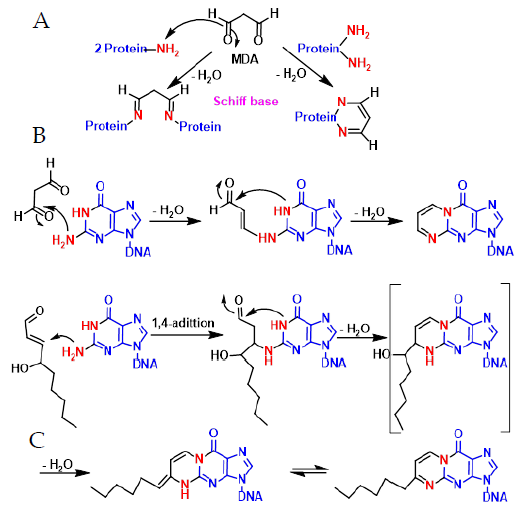
4. ROS Damage on DNA
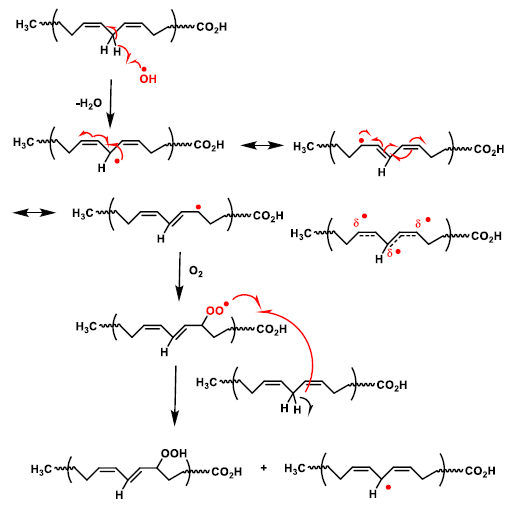
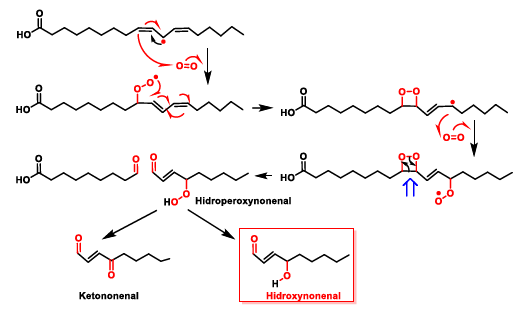
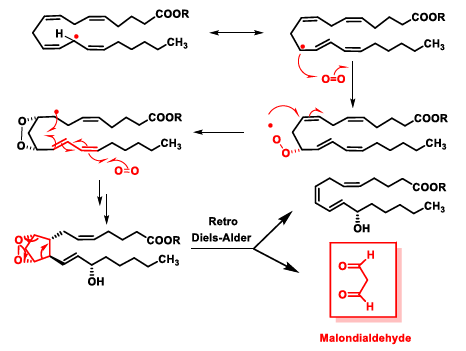
5. Lipid Peroxidation Caused by ROS
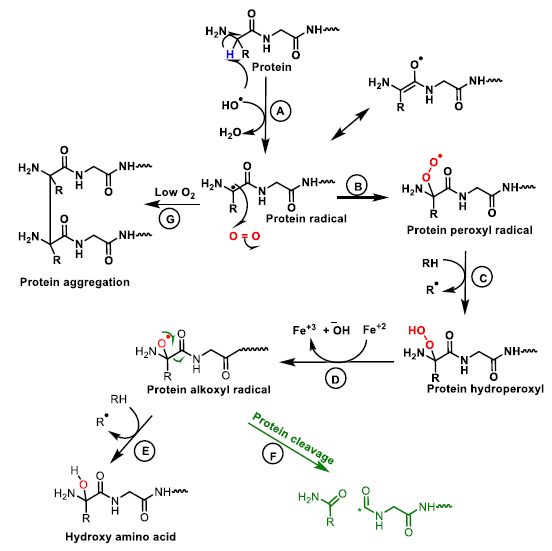
6. Protein and Enzyme Damage Caused by ROS
7. Neutrophil Oxidative DNA Damage: Consequences for Human Health
8. ROS-Mediated Activity of Antimicrobial Peptides
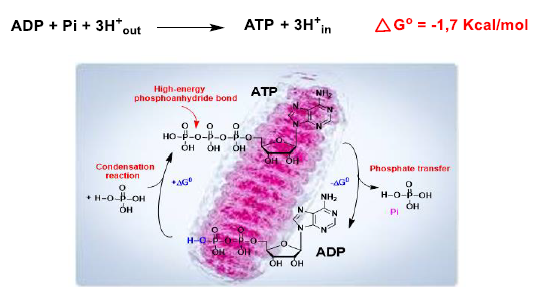
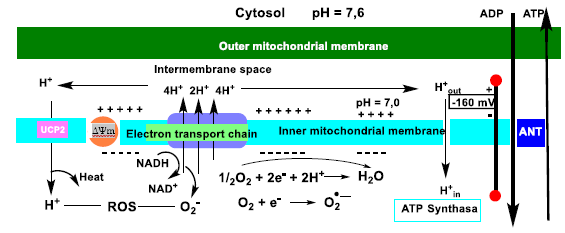
9. Oxidative Phosphorylation and Mitochondrial Uncoupling
10. ROS-Mediated Damage in Endoplasmic Reticulum (ER) and in Haemolytic Diseases
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mailloux, R.J. An update on mitochondrial reactive oxygen species production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Springer: Singapore, 2018; pp. 167–227. [Google Scholar]
- Valenta, H.; Erard, M.; Dupré-Crochet, S.; Nüβe, O. The NADPH Oxidase and the Phagosome. In Molecular and Cellular Biology of Phagocytosis; Hallett, M.B., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 153–177. [Google Scholar]
- Kehrer, J.P. The haber–weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Chen, H.-Y. Why the reactive oxygen species of the fenton reaction switches from oxoiron(IV) species to hydroxyl radical in phosphate buffer solutions? A computational rationale. ACS Omega 2019, 4, 14105–14113. [Google Scholar] [CrossRef]
- Fujiwara, O.; Fukuda, S.; Lopez, E.; Zeng, Y.; Niimi, Y.; DeWitt, D.S.; Herndon, D.N.; Prough, D.S.; Enkhbaatar, P. Peroxynitrite decomposition catalyst reduces vasopressin requirement in ovine MRSA sepsis. Intensive Care Med. Exp. 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xue, A.; Wang, Z.; Xia, R.; Wang, L.; Tang, Y.; Wan, P.; Chen, Y. Electrochemical degradation of lignin by ROS. Sustain. Chem. 2020, 1, 345–360. [Google Scholar] [CrossRef]
- Fisher, A.B.; Zhang, Q. Nadph and Nadph Oxidase. In Encyclopedia of Respiratory Medicine; Laurent, G.J., Shapiro, S.D., Eds.; Academic Press: Oxford, UK, 2006; pp. 77–84. [Google Scholar]
- Wen, J.J.; Garg, N.J. Manganese superoxide dismutase deficiency exacerbates the mitochondrial ROS production and oxidative damage in Chagas disease. PLoS Negl. Trop. Dis. 2018, 12, e0006687. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting oxidatively induced DNA damage response in cancer: Opportunities for novel cancer therapies. Oxidative Med. Cell. Longev. 2018, 2018, 2389523. [Google Scholar] [CrossRef]
- Yan, L.L.; Zaher, H.S. How do cells cope with RNA damage and its consequences? J. Biol. Chem. 2019, 294, 15158–15171. [Google Scholar] [CrossRef]
- Ito, F.; Sono, Y.; Ito, T. Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: Oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef]
- Theurey, P.; Pizzo, P. The aging mitochondria. Genes 2018, 9, 22. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in health and disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef]
- Auboeuf, D. Physicochemical foundations of life that direct evolution: Chance and natural selection are not evolutionary driving forces. Life 2020, 10, 7. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Bui, A.D.; Sharma, R.; Henkel, R.; Agarwal, A. Reactive oxygen species impact on sperm DNA and its role in male infertility. Andrologia 2018, 50, e13012. [Google Scholar] [CrossRef]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, B.; Pogozelski, W.K.; Tullius, T.D. DNA strand breaking by the hydroxyl radical is governed by the accessible surface areas of the hydrogen atoms of the DNA backbone. Proc. Natl. Acad. Sci. USA 1998, 95, 9738–9743. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.A.O.; Demple, B. DNA Base Excision Repair Pathways. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 1–8. [Google Scholar]
- Avendaño, C.; Menéndez, J.C. Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Elsevier: Boston, MA, USA, 2015. [Google Scholar]
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef] [PubMed]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 13902. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Shi, L.; Liang, F.; Xu, W.; Li, T.; Gao, L.; Sun, Z.; Yu, J.; Zhang, J. Sirt3 ameliorates oxidative stress and mitochondrial dysfunction after intracerebral hemorrhage in diabetic rats. Front. Neurosci. 2018, 12, 414. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular mechanisms and genetics of oxidative stress in Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The continuum of aging and age-related diseases: Common mechanisms but different rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef]
- Hahn, A.; Zuryn, S. Mitochondrial genome (mtDNA) mutations that generate reactive oxygen species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef]
- Yadav, D.K.; Kumar, S.; Choi, E.-H.; Chaudhary, S.; Kim, M.-H. Molecular dynamic simulations of oxidized skin lipid bilayer and permeability of reactive oxygen species. Sci. Rep. 2019, 9, 4496. [Google Scholar] [CrossRef]
- Höhn, A.; Tramutola, A.; Cascella, R. Proteostasis failure in neurodegenerative diseases: Focus on oxidative stress. Oxidative Med. Cell. Longev. 2020, 2020, 5497046. [Google Scholar] [CrossRef]
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid peroxidation in atherosclerotic cardiovascular diseases. Antioxid. Redox Signal. 2020, 34, 49–98. [Google Scholar] [CrossRef]
- Kao, Y.-C.; Ho, P.-C.; Tu, Y.-K.; Jou, I.-M.; Tsai, K.-J. Lipids and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Yang, B.; Fritsche, K.L.; Beversdorf, D.Q.; Gu, Z.; Lee, J.C.; Folk, W.R.; Greenlief, C.M.; Sun, G.Y. Yin-Yang Mechanisms regulating lipid peroxidation of docosahexaenoic acid and arachidonic acid in the central nervous system. Front. Neurol. 2019, 10, 642. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short overview of ROS as cell function regulators and their implications in therapy concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Imlay, J.A. Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 2008, 77, 755–776. [Google Scholar] [CrossRef]
- Imlay, J.A. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006, 59, 1073–1082. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, X.; Templeton, L.J.; Smulski, D.R.; LaRossa, R.A.; Storz, G. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 2001, 183, 4562–4570. [Google Scholar] [CrossRef]
- Matilla, M.A.; Krell, T. The effect of bacterial chemotaxis on host infection and pathogenicity. FEMS Microbiol. Rev. 2017, 42. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive oxygen species and neutrophil function. Annu. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef]
- Dahlgren, C.; Karlsson, A.; Bylund, J. Intracellular neutrophil oxidants: From laboratory curiosity to clinical reality. J. Immunol. 2019, 202, 3127–3134. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid peroxidation-derived aldehydes, 4-hydroxynonenal and malondialdehyde in aging-related disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef]
- Buvelot, H.; Jaquet, V.; Krause, K.-H. Mammalian NADPH Oxidases. In NADPH Oxidases: Methods and Protocols; Knaus, U.G., Leto, T.L., Eds.; Springer: New York, NY, USA, 2019; pp. 17–36. [Google Scholar]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef]
- Datta, S.; Roy, A. Antimicrobial peptides as potential therapeutic agents: A review. Int. J. Pept. Res. Ther. 2021, 27, 555–577. [Google Scholar] [CrossRef]
- Boto, A.; Pérez de la Lastra, J.M.; González, C.C. The road from host-defense peptides to a new generation of antimicrobial drugs. Molecules 2018, 23, 311. [Google Scholar] [CrossRef] [PubMed]
- Jaspreet Kaur, B.; Pushpender Kumar, S. Mini review on antimicrobial peptides, sources, mechanism and recent applications. Protein Pept. Lett. 2020, 27, 4–16. [Google Scholar]
- Lam, P.L.; Wong, R.S.M.; Lam, K.H.; Hung, L.K.; Wong, M.M.; Yung, L.H.; Ho, Y.W.; Wong, W.Y.; Hau, D.K.P.; Gambari, R.; et al. The role of reactive oxygen species in the biological activity of antimicrobial agents: An updated mini review. Chem. Biol. Interact. 2020, 320, 109023. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.K.; O’Neil, D.A. Innate inspiration: Antifungal peptides and other immunotherapeutics from the host immune response. Front. Immunol. 2020, 11, 2177. [Google Scholar] [CrossRef]
- Huang, J.; Peng, W.; Zheng, Y.; Hao, H.; Li, S.; Yao, Y.; Ding, Y.; Zhang, J.; Lyu, J.; Zeng, Q. Upregulation of UCP2 expression protects against LPS-induced oxidative stress and apoptosis in cardiomyocytes. Oxidative Med. Cell. Longev. 2019, 2019, 2758262. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Perez-Samper, G.; Cerulus, B.; Jariani, A.; Vermeersch, L.; Barrajón Simancas, N.; Bisschops, M.M.M.; van den Brink, J.; Solis-Escalante, D.; Gallone, B.; De Maeyer, D.; et al. The crabtree effect shapes the Saccharomyces cerevisiae Lag Phase during the Switch between different carbon sources. mBio 2018, 9, e01331-18. [Google Scholar] [CrossRef]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef]
- Raghav, K.; Bailey, A.M.; Loree, J.M.; Kopetz, S.; Holla, V.; Yap, T.A.; Wang, F.; Chen, K.; Salgia, R.; Hong, D. Untying the gordion knot of targeting MET in cancer. Cancer Treat. Rev. 2018, 66, 95–103. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is mitochondrial dysfunction a common root of noncommunicable chronic diseases? Endocr. Rev. 2020, 41, 491–517. [Google Scholar] [CrossRef]
- Hamblin, M.R. Mechanisms and mitochondrial redox signaling in photobiomodulation. Photochem. Photobiol. 2018, 94, 199–212. [Google Scholar] [CrossRef]
- Brand, K.A.; Hermfisse, U. Aerobic glycolysis by proliferating cells: A protective strategy against reactive oxygen species1. FASEB J. 1997, 11, 388–395. [Google Scholar] [CrossRef]
- Baffy, G. Uncoupling protein-2 and cancer. Mitochondrion 2010, 10, 243–252. [Google Scholar] [CrossRef]
- Vallejo, F.A.; Vanni, S.; Graham, R.M. UCP2 as a potential biomarker for adjunctive metabolic therapies in tumor management. Front. Oncol. 2021, 11, 640720. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, Z.; Zhang, G.; Ng, T.K.; Wu, Z. Association of UCP1 and UCP2 variants with diabetic retinopathy susceptibility in type-2 diabetes mellitus patients: A meta-analysis. BMC Ophthalmol. 2021, 21, 81. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Parton, L.E.; Ye, C.P.; Krauss, S.; Shen, R.; Lin, C.-T.; Porco, J.A., Jr.; Lowell, B.B. Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced β cell dysfunction in isolated pancreatic islets. Cell Metab. 2006, 3, 417–427. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Kritsiligkou, P.; Rand, J.D.; Weids, A.J.; Wang, X.; Kershaw, C.J.; Grant, C.M. Endoplasmic reticulum (ER) stress–induced reactive oxygen species (ROS) are detrimental for the fitness of a thioredoxin reductase mutant. J. Biol. Chem. 2018, 293, 11984–11995. [Google Scholar] [CrossRef]
- Fibach, E.; Rachmilewitz, E. The role of oxidative stress in hemolytic anemia. Curr. Mol. Med. 2008, 8, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Dana, M. Red Blood Cells as Redox Modulators in Hemolytic Anemia. In Erythrocyte; Tombak, A., Ed.; IntechOpen: London, UK, 2019. [Google Scholar]



















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. https://doi.org/10.3390/ijms22094642
Juan CA, Pérez de la Lastra JM, Plou FJ, Pérez-Lebeña E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. International Journal of Molecular Sciences. 2021; 22(9):4642. https://doi.org/10.3390/ijms22094642
Chicago/Turabian StyleJuan, Celia Andrés, José Manuel Pérez de la Lastra, Francisco J. Plou, and Eduardo Pérez-Lebeña. 2021. "The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies" International Journal of Molecular Sciences 22, no. 9: 4642. https://doi.org/10.3390/ijms22094642
APA StyleJuan, C. A., Pérez de la Lastra, J. M., Plou, F. J., & Pérez-Lebeña, E. (2021). The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. International Journal of Molecular Sciences, 22(9), 4642. https://doi.org/10.3390/ijms22094642