
Inherited Platelet Disorders: An Updated Overview

 , , , and
, , , and 
Abstract
1. Introduction
- i.
- ii.
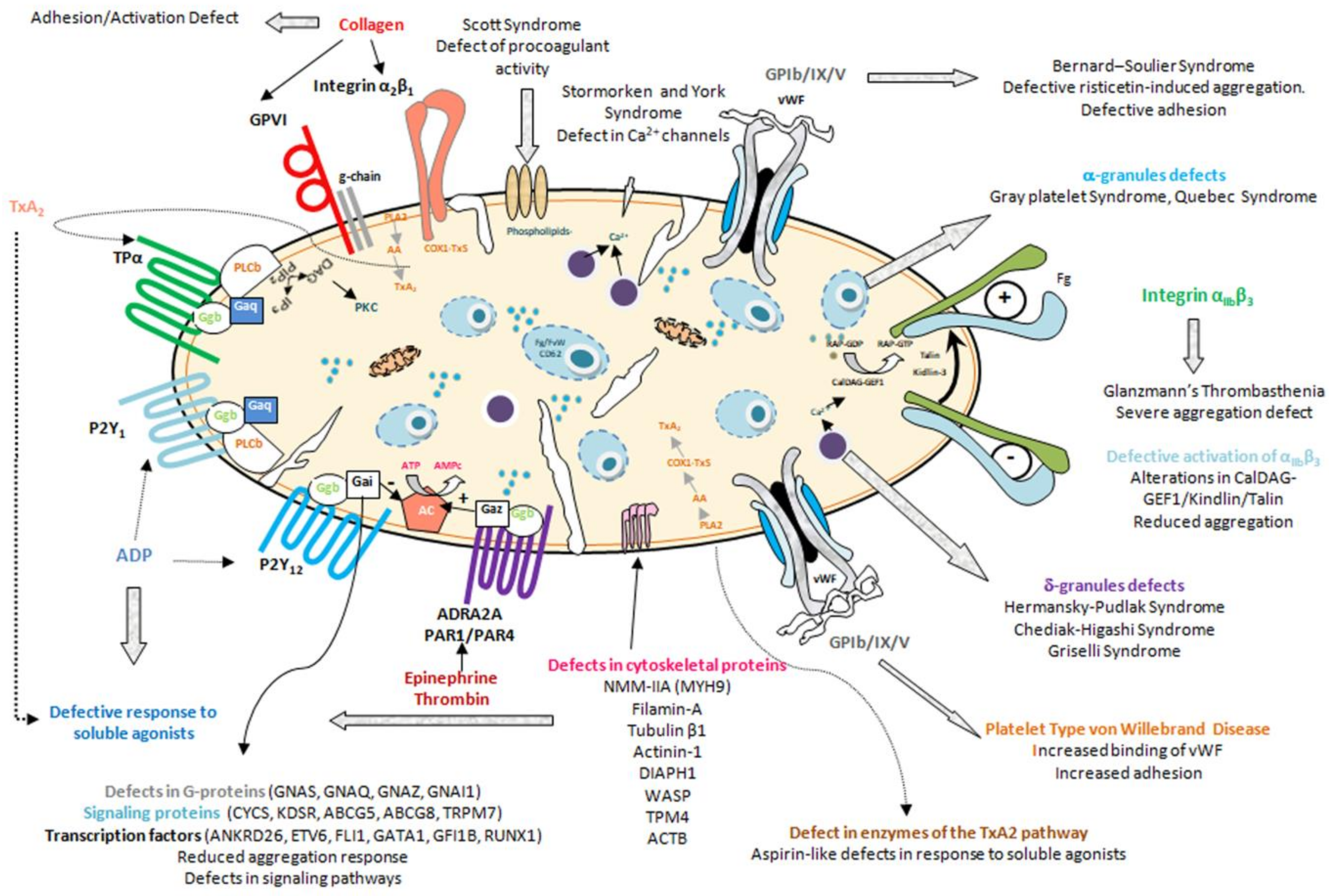
- Inherited platelet function disorders, characterized by dysfunctional, typically hypofunctional, platelets resulting from defects of the membrane receptors, granules, elements involved in signal transduction, or other defects of the biochemical platelet machinery [7,8] (Figure 1, Table 2, Table 3 and Table 4)
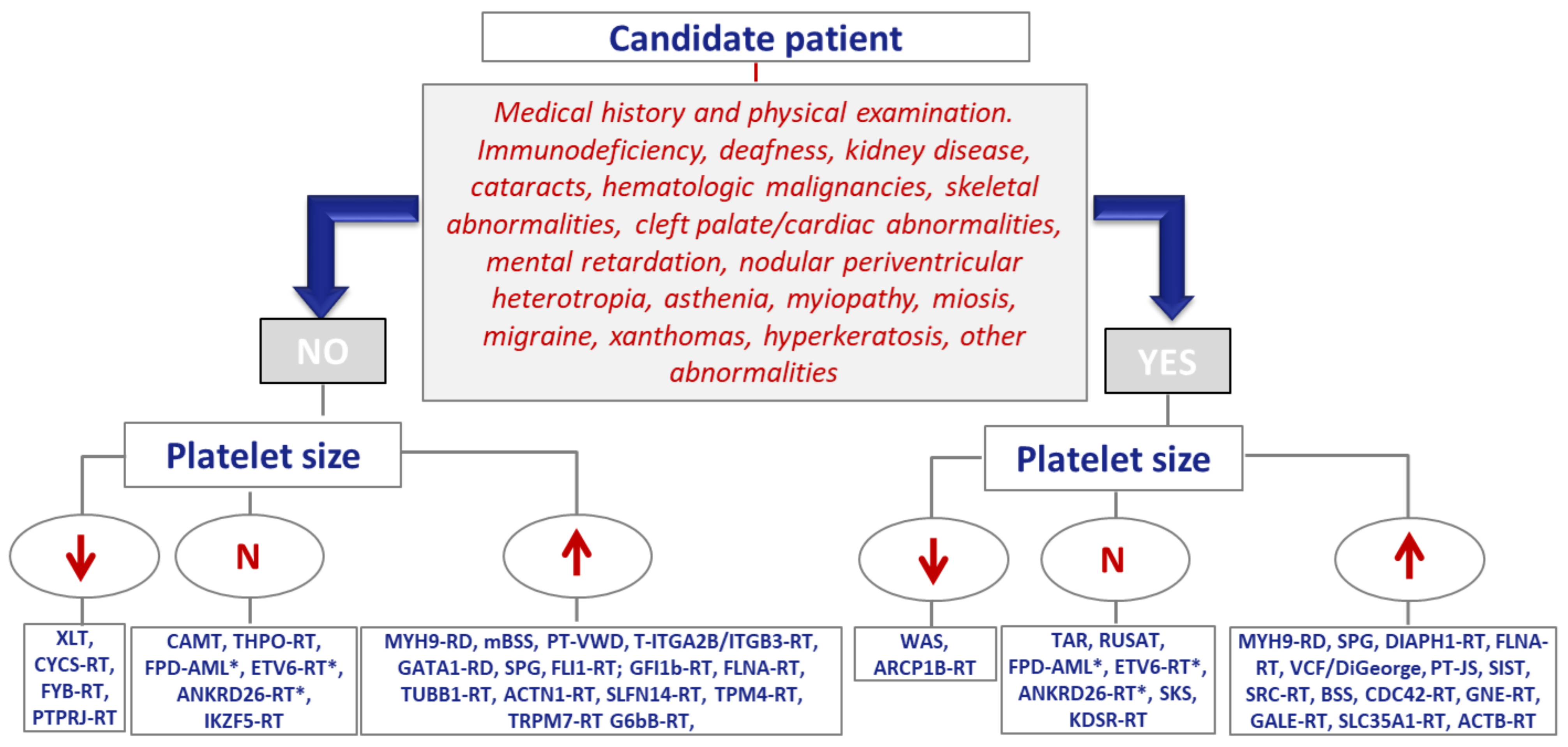
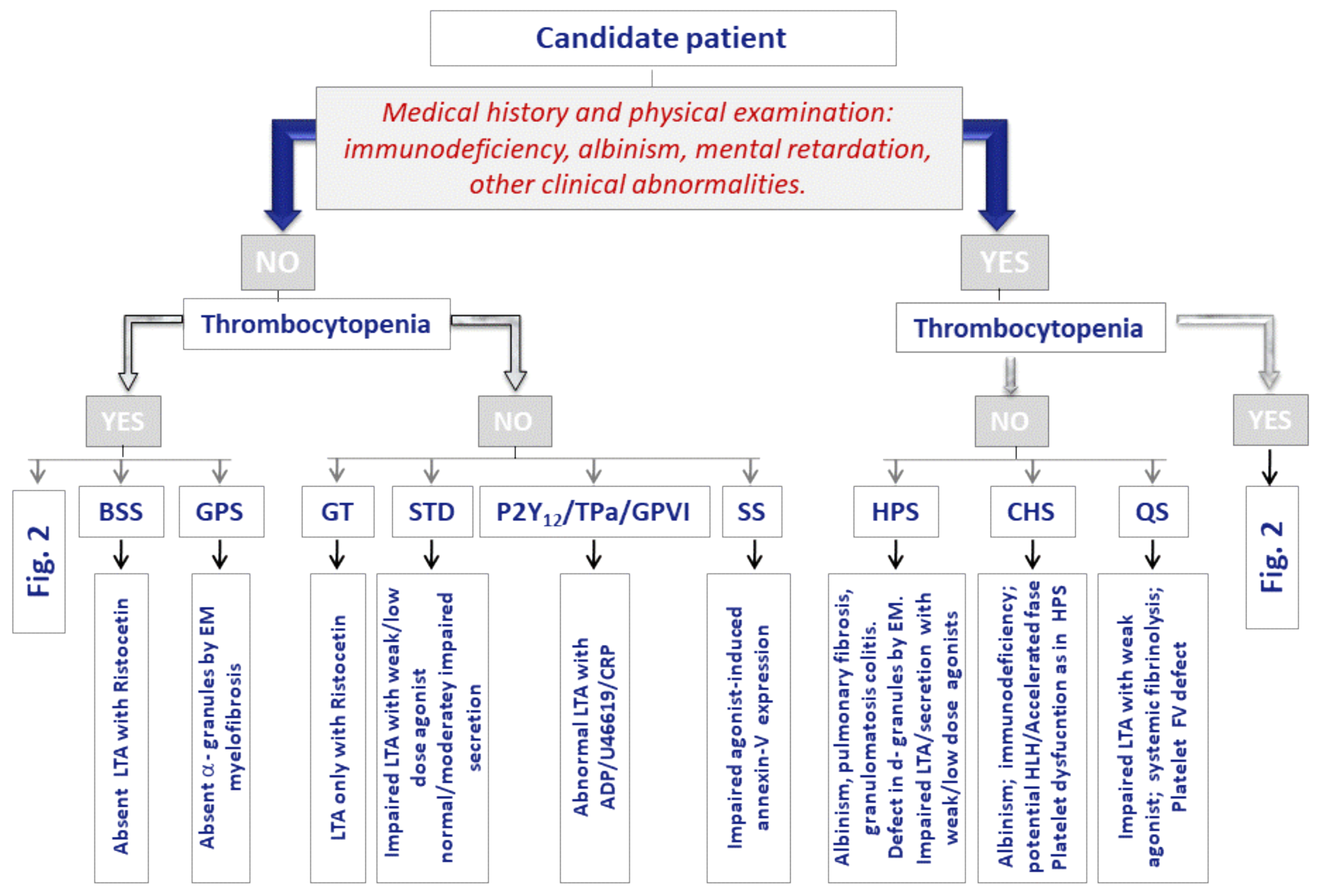
2. Inherited Platelet Disorder Diagnosis
3. Inherited Platelet Disorders of Particular Clinical Relevance
3.1. Syndromic Platelet Diseases
3.1.1. Congenital Amegakaryocytic Thrombocytopenia (CAMT)
3.1.2. MYH9-Related Disease (MYH9-RD)
3.1.3. Wiskott-Aldrich Syndrome (WAS)
3.1.4. Sitosterolemia (STSL)
3.1.5. DIAPH1-Related Thrombocytopenia (DIAPH1-RT)
3.2. Inherited Platelet Disorders with Predisposition to Hematological Neoplasms
3.3. Syndromic Disorders Due to Congenital Defects of Platelet Granules
3.4. Platelet Disorders with High Risk of Severe Bleeding
3.4.1. Defects of the GPIb/IX/V Complex
3.4.2. Glanzmann Thrombasthenia
4. Management of Patients with Inherited Platelet Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolton-Maggs, P.H.; Chalmers, E.A.; Collins, P.W.; Harrison, P.; Kitchen, S.; Liesner, R.J.; Minford, A.; Mumford, A.D.; Parapia, L.A.; Perry, D.J.; et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br. J. Haematol. 2006, 135, 603–633. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.M.; Benito, R.; Lozano, M.L.; Marin-Quilez, A.; Janusz, K.; Martin-Izquierdo, M.; Hernandez-Sanchez, J.; Palma-Barqueros, V.; Hernandez-Rivas, J.M.; Rivera, J.; et al. Molecular Diagnosis of Inherited Coagulation and Bleeding Disorders. Semin. Thromb. Hemost. 2019. [Google Scholar] [CrossRef] [PubMed]
- Oved, J.H.; Lambert, M.P.; Kowalska, M.A.; Poncz, M.; Karczewski, K.J. Population based frequency of naturally occurring loss-of-function variants in genes associated with platelet disorders. J. Thromb. Haemost. JTH 2021, 19, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Pecci, A. Hereditary thrombocytopenias: A growing list of disorders. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 385–399. [Google Scholar] [CrossRef]
- Pecci, A.; Balduini, C.L. Inherited thrombocytopenias: An updated guide for clinicians. Blood Rev. 2020, 100784. [Google Scholar] [CrossRef]
- Bury, L.; Falcinelli, E.; Gresele, P. Learning the Ropes of Platelet Count Regulation: Inherited Thrombocytopenias. J. Clin. Med. 2021, 10, 533. [Google Scholar] [CrossRef]
- Al-Huniti, A.; Kahr, W.H. Inherited Platelet Disorders: Diagnosis and Management. Transfus. Med. Rev. 2020, 34, 277–285. [Google Scholar] [CrossRef]
- Nurden, P.; Stritt, S.; Favier, R.; Nurden, A.T. Inherited platelet diseases with normal platelet count: Phenotypes, genotypes and diagnostic strategy. Haematologica 2021, 106, 337–350. [Google Scholar] [CrossRef]
- Ballmaier, M.; Germeshausen, M. Congenital amegakaryocytic thrombocytopenia: Clinical presentation, diagnosis, and treatment. Semin. Thromb. Hemost. 2011, 37, 673–681. [Google Scholar] [CrossRef]
- Germeshausen, M.; Ballmaier, M. CAMT-MPL: Congenital Amegakaryocytic Thrombocytopenia caused by MPL mutations - Heterogeneity of a monogenic disorder—Comprehensive analysis of 56 patients. Haematologica 2020. [Google Scholar] [CrossRef]
- Noris, P.; Marconi, C.; De Rocco, D.; Melazzini, F.; Pippucci, T.; Loffredo, G.; Giangregorio, T.; Pecci, A.; Seri, M.; Savoia, A. A new form of inherited thrombocytopenia due to monoallelic loss of function mutation in the thrombopoietin gene. Br. J. Haematol. 2018, 181, 698–701. [Google Scholar] [CrossRef]
- Cornish, N.; Aungraheeta, M.R.; FitzGibbon, L.; Burley, K.; Alibhai, D.; Collins, J.; Greene, D.; Downes, K.; Westbury, S.K.; Turro, E.; et al. Monoallelic loss-of-function THPO variants cause heritable thrombocytopenia. Blood Adv. 2020, 4, 920–924. [Google Scholar] [CrossRef]
- Pecci, A.; Ragab, I.; Bozzi, V.; De Rocco, D.; Barozzi, S.; Giangregorio, T.; Ali, H.; Melazzini, F.; Sallam, M.; Alfano, C.; et al. Thrombopoietin mutation in congenital amegakaryocytic thrombocytopenia treatable with romiplostim. EMBO Mol. Med. 2018, 10, 63–75. [Google Scholar] [CrossRef]
- Thompson, A.A.; Woodruff, K.; Feig, S.A.; Nguyen, L.T.; Schanen, N.C. Congenital thrombocytopenia and radio-ulnar synostosis: A new familial syndrome. Br. J. Haematol. 2001, 113, 866–870. [Google Scholar] [CrossRef]
- Germeshausen, M.; Ancliff, P.; Estrada, J.; Metzler, M.; Ponstingl, E.; Rutschle, H.; Schwabe, D.; Scott, R.H.; Unal, S.; Wawer, A.; et al. MECOM-associated syndrome: A heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Adv. 2018, 2, 586–596. [Google Scholar] [CrossRef]
- Niihori, T.; Ouchi-Uchiyama, M.; Sasahara, Y.; Kaneko, T.; Hashii, Y.; Irie, M.; Sato, A.; Saito-Nanjo, Y.; Funayama, R.; Nagashima, T.; et al. Mutations in MECOM, Encoding Oncoprotein EVI1, Cause Radioulnar Synostosis with Amegakaryocytic Thrombocytopenia. Am. J. Hum. Genet. 2015, 97, 848–854. [Google Scholar] [CrossRef]
- Albers, C.A.; Paul, D.S.; Schulze, H.; Freson, K.; Stephens, J.C.; Smethurst, P.A.; Jolley, J.D.; Cvejic, A.; Kostadima, M.; Bertone, P.; et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat. Genet. 2021, 44, 435–439. [Google Scholar] [CrossRef]
- Galera, P.; Dulau-Florea, A.; Calvo, K.R. Inherited thrombocytopenia and platelet disorders with germline predisposition to myeloid neoplasia. Int. J. Lab. Hematol. 2019, 41 (Suppl. 1), 131–141. [Google Scholar] [CrossRef]
- Schlegelberger, B.; Heller, P.G. RUNX1 deficiency (familial platelet disorder with predisposition to myeloid leukemia, FPDMM). Semin. Hematol. 2017, 54, 75–80. [Google Scholar] [CrossRef]
- Ferrari, S.; Lombardi, A.M.; Putti, M.C.; Bertomoro, A.; Cortella, I.; Barzon, I.; Girolami, A.; Fabris, F. Spectrum of 5’UTR mutations in ANKRD26 gene in patients with inherited thrombocytopenia: C.-140C>G mutation is more frequent than expected. Platelets 2017, 28, 621–624. [Google Scholar] [CrossRef]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: Analysis of 78 patients from 21 families. Blood 2011, 117, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, J.; Porter, C.C. ETV6-related thrombocytopenia and leukemia predisposition. Blood 2019, 134, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.; et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat. Genet. 2015, 47, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Lentaigne, C.; Greene, D.; Sivapalaratnam, S.; Favier, R.; Seyres, D.; Thys, C.; Grassi, L.; Mangles, S.; Sibson, K.; Stubbs, M.; et al. Germline mutations in the transcription factor IKZF5 cause thrombocytopenia. Blood 2019, 134, 2070–2081. [Google Scholar] [CrossRef]
- Morison, I.M.; Cramer Borde, E.M.; Cheesman, E.J.; Cheong, P.L.; Holyoake, A.J.; Fichelson, S.; Weeks, R.J.; Lo, A.; Davies, S.M.; Wilbanks, S.M.; et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat. Genet. 2008, 40, 387–389. [Google Scholar] [CrossRef]
- Nurden, A.T.; Nurden, P. Inherited thrombocytopenias: History, advances and perspectives. Haematologica 2020, 105, 2004–2019. [Google Scholar] [CrossRef]
- Bariana, T.K.; Labarque, V.; Heremans, J.; Thys, C.; De Reys, M.; Greene, D.; Jenkins, B.; Grassi, L.; Seyres, D.; Burden, F.; et al. Sphingolipid dysregulation due to lack of functional KDSR impairs proplatelet formation causing thrombocytopenia. Haematologica 2019, 104, 1036–1045. [Google Scholar] [CrossRef]
- Takeichi, T.; Torrelo, A.; Lee, J.Y.W.; Ohno, Y.; Lozano, M.L.; Kihara, A.; Liu, L.; Yasuda, Y.; Ishikawa, J.; Murase, T.; et al. Biallelic Mutations in KDSR Disrupt Ceramide Synthesis and Result in a Spectrum of Keratinization Disorders Associated with Thrombocytopenia. J. Investig. Dermatol. 2017, 137, 2344–2353. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef]
- Markello, T.; Chen, D.; Kwan, J.Y.; Horkayne-Szakaly, I.; Morrison, A.; Simakova, O.; Maric, I.; Lozier, J.; Cullinane, A.R.; Kilo, T.; et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol. Genet. Metab. 2015, 114, 474–482. [Google Scholar] [CrossRef]
- Savoia, A.; Kunishima, S.; De Rocco, D.; Zieger, B.; Rand, M.L.; Pujol-Moix, N.; Caliskan, U.; Tokgoz, H.; Pecci, A.; Noris, P.; et al. Spectrum of the mutations in Bernard-Soulier syndrome. Hum. Mutat. 2014, 35, 1033–1045. [Google Scholar] [CrossRef]
- Berndt, M.C.; Andrews, R.K. Bernard-Soulier syndrome. Haematologica 2011, 96, 355–359. [Google Scholar] [CrossRef]
- Ferrari, S.; Lombardi, A.M.; Cortella, I.; Businaro, M.A.; Bertomoro, A.; Di Pasquale, I.; Fabris, F. New heterozygous variant in GP1BB gene is responsible for an inherited form of macrothrombocytopenia. Br. J. Haematol. 2018. [Google Scholar] [CrossRef]
- Noris, P.; Perrotta, S.; Bottega, R.; Pecci, A.; Melazzini, F.; Civaschi, E.; Russo, S.; Magrin, S.; Loffredo, G.; Di Salvo, V.; et al. Clinical and laboratory features of 103 patients from 42 Italian families with inherited thrombocytopenia derived from the monoallelic Ala156Val mutation of GPIbalpha (Bolzano mutation). Haematologica 2012, 97, 82–88. [Google Scholar] [CrossRef]
- Sivapalaratnam, S.; Westbury, S.K.; Stephens, J.C.; Greene, D.; Downes, K.; Kelly, A.M.; Lentaigne, C.; Astle, W.J.; Huizinga, E.G.; Nurden, P.; et al. Rare variants in GP1BB are responsible for autosomal dominant macrothrombocytopenia. Blood 2017, 129, 520–524. [Google Scholar] [CrossRef]
- Othman, M.; Gresele, P. Guidance on the diagnosis and management of platelet-type von Willebrand disease: A communication from the Platelet Physiology Subcommittee of the ISTH. J. Thromb. Haemost. JTH 2020, 18, 1855–1858. [Google Scholar] [CrossRef]
- Favier, M.; Bordet, J.C.; Favier, R.; Gkalea, V.; Pillois, X.; Rameau, P.; Debili, N.; Alessi, M.C.; Nurden, P.; Raslova, H.; et al. Mutations of the integrin alphaIIb/beta3 intracytoplasmic salt bridge cause macrothrombocytopenia and enlarged platelet alpha-granules. Am. J. Hematol. 2018, 93, 195–204. [Google Scholar] [CrossRef]
- Kunishima, S.; Kashiwagi, H.; Otsu, M.; Takayama, N.; Eto, K.; Onodera, M.; Miyajima, Y.; Takamatsu, Y.; Suzumiya, J.; Matsubara, K.; et al. Heterozygous ITGA2B R995W mutation inducing constitutive activation of the alphaIIbbeta3 receptor affects proplatelet formation and causes congenital macrothrombocytopenia. Blood 2011, 117, 5479–5484. [Google Scholar] [CrossRef]
- Morais, S.; Oliveira, J.; Lau, C.; Pereira, M.; Goncalves, M.; Monteiro, C.; Goncalves, A.R.; Matos, R.; Sampaio, M.; Cruz, E.; et al. alphaIIbbeta3 variants in ten families with autosomal dominant macrothrombocytopenia: Expanding the mutational and clinical spectrum. PLoS ONE 2020, 15, e0235136. [Google Scholar] [CrossRef]
- Lambert, M.P.; Arulselvan, A.; Schott, A.; Markham, S.J.; Crowley, T.B.; Zackai, E.H.; McDonald-McGinn, D.M. The 22q11.2 deletion syndrome: Cancer predisposition, platelet abnormalities and cytopenias. Am. J. Med Genet. Part. A 2017. [Google Scholar] [CrossRef]
- Favier, R.; Akshoomoff, N.; Mattson, S.; Grossfeld, P. Jacobsen syndrome: Advances in our knowledge of phenotype and genotype. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2015, 169, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Mattina, T.; Perrotta, C.S.; Grossfeld, P. Jacobsen syndrome. Orphanet J. Rare Dis. 2009, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Saultier, P.; Vidal, L.; Canault, M.; Bernot, D.; Falaise, C.; Pouymayou, C.; Bordet, J.C.; Saut, N.; Rostan, A.; Baccini, V.; et al. Macrothrombocytopenia and dense granule deficiency associated with FLI1 variants: Ultrastructural and pathogenic features. Haematologica 2017, 102, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, W.S.; Rabbolini, D.J.; Beutler, L.; Chen, Q.; Gabrielli, S.; Mackay, J.P.; Brighton, T.A.; Ward, C.M.; Morel-Kopp, M.C. Paris-Trousseau thrombocytopenia is phenocopied by the autosomal recessive inheritance of a DNA-binding domain mutation in FLI1. Blood 2015, 126, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Millikan, P.D.; Balamohan, S.M.; Raskind, W.H.; Kacena, M.A. Inherited thrombocytopenia due to GATA-1 mutations. Semin. Thromb. Hemost. 2011, 37, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Crispino, J.D.; Horwitz, M.S. GATA factor mutations in hematologic disease. Blood 2017, 129, 2103–2110. [Google Scholar] [CrossRef]
- Marneth, A.E.; van Heerde, W.L.; Hebeda, K.M.; Laros-van Gorkom, B.A.; Barteling, W.; Willemsen, B.; de Graaf, A.O.; Simons, A.; Jansen, J.H.; Preijers, F.; et al. Platelet CD34 expression and alpha/delta-granule abnormalities in GFI1B- and RUNX1-related familial bleeding disorders. Blood 2017, 129, 1733–1736. [Google Scholar] [CrossRef]
- Rabbolini, D.J.; Morel-Kopp, M.C.; Ward, C.M.; Stevenson, W.S. GFI1B variants associated with thrombocytopenia. Platelets 2017, 28, 525–527. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Chen, D.; Abraham, S.M.; Adams, D.R.; Simon, K.L.; Malicdan, M.C.; Markello, T.C.; Gunay-Aygun, M.; Gahl, W.A. Combined alpha-delta platelet storage pool deficiency is associated with mutations in GFI1B. Mol. Genet. Metab. 2017, 120, 288–294. [Google Scholar] [CrossRef]
- Bariana, T.K.; Ouwehand, W.H.; Guerrero, J.A.; Gomez, K. Dawning of the age of genomics for platelet granule disorders: Improving insight, diagnosis and management. Br. J. Haematol. 2017, 176, 705–720. [Google Scholar] [CrossRef]
- Sims, M.C.; Mayer, L.; Collins, J.H.; Bariana, T.K.; Megy, K.; Lavenu-Bombled, C.; Seyres, D.; Kollipara, L.; Burden, F.S.; Greene, D.; et al. Novel manifestations of immune dysregulation and granule defects in gray platelet syndrome. Blood 2020, 136, 1956–1967. [Google Scholar] [CrossRef]
- Pluthero, F.G.; Di Paola, J.; Carcao, M.D.; Kahr, W.H.A. NBEAL2 mutations and bleeding in patients with gray platelet syndrome. Platelets 2018, 29, 632–635. [Google Scholar] [CrossRef]
- Pluthero, F.G.; Kahr, W.H.A. Gray platelet syndrome: NBEAL2 mutations are associated with pathology beyond megakaryocyte and platelet function defects. J. Thromb. Haemost. JTH 2021, 19, 318–322. [Google Scholar] [CrossRef]
- Hofmann, I.; Geer, M.J.; Vogtle, T.; Crispin, A.; Campagna, D.R.; Barr, A.; Calicchio, M.L.; Heising, S.; van Geffen, J.P.; Kuijpers, M.J.E.; et al. Congenital macrothrombocytopenia with focal myelofibrosis due to mutations in human G6b-B is rescued in humanized mice. Blood 2018. [Google Scholar] [CrossRef]
- Pecci, A.; Klersy, C.; Gresele, P.; Lee, K.J.; De Rocco, D.; Bozzi, V.; Russo, G.; Heller, P.G.; Loffredo, G.; Ballmaier, M.; et al. MYH9-related disease: A novel prognostic model to predict the clinical evolution of the disease based on genotype-phenotype correlations. Hum. Mutat. 2014, 35, 236–247. [Google Scholar] [CrossRef]
- Savoia, A.; De Rocco, D.; Pecci, A. MYH9 gene mutations associated with bleeding. Platelets 2017, 28, 312–315. [Google Scholar] [CrossRef]
- Bastida, J.M.; Benito, R.; Gonzalez-Porras, J.R.; Rivera, J. ABCG5 and ABCG8 gene variations associated with sitosterolemia and platelet dysfunction. Platelets 2020, 1–5. [Google Scholar] [CrossRef]
- Bastida, J.M.; Benito, R.; Janusz, K.; Diez-Campelo, M.; Hernandez-Sanchez, J.M.; Marcellini, S.; Giros, M.; Rivera, J.; Lozano, M.L.; Hortal, A.; et al. Two novel variants of the ABCG5 gene cause xanthelasmas and macrothrombocytopenia: A brief review of hematologic abnormalities of sitosterolemia. J. Thromb. Haemost. JTH 2017, 15, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, Z.A.; Macharg, A.; Chang, C.Y.; van Kogelenberg, M.; Morgan, T.; Frentz, S.; Wei, W.; Pilch, J.; Hannibal, M.; Foulds, N.; et al. Differential regulation of two FLNA transcripts explains some of the phenotypic heterogeneity in the loss-of-function filaminopathies. Hum. Mutat. 2018, 39, 103–113. [Google Scholar] [CrossRef]
- Nurden, P.; Debili, N.; Coupry, I.; Bryckaert, M.; Youlyouz-Marfak, I.; Sole, G.; Pons, A.C.; Berrou, E.; Adam, F.; Kauskot, A.; et al. Thrombocytopenia resulting from mutations in filamin A can be expressed as an isolated syndrome. Blood 2011, 118, 5928–5937. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, P.; Westbury, S.K.; Mumford, A.D. FLNA variants associated with disorders of platelet number or function. Platelets 2020, 31, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Burley, K.; Westbury, S.K.; Mumford, A.D. TUBB1 variants and human platelet traits. Platelets 2018, 29, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Kunishima, S.; Kobayashi, R.; Itoh, T.J.; Hamaguchi, M.; Saito, H. Mutation of the beta1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood 2009, 113, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Westbury, S.K.; Shoemark, D.K.; Mumford, A.D. ACTN1 variants associated with thrombocytopenia. Platelets 2017, 28, 625–627. [Google Scholar] [CrossRef]
- Kunishima, S.; Okuno, Y.; Yoshida, K.; Shiraishi, Y.; Sanada, M.; Muramatsu, H.; Chiba, K.; Tanaka, H.; Miyazaki, K.; Sakai, M.; et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am. J. Hum. Genet. 2013, 92, 431–438. [Google Scholar] [CrossRef]
- Westbury, S.K.; Downes, K.; Burney, C.; Lozano, M.L.; Obaji, S.G.; Toh, C.H.; Sevivas, T.; Morgan, N.V.; Erber, W.N.; Kempster, C.; et al. Phenotype description and response to thrombopoietin receptor agonist in DIAPH1-related disorder. Blood Adv. 2018, 2, 2341–2346. [Google Scholar] [CrossRef]
- Fletcher, S.J.; Johnson, B.; Lowe, G.C.; Bem, D.; Drake, S.; Lordkipanidze, M.; Guiu, I.S.; Dawood, B.; Rivera, J.; Simpson, M.A.; et al. SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J. Clin. Investig. 2015, 125, 3600–3605. [Google Scholar] [CrossRef]
- Stapley, R.J.; Pisareva, V.P.; Pisarev, A.V.; Morgan, N.V. SLFN14 gene mutations associated with bleeding. Platelets 2020, 31, 407–410. [Google Scholar] [CrossRef]
- Barozzi, S.; Di Buduo, C.A.; Marconi, C.; Bozzi, V.; Seri, M.; Romano, F.; Balduini, A.; Pecci, A. Pathogenetic and clinical study of a patient with thrombocytopenia due to the p.E527K gain-of-function variant of SRC. Haematologica 2020, 918–922, Online ahead of print. [Google Scholar] [CrossRef]
- De Kock, L.; Thys, C.; Downes, K.; Duarte, D.; Megy, K.; Van Geet, C.; Freson, K. De novo variant in tyrosine kinase SRC causes thrombocytopenia: Case report of a second family. Platelets 2019, 30, 931–934. [Google Scholar] [CrossRef]
- Turro, E.; Greene, D.; Wijgaerts, A.; Thys, C.; Lentaigne, C.; Bariana, T.K.; Westbury, S.K.; Kelly, A.M.; Selleslag, D.; Stephens, J.C.; et al. A dominant gain-of-function mutation in universal tyrosine kinase SRC causes thrombocytopenia, myelofibrosis, bleeding, and bone pathologies. Sci. Transl. Med. 2016, 8, 328ra330. [Google Scholar] [CrossRef]
- Pleines, I.; Woods, J.; Chappaz, S.; Kew, V.; Foad, N.; Ballester-Beltran, J.; Aurbach, K.; Lincetto, C.; Lane, R.M.; Schevzov, G.; et al. Mutations in tropomyosin 4 underlie a rare form of human macrothrombocytopenia. J. Clin. Investig. 2017, 127, 814–829. [Google Scholar] [CrossRef]
- Stritt, S.; Nurden, P.; Favier, R.; Favier, M.; Ferioli, S.; Gotru, S.K.; van Eeuwijk, J.M.; Schulze, H.; Nurden, A.T.; Lambert, M.P.; et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg(2+) homeostasis and cytoskeletal architecture. Nat. Commun. 2016, 7, 11097. [Google Scholar] [CrossRef]
- Manchev, V.T.; Hilpert, M.; Berrou, E.; Elaib, Z.; Aouba, A.; Boukour, S.; Souquere, S.; Pierron, G.; Rameau, P.; Andrews, R.; et al. A new form of macrothrombocytopenia induced by a germ-line mutation in the PRKACG gene. Blood 2014, 124, 2554–2563. [Google Scholar] [CrossRef]
- Martinelli, S.; Krumbach, O.H.F.; Pantaleoni, F.; Coppola, S.; Amin, E.; Pannone, L.; Nouri, K.; Farina, L.; Dvorsky, R.; Lepri, F.; et al. Functional Dysregulation of CDC42 Causes Diverse Developmental Phenotypes. Am. J. Hum. Genet. 2018, 102, 309–320. [Google Scholar] [CrossRef]
- Takenouchi, T.; Okamoto, N.; Ida, S.; Uehara, T.; Kosaki, K. Further evidence of a mutation in CDC42 as a cause of a recognizable syndromic form of thrombocytopenia. Am. J. Med. Genet. Part. A 2016, 170A, 852–855. [Google Scholar] [CrossRef]
- Futterer, J.; Dalby, A.; Lowe, G.C.; Johnson, B.; Simpson, M.A.; Motwani, J.; Williams, M.; Watson, S.P.; Morgan, N.V. Mutation in GNE is associated with severe congenital thrombocytopenia. Blood 2018, 132, 1855–1858. [Google Scholar] [CrossRef]
- Lee-Sundlov, M.M.; Stowell, S.R.; Hoffmeister, K.M. Multifaceted role of glycosylation in transfusion medicine, platelets, and red blood cells. J. Thromb. Haemost. JTH 2020, 18, 1535–1547. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Shai, E.; Turro, E.; Jahshan, N.; Hi-Am, E.; Spectre, G.; Daum, H.; Kalish, Y.; Althaus, K.; Greinacher, A.; et al. GNE variants causing autosomal recessive macrothrombocytopenia without associated muscle wasting. Blood 2018, 132, 1851–1854. [Google Scholar] [CrossRef]
- Seo, A.; Gulsuner, S.; Pierce, S.; Ben-Harosh, M.; Shalev, H.; Walsh, T.; Krasnov, T.; Dgany, O.; Doulatov, S.; Tamary, H.; et al. Inherited thrombocytopenia associated with mutation of UDP-galactose-4-epimerase (GALE). Hum. Mol. Genet. 2019, 28, 133–142. [Google Scholar] [CrossRef]
- Kauskot, A.; Pascreau, T.; Adam, F.; Bruneel, A.; Reperant, C.; Lourenco-Rodrigues, M.D.; Rosa, J.P.; Petermann, R.; Maurey, H.; Auditeau, C.; et al. A mutation in the gene coding for the sialic acid transporter SLC35A1 is required for platelet life span but not proplatelet formation. Haematologica 2018, 103, e613–e617. [Google Scholar] [CrossRef]
- Latham, S.L.; Ehmke, N.; Reinke, P.Y.A.; Taft, M.H.; Eicke, D.; Reindl, T.; Stenzel, W.; Lyons, M.J.; Friez, M.J.; Lee, J.A.; et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat. Commun. 2018, 9, 4250. [Google Scholar] [CrossRef]
- Candotti, F. Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrome. J. Clin. Immunol. 2018, 38, 13–27. [Google Scholar] [CrossRef]
- Rivers, E.; Worth, A.; Thrasher, A.J.; Burns, S.O. How I manage patients with Wiskott Aldrich syndrome. Br. J. Haematol. 2019, 185, 647–655. [Google Scholar] [CrossRef]
- Levin, C.; Koren, A.; Pretorius, E.; Rosenberg, N.; Shenkman, B.; Hauschner, H.; Zalman, L.; Khayat, M.; Salama, I.; Elpeleg, O.; et al. Deleterious mutation in the FYB gene is associated with congenital autosomal recessive small-platelet thrombocytopenia. J. Thromb. Haemost. JTH 2015, 13, 1285–1292. [Google Scholar] [CrossRef]
- Spindler, M.; van Eeuwijk, J.M.M.; Schurr, Y.; Nurden, P.; Nieswandt, B.; Stegner, D.; Reinhold, A.; Bender, M. ADAP deficiency impairs megakaryocyte polarization with ectopic proplatelet release and causes microthrombocytopenia. Blood 2018, 132, 635–646. [Google Scholar] [CrossRef]
- Brigida, I.; Zoccolillo, M.; Cicalese, M.P.; Pfajfer, L.; Barzaghi, F.; Scala, S.; Oleaga-Quintas, C.; Alvarez-Alvarez, J.A.; Sereni, L.; Giannelli, S.; et al. T-cell defects in patients with ARPC1B germline mutations account for combined immunodeficiency. Blood 2018, 132, 2362–2374. [Google Scholar] [CrossRef]
- Kahr, W.H.; Pluthero, F.G.; Elkadri, A.; Warner, N.; Drobac, M.; Chen, C.H.; Lo, R.W.; Li, L.; Li, R.; Li, Q.; et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat. Commun. 2017, 8, 14816. [Google Scholar] [CrossRef]
- Marconi, C.; Di Buduo, C.A.; LeVine, K.; Barozzi, S.; Faleschini, M.; Bozzi, V.; Palombo, F.; McKinstry, S.; Lassandro, G.; Giordano, P.; et al. Loss-of-function mutations in PTPRJ cause a new form of inherited thrombocytopenia. Blood 2019, 133, 1346–1357. [Google Scholar] [CrossRef]
- Nurden, A.T. Should studies on Glanzmann thrombasthenia not be telling us more about cardiovascular disease and other major illnesses? Blood Rev. 2017, 31, 287–299. [Google Scholar] [CrossRef]
- Nurden, A.T.; Pillois, X. ITGA2B and ITGB3 gene mutations associated with Glanzmann thrombasthenia. Platelets 2018, 29, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T.; Pillois, X.; Fiore, M.; Alessi, M.C.; Bonduel, M.; Dreyfus, M.; Goudemand, J.; Gruel, Y.; Benabdallah-Guerida, S.; Latger-Cannard, V.; et al. Expanding the Mutation Spectrum Affecting alphaIIbbeta3 Integrin in Glanzmann Thrombasthenia: Screening of the ITGA2B and ITGB3 Genes in a Large International Cohort. Hum. Mutat. 2015, 36, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M. The platelet P2Y(1)(2) receptor for adenosine diphosphate: Congenital and drug-induced defects. Blood 2011, 117, 2102–2112. [Google Scholar] [CrossRef] [PubMed]
- Scavone, M.; Femia, E.A.; Cattaneo, M. P2Y(1)(2) receptor gene mutations associated with bleeding. Platelets 2017, 28, 421–423. [Google Scholar] [CrossRef]
- Mundell, S.J.; Mumford, A. TBXA2R gene variants associated with bleeding. Platelets 2018, 1–4. [Google Scholar] [CrossRef]
- Jandrot-Perrus, M.; Hermans, C.; Mezzano, D. Platelet glycoprotein VI genetic quantitative and qualitative defects. Platelets 2019, 30, 708–713. [Google Scholar] [CrossRef]
- Matus, V.; Valenzuela, G.; Saez, C.G.; Hidalgo, P.; Lagos, M.; Aranda, E.; Panes, O.; Pereira, J.; Pillois, X.; Nurden, A.T.; et al. An adenine insertion in exon 6 of human GP6 generates a truncated protein associated with a bleeding disorder in four Chilean families. J. Thromb. Haemost. JTH 2013, 11, 1751–1759. [Google Scholar] [CrossRef]
- Berrou, E.; Soukaseum, C.; Favier, R.; Adam, F.; Elaib, Z.; Kauskot, A.; Bordet, J.C.; Ballerini, P.; Loyau, S.; Feng, M.; et al. A mutation of the human EPHB2 gene leads to a major platelet functional defect. Blood 2018, 132, 2067–2077. [Google Scholar] [CrossRef]
- Dupuis, A.; Bordet, J.C.; Eckly, A.; Gachet, C. Platelet delta-Storage Pool Disease: An Update. J. Clin. Med. 2020, 9, 2508. [Google Scholar] [CrossRef]
- Hayward, C.P.; Rivard, G.E. Quebec platelet disorder. Expert Rev. Hematol. 2011, 4, 137–141. [Google Scholar] [CrossRef]
- Liang, M.; Soomro, A.; Tasneem, S.; Abatti, L.E.; Alizada, A.; Yuan, X.; Uuskula-Reimand, L.; Antounians, L.; Alvi, S.A.; Paterson, A.D.; et al. Enhancer-gene rewiring in the pathogenesis of Quebec platelet disorder. Blood 2020, 136, 2679–2690. [Google Scholar] [CrossRef]
- Bastida, J.M.; Morais, S.; Palma-Barqueros, V.; Benito, R.; Bermejo, N.; Karkucak, M.; Trapero-Marugan, M.; Bohdan, N.; Pereira, M.; Marin-Quilez, A.; et al. Identification of novel variants in ten patients with Hermansky-Pudlak syndrome by high-throughput sequencing. Ann. Med. 2019, 51, 141–148. [Google Scholar] [CrossRef]
- Merideth, M.A.; Introne, W.J.; Wang, J.A.; O’Brien, K.J.; Huizing, M.; Gochuico, B.R. Genetic variants associated with Hermansky-Pudlak syndrome. Platelets 2020, 31, 544–547. [Google Scholar] [CrossRef]
- Sanchez-Guiu, I.; Torregrosa, J.M.; Velasco, F.; Anton, A.I.; Lozano, M.L.; Vicente, V.; Rivera, J. Hermansky-Pudlak syndrome. Overview of clinical and molecular features and case report of a new HPS-1 variant. Hamostaseologie 2014, 34, 301–309. [Google Scholar] [CrossRef]
- Huizing, M.; Malicdan, M.C.V.; Wang, J.A.; Pri-Chen, H.; Hess, R.A.; Fischer, R.; O’Brien, K.J.; Merideth, M.A.; Gahl, W.A.; Gochuico, B.R. Hermansky-Pudlak syndrome: Mutation update. Hum. Mutat. 2020, 41, 543–580. [Google Scholar] [CrossRef]
- Lozano, M.L.; Rivera, J.; Sanchez-Guiu, I.; Vicente, V. Towards the targeted management of Chediak-Higashi syndrome. Orphanet J. Rare Dis. 2014, 9, 132. [Google Scholar] [CrossRef]
- Castano-Jaramillo, L.M.; Lugo-Reyes, S.O.; Cruz Munoz, M.E.; Scheffler-Mendoza, S.C.; Duran McKinster, C.; Yamazaki-Nakashimada, M.A.; Espinosa-Padilla, S.E.; Saez-de-Ocariz Gutierrez, M.D.M. Diagnostic and therapeutic caveats in Griscelli syndrome. Scand. J. Immunol. 2021, e13034. [Google Scholar] [CrossRef]
- Chan, M.V.; Hayman, M.A.; Sivapalaratnam, S.; Crescente, M.; Allan, H.E.; Edin, M.L.; Zeldin, D.C.; Milne, G.L.; Stephens, J.; Greene, D.; et al. Identification of a homozygous recessive variant in PTGS1 resulting in a congenital aspirin-like defect in platelet function. Haematologica 2020. [Google Scholar] [CrossRef]
- Palma-Barqueros, V.; Bohdan, N.; Revilla, N.; Vicente, V.; Bastida, J.M.; Rivera, J. PTGS1 gene variations associated with bleeding and platelet dysfunction. Platelets 2020, 1–7. [Google Scholar] [CrossRef]
- Palma-Barqueros, V.; Crescente, M.; de la Morena, M.E.; Chan, M.V.; Almarza, E.; Revilla, N.; Bohdan, N.; Minano, A.; Padilla, J.; Allan, H.E.; et al. A novel genetic variant in PTGS1 affects N-glycosylation of cyclooxygenase-1 causing a dominant-negative effect on platelet function and bleeding diathesis. Am. J. Hematol. 2021, 96, E83–E88. [Google Scholar] [CrossRef]
- Nurden, A.T.; Nurden, P. Congenital platelet disorders and understanding of platelet function. Br. J. Haematol. 2014, 165, 165–178. [Google Scholar] [CrossRef]
- Rao, A.K. Inherited platelet function disorders: Overview and disorders of granules, secretion, and signal transduction. Hematol. Oncol. Clin. N. Am. 2013, 27, 585–611. [Google Scholar] [CrossRef]
- Canault, M.; Alessi, M.C. RasGRP2 Structure, Function and Genetic Variants in Platelet Pathophysiology. Int. J. Mol. Sci. 2020, 21, 1075. [Google Scholar] [CrossRef]
- Lozano, M.L.; Cook, A.; Bastida, J.M.; Paul, D.S.; Iruin, G.; Cid, A.R.; Adan-Pedroso, R.; Ramon Gonzalez-Porras, J.; Hernandez-Rivas, J.M.; Fletcher, S.J.; et al. Novel mutations in RASGRP2, which encodes CalDAG-GEFI, abrogate Rap1 activation, causing platelet dysfunction. Blood 2016, 128, 1282–1289. [Google Scholar] [CrossRef]
- Palma-Barqueros, V.; Ruiz-Pividal, J.; Bohdan, N.; Vicente, V.; Bastida, J.M.; Lozano, M.; Rivera, J. RASGRP2 gene variations associated with platelet dysfunction and bleeding. Platelets 2019, 30, 535–539. [Google Scholar] [CrossRef]
- Rognoni, E.; Ruppert, R.; Fassler, R. The kindlin family: Functions, signaling properties and implications for human disease. J. Cell Sci. 2016, 129, 17–27. [Google Scholar] [CrossRef]
- Van de Vijver, E.; De Cuyper, I.M.; Gerrits, A.J.; Verhoeven, A.J.; Seeger, K.; Gutierrez, L.; van den Berg, T.K.; Kuijpers, T.W. Defects in Glanzmann thrombasthenia and LAD-III (LAD-1/v) syndrome: The role of integrin beta1 and beta3 in platelet adhesion to collagen. Blood 2012, 119, 583–586. [Google Scholar] [CrossRef][Green Version]
- Millington-Burgess, S.L.; Harper, M.T. Gene of the issue: ANO6 and Scott Syndrome. Platelets 2020, 31, 964–967. [Google Scholar] [CrossRef]
- Sanchez-Guiu, I.; Anton, A.I.; Padilla, J.; Velasco, F.; Lucia, J.F.; Lozano, M.; Cid, A.R.; Sevivas, T.; Lopez-Fernandez, M.F.; Vicente, V.; et al. Functional and molecular characterization of inherited platelet disorders in the Iberian Peninsula: Results from a collaborative study. Orphanet J. Rare Dis. 2014, 9, 213. [Google Scholar] [CrossRef]
- Gresele, P.; Orsini, S.; Noris, P.; Falcinelli, E.; Alessi, M.C.; Bury, L.; Borhany, M.; Santoro, C.; Glembotsky, A.C.; Cid, A.R.; et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: A communication from the Platelet Physiology SSC. J. Thromb. Haemost. JTH 2020, 18, 732–739. [Google Scholar] [CrossRef]
- Melazzini, F.; Zaninetti, C.; Balduini, C.L. Bleeding is not the main clinical issue in many patients with inherited thrombocytopaenias. Haemoph. Off. J. World Fed. Hemoph. 2017, 23, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, J.; Fisher, M.H. ETV6-related thrombocytopenia and platelet dysfunction. Platelets 2021, 32, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Nava, T.; Rivard, G.E.; Bonnefoy, A. Challenges on the diagnostic approach of inherited platelet function disorders: Is a paradigm change necessary? Platelets 2018, 29, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernandez-Sanchez, J.M.; Riesco, S.; Bermejo, N.; Gonzalez-Garcia, H.; et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica 2018, 103, 148–162. [Google Scholar] [CrossRef]
- Downes, K.; Megy, K.; Duarte, D.; Vries, M.; Gebhart, J.; Hofer, S.; Shamardina, O.; Deevi, S.V.V.; Stephens, J.; Mapeta, R.; et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood 2019, 134, 2082–2091. [Google Scholar] [CrossRef]
- Johnson, B.; Lowe, G.C.; Futterer, J.; Lordkipanidze, M.; MacDonald, D.; Simpson, M.A.; Sanchez-Guiu, I.; Drake, S.; Bem, D.; Leo, V.; et al. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica 2016, 101, 1170–1179. [Google Scholar] [CrossRef]
- Downes, K.; Borry, P.; Ericson, K.; Gomez, K.; Greinacher, A.; Lambert, M.; Leinoe, E.; Noris, P.; Van Geet, C.; Freson, K. Clinical management, ethics and informed consent related to multi-gene panel-based high throughput sequencing testing for platelet disorders: Communication from the SSC of the ISTH. J. Thromb. Haemost. JTH 2020, 18, 2751–2758. [Google Scholar] [CrossRef]
- Greinacher, A.; Eekels, J.J.M. Simplifying the diagnosis of inherited platelet disorders? The new tools do not make it any easier. Blood 2019, 133, 2478–2483. [Google Scholar] [CrossRef]
- Dovlatova, N. Current status and future prospects for platelet function testing in the diagnosis of inherited bleeding disorders. Br. J. Haematol. 2015, 170, 150–161. [Google Scholar] [CrossRef]
- Gresele, P. Diagnosis of inherited platelet function disorders: Guidance from the SSC of the ISTH. J. Thromb. Haemost. JTH 2015, 13, 314–322. [Google Scholar] [CrossRef]
- Gresele, P.; Bury, L.; Falcinelli, E. Inherited Platelet Function Disorders: Algorithms for Phenotypic and Genetic Investigation. Semin. Thromb. Hemost. 2016, 42, 292–305. [Google Scholar] [CrossRef]
- Greinacher, A.; Pecci, A.; Kunishima, S.; Althaus, K.; Nurden, P.; Balduini, C.L.; Bakchoul, T. Diagnosis of inherited platelet disorders on a blood smear: A tool to facilitate worldwide diagnosis of platelet disorders. J. Thromb. Haemost. JTH 2017, 15, 1511–1521. [Google Scholar] [CrossRef]
- Zaninetti, C.; Greinacher, A. Diagnosis of Inherited Platelet Disorders on a Blood Smear. J. Clin. Med. 2020, 9, 539. [Google Scholar] [CrossRef]
- Harker, L.A.; Slichter, S.J. The bleeding time as a screening test for evaluation of platelet function. New Engl. J. Med. 1972, 287, 155–159. [Google Scholar] [CrossRef]
- Quiroga, T.; Goycoolea, M.; Munoz, B.; Morales, M.; Aranda, E.; Panes, O.; Pereira, J.; Mezzano, D. Template bleeding time and PFA-100 have low sensitivity to screen patients with hereditary mucocutaneous hemorrhages: Comparative study in 148 patients. J. Thromb. Haemost. JTH 2004, 2, 892–898. [Google Scholar] [CrossRef]
- Hayward, C.P.; Harrison, P.; Cattaneo, M.; Ortel, T.L.; Rao, A.K. Platelet function analyzer (PFA)-100 closure time in the evaluation of platelet disorders and platelet function. J. Thromb. Haemost. JTH 2006, 4, 312–319. [Google Scholar] [CrossRef]
- Le Blanc, J.; Mullier, F.; Vayne, C.; Lordkipanidze, M. Advances in Platelet Function Testing-Light Transmission Aggregometry and Beyond. J. Clin. Med. 2020, 9, 2636. [Google Scholar] [CrossRef]
- Rivera, J.; Lozano, M.L. Performance and usefulness of platelet aggregation testing. Platelets 2018, 29, 637. [Google Scholar] [CrossRef]
- Alessi, M.C.; Sie, P.; Payrastre, B. Strengths and Weaknesses of Light Transmission Aggregometry in Diagnosing Hereditary Platelet Function Disorders. J. Clin. Med. 2020, 9, 763. [Google Scholar] [CrossRef]
- Cattaneo, M.; Cerletti, C.; Harrison, P.; Hayward, C.P.; Kenny, D.; Nugent, D.; Nurden, P.; Rao, A.K.; Schmaier, A.H.; Watson, S.P.; et al. Recommendations for the Standardization of Light Transmission Aggregometry: A Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J. Thromb. Haemost. JTH 2013. [Google Scholar] [CrossRef]
- Van Asten, I.; Schutgens, R.E.G.; Baaij, M.; Zandstra, J.; Roest, M.; Pasterkamp, G.; Huisman, A.; Korporaal, S.J.A.; Urbanus, R.T. Validation of flow cytometric analysis of platelet function in patients with a suspected platelet function defect. J. Thromb. Haemost. JTH 2018, 16, 689–698. [Google Scholar] [CrossRef]
- White, J.G. Electron microscopy methods for studying platelet structure and function. Methods Mol. Biol 2004, 272, 47–63. [Google Scholar] [CrossRef]
- Leinoe, E.; Zetterberg, E.; Kinalis, S.; Ostrup, O.; Kampmann, P.; Norstrom, E.; Andersson, N.; Klintman, J.; Qvortrup, K.; Nielsen, F.C.; et al. Application of whole-exome sequencing to direct the specific functional testing and diagnosis of rare inherited bleeding disorders in patients from the Oresund Region, Scandinavia. Br. J. Haematol. 2017, 179, 308–322. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.; Arnold, D.M.; Coller, B.; James, P.; Neunert, C.; Lillicrap, D. ISTH/SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J. Thromb. Haemost. JTH 2010, 8, 2063–2065. [Google Scholar] [CrossRef]
- Bastida, J.M.; Del Rey, M.; Revilla, N.; Benito, R.; Perez-Andres, M.; Gonzalez, B.; Riesco, S.; Janusz, K.; Padilla, J.; Benito-Sendin, A.H.; et al. Wiskott-Aldrich syndrome in a child presenting with macrothrombocytopenia. Platelets 2016, 1–4. [Google Scholar] [CrossRef]
- Heremans, J.; Freson, K. High-throughput sequencing for diagnosing platelet disorders: Lessons learned from exploring the causes of bleeding disorders. Int. J. Lab. Hematol. 2018, 40 (Suppl. 1), 89–96. [Google Scholar] [CrossRef]
- Megy, K.; Downes, K.; Simeoni, I.; Bury, L.; Morales, J.; Mapeta, R.; Bellissimo, D.B.; Bray, P.F.; Goodeve, A.C.; Gresele, P.; et al. Curated disease-causing genes for bleeding, thrombotic, and platelet disorders: Communication from the SSC of the ISTH. J. Thromb. Haemost. JTH 2019, 17, 1253–1260. [Google Scholar] [CrossRef]
- Sivapalaratnam, S.; Collins, J.; Gomez, K. Diagnosis of inherited bleeding disorders in the genomic era. Br. J. Haematol. 2017, 179, 363–376. [Google Scholar] [CrossRef]
- Ver Donck, F.; Downes, K.; Freson, K. Strengths and limitations of high-throughput sequencing for the diagnosis of inherited bleeding and platelet disorders. J. Thromb. Haemost. JTH 2020, 18, 1839–1845. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Greinacher, A.; Eekels, J.J.M. Diagnosis of hereditary platelet disorders in the era of next-generation sequencing: “primum non nocere”. J. Thromb. Haemost. JTH 2019, 17, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Bury, L.; Megy, K.; Stephens, J.C.; Grassi, L.; Greene, D.; Gleadall, N.; Althaus, K.; Allsup, D.; Bariana, T.K.; Bonduel, M.; et al. Next-generation sequencing for the diagnosis of MYH9-RD: Predicting pathogenic variants. Hum. Mutat. 2020, 41, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Kunishima, S.; Kitamura, K.; Matsumoto, T.; Sekine, T.; Saito, H. Somatic mosaicism in MYH9 disorders: The need to carefully evaluate apparently healthy parents. Br. J. Haematol. 2014, 165, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Rodeghiero, F.; Pecci, A.; Balduini, C.L. Thrombopoietin receptor agonists in hereditary thrombocytopenias. J. Thromb. Haemost. JTH 2018. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Morgan, N.V.; Daly, M.E. Gene of the issue: RUNX1 mutations and inherited bleeding. Platelets 2017, 28, 208–210. [Google Scholar] [CrossRef]
- Luo, X.; Feurstein, S.; Mohan, S.; Porter, C.C.; Jackson, S.A.; Keel, S.; Chicka, M.; Brown, A.L.; Kesserwan, C.; Agarwal, A.; et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 2019, 3, 2962–2979. [Google Scholar] [CrossRef]
- Aarts, C.E.M.; Downes, K.; Hoogendijk, A.J.; Sprenkeler, E.G.G.; Gazendam, R.P.; Favier, R.; Favier, M.; Tool, A.T.J.; van Hamme, J.L.; Kostadima, M.A.; et al. Neutrophil specific granule and NETosis defects in gray platelet syndrome. Blood Adv. 2021, 5, 549–564. [Google Scholar] [CrossRef]
- Tariq, H.; Perez Botero, J.; Higgins, R.A.; Medina, E.A. Gray Platelet Syndrome Presenting With Pancytopenia, Splenomegaly, and Bone Marrow Fibrosis. Am. J. Clin. Pathol. 2021. [Google Scholar] [CrossRef]
- Nurden, A.T.; Nurden, P. Should any genetic defect affecting alpha-granules in platelets be classified as gray platelet syndrome? Am. J. Hematol. 2016, 91, 714–718. [Google Scholar] [CrossRef]
- Morrow, B.E.; McDonald-McGinn, D.M.; Emanuel, B.S.; Vermeesch, J.R.; Scambler, P.J. Molecular genetics of 22q11.2 deletion syndrome. Am. J. Med Genet. Part. A 2018, 176, 2070–2081. [Google Scholar] [CrossRef]
- Rivera, J.; Lozano, M.L.; Navarro-Nunez, L.; Vicente, V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica 2009, 94, 700–711. [Google Scholar] [CrossRef]
- Botero, J.P.; Lee, K.; Branchford, B.R.; Bray, P.F.; Freson, K.; Lambert, M.P.; Luo, M.; Mohan, S.; Ross, J.E.; Bergmeier, W.; et al. Glanzmann thrombasthenia: Genetic basis and clinical correlates. Haematologica 2020, 105, 888–894. [Google Scholar] [CrossRef]
- Alamelu, J.; Liesner, R. Modern management of severe platelet function disorders. Br. J. Haematol. 2010, 149, 813–823. [Google Scholar] [CrossRef]
- Grainger, J.D.; Thachil, J.; Will, A.M. How we treat the platelet glycoprotein defects; Glanzmann thrombasthenia and Bernard Soulier syndrome in children and adults. Br. J. Haematol. 2018, 182, 621–632. [Google Scholar] [CrossRef]
- Poon, M.C.; Di Minno, G.; d’Oiron, R.; Zotz, R. New Insights Into the Treatment of Glanzmann Thrombasthenia. Transfus. Med. Rev. 2016, 30, 92–99. [Google Scholar] [CrossRef]
- Orsini, S.; Noris, P.; Bury, L.; Heller, P.G.; Santoro, C.; Kadir, R.A.; Butta, N.C.; Falcinelli, E.; Cid, A.R.; Fabris, F.; et al. Bleeding risk of surgery and its prevention in patients with inherited platelet disorders. The Surgery in Platelet disorders And Therapeutic Approach (SPATA) study. Haematologica 2017. [Google Scholar] [CrossRef]
- Coppola, A.; Simone, C.D.; Palmieri, N.M.; Coppola, D.; Lanza, F.; Ruosi, C.; Amoriello, A.; Di Minno, G. Recombinant activated factor VII for hemostatic cover of orthopedic interventions in a girl with thrombocytopenia with absent radii syndrome. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2007, 18, 199–201. [Google Scholar] [CrossRef]
- Tefre, K.L.; Ingerslev, J.; Sorensen, B. Clinical benefit of recombinant factor VIIa in management of bleeds and surgery in two brothers suffering from the Bernard-Soulier syndrome. Haemoph. Off. J. World Fed. Hemoph. 2009, 15, 281–284. [Google Scholar] [CrossRef]
- Lee, A.; Poon, M.C. Inherited platelet functional disorders: General principles and practical aspects of management. Transfus. Apher. Sci. Off. J. World Apher. Assoc. Off. J. Eur. Soc. Haemapheresis 2018. [Google Scholar] [CrossRef]
- Lee, R.H.; Piatt, R.; Dhenge, A.; Lozano, M.L.; Palma-Barqueros, V.; Rivera, J.; Bergmeier, W. Impaired hemostatic activity of healthy transfused platelets in inherited and acquired platelet disorders: Mechanisms and implications. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Dupuis, A.; Gachet, C. Inherited platelet disorders: Management of the bleeding risk. Transfus. Clin. Biol. J. Soc. Fr. Transfus. Sang. 2018. [Google Scholar] [CrossRef]
- Zaninetti, C.; Gresele, P.; Bertomoro, A.; Klersy, C.; De Candia, E.; Veneri, D.; Barozzi, S.; Fierro, T.; Alberelli, M.A.; Musella, V.; et al. Eltrombopag for the treatment of inherited thrombocytopenias: A phase II clinical trial. Haematologica 2020, 105, 820–828. [Google Scholar] [CrossRef]
- Pecci, A. Diagnosis and treatment of inherited thrombocytopenias. Clin. Genet. 2016, 89, 141–153. [Google Scholar] [CrossRef]
- Pecci, A.; Verver, E.J.; Schlegel, N.; Canzi, P.; Boccio, C.M.; Platokouki, H.; Krause, E.; Benazzo, M.; Topsakal, V.; Greinacher, A. Cochlear implantation is safe and effective in patients with MYH9-related disease. Orphanet J. Rare Dis. 2014, 9, 100. [Google Scholar] [CrossRef]
- Cid, A.R.; Montesinos, P.; Sanchez-Guiu, I.; Haya, S.; Lorenzo, J.I.; Sanz, J.; Moscardo, F.; Puig, N.; Planelles, D.; Bonanad, S.; et al. Allogeneic hematopoietic cell transplantation in an adult patient with Glanzmann thrombasthenia. Clin. Case Rep. 2017, 5, 1887–1890. [Google Scholar] [CrossRef]
- Woods, G.; Bajwa, R.P.; Rose, M.J. Reduced intensity transplantation for congenital amegakaryocytic thrombocytopenia: Report of a case and review of the literature. Pediatric Transplant. 2014, 18, E31–E34. [Google Scholar] [CrossRef] [PubMed]
- Moratto, D.; Giliani, S.; Bonfim, C.; Mazzolari, E.; Fischer, A.; Ochs, H.D.; Cant, A.J.; Thrasher, A.J.; Cowan, M.J.; Albert, M.H.; et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: An international collaborative study. Blood 2011, 118, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, L.M.; Petrovic, A.; Brazauskas, R.; Liu, X.; Griffith, L.M.; Ochs, H.D.; Bleesing, J.J.; Edwards, S.; Dvorak, C.C.; Chaudhury, S.; et al. Excellent outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome: A PIDTC report. Blood 2020, 135, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Ferrua, F.; Cicalese, M.P.; Galimberti, S.; Giannelli, S.; Dionisio, F.; Barzaghi, F.; Migliavacca, M.; Bernardo, M.E.; Calbi, V.; Assanelli, A.A.; et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: Interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet. Haematol. 2019, 6, e239–e253. [Google Scholar] [CrossRef]
- Hacein-Bey Abina, S.; Gaspar, H.B.; Blondeau, J.; Caccavelli, L.; Charrier, S.; Buckland, K.; Picard, C.; Six, E.; Himoudi, N.; Gilmour, K.; et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA 2015, 313, 1550–1563. [Google Scholar] [CrossRef]
- Morris, E.C.; Fox, T.; Chakraverty, R.; Tendeiro, R.; Snell, K.; Rivat, C.; Grace, S.; Gilmour, K.; Workman, S.; Buckland, K.; et al. Gene therapy for Wiskott-Aldrich syndrome in a severely affected adult. Blood 2017, 130, 1327–1335. [Google Scholar] [CrossRef]
- Sereni, L.; Castiello, M.C.; Di Silvestre, D.; Della Valle, P.; Brombin, C.; Ferrua, F.; Cicalese, M.P.; Pozzi, L.; Migliavacca, M.; Bernardo, M.E.; et al. Lentiviral gene therapy corrects platelet phenotype and function in patients with Wiskott-Aldrich syndrome. J. Allergy Clin. Immunol. 2019, 144, 825–838. [Google Scholar] [CrossRef]
- Wilcox, D.A. Megakaryocyte- and megakaryocyte precursor-related gene therapies. Blood 2016, 127, 1260–1268. [Google Scholar] [CrossRef]
- Fang, J.; Hodivala-Dilke, K.; Johnson, B.D.; Du, L.M.; Hynes, R.O.; White, G.C., 2nd; Wilcox, D.A. Therapeutic expression of the platelet-specific integrin, alphaIIbbeta3, in a murine model for Glanzmann thrombasthenia. Blood 2005, 106, 2671–2679. [Google Scholar] [CrossRef]
- Fang, J.; Jensen, E.S.; Boudreaux, M.K.; Du, L.M.; Hawkins, T.B.; Koukouritaki, S.B.; Cornetta, K.; Wilcox, D.A. Platelet gene therapy improves hemostatic function for integrin alphaIIbbeta3-deficient dogs. Proc. Natl. Acad. Sci. USA 2011, 108, 9583–9588. [Google Scholar] [CrossRef]
- Wilcox, D.A.; White, G.C., 2nd. Gene therapy for platelet disorders: Studies with Glanzmann’s thrombasthenia. J. Thromb. Haemost. JTH 2003, 1, 2300–2311. [Google Scholar] [CrossRef]
- Kanaji, S.; Kuether, E.L.; Fahs, S.A.; Schroeder, J.A.; Ware, J.; Montgomery, R.R.; Shi, Q. Correction of murine Bernard-Soulier syndrome by lentivirus-mediated gene therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 625–632. [Google Scholar] [CrossRef]
- Cleyrat, C.; Girard, R.; Choi, E.H.; Jeziorski, E.; Lavabre-Bertrand, T.; Hermouet, S.; Carillo, S.; Wilson, B.S. Gene editing rescue of a novel MPL mutant associated with congenital amegakaryocytic thrombocytopenia. Blood Adv. 2017, 1, 1815–1826. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease (OMIM Code) | Inh. | Gene | Type Tcp/S/HM | Clinical & Laboratory Phenotype in Most Reported Cases | Ref. |
|---|---|---|---|---|---|
| INHERITED THROMBOCYTOPENIAS WITH NORMAL PLATELET SIZE | |||||
| Congenital amegakaryocytic thrombocytopenia (CAMT) (604498) | AR | MPL | S | Very severe neonatal thrombocytopenia, amegakaryocytic; progression to aplastic anemia in childhood. Severe bleeding tendency. | [9,10] |
| THPO-related thrombocytopenia | AD/AR | THPO | S | Mono-allelic mutations are associated with mild thrombocytopenia. Bialellic mutations resemble CAMT. It does not respond to allo-HSCTH but it responds to Romiplostim. Severe bleeding tendency. | [11,12,13] |
| Radioulnar synostosis with amegakaryocytic thrombocytopenia 1 (RUSAT1, 605432) | AD | HOXA11 | S | Severe neonatal thrombocytopenia; reduced/absent megakaryocytes. Possible evolution to aplastic anemia in childhood. Radius and ulna synostosis with/without other skeletal alterations; probable sensorineural hearing loss. Severe bleeding tendency. | [5,14] |
| MECOM-related thrombocytopenia (including amegakaryocytic thrombocytopenia 2 with Radioulnar synostosis, (RUSAT2) (616738) | AD | MECOM | S | Severe neonatal thrombocytopenia. Reduced/absent megakaryocytes and/or hyporegenerative anemia. Radius and ulna synostosis with/without other skeletal alterations; B-cell deficiency. Possible renal or cardiac malformations and probable sensorineural hearing loss. Severe bleeding tendency. | [15,16] |
| Thrombocytopenia with absent radii (TAR) (274000) | AR | RBM8A (Microdeletion & rs139428292/ rs201779890) | S | Moderate-severe central neonatal thrombocytopenia that improves with age; bilateral absence of radius with or without other skeletal abnormalities. Potential kidney, cardiac or central nervous system anomalies. Severe bleeding tendency. | [17] |
| Familial platelet disorder with propensity to acute myelogenous leukemia (FPD-AML) (601399) | AD | RUNX1 | HM | Mild-moderate neonatal thrombocytopenia. Platelet function defect “Aspirin-like”. Platelet granule deficiency. High risk (>40%) of acute myeloblastic leukemia or myelodysplastic syndrome at a young age; increases risk or lymphoblastic leukemia and solid tumor. Absent to moderate bleeding tendency. | [18,19] |
| ANKRD26-related thrombocytopenia (thrombocytopenia Type-2) (188000) | AD | ANKRD26 | HM | Mild-moderate neonatal thrombocytopenia. Some patients with high levels of hemoglobin and/or leukocytosis. Approximately 10% of patients acquire myeloid neoplasms. Absent to mild bleeding tendency. | [18,20,21] |
| ETV6-related thrombocytopenia (thrombocytopenia Type-5) (616216) | AD | ETV6 | HM | Red blood cells with high mean corpuscular volume. Platelets may show elongated α granules, impaired spreading and clot retraction. High number of circulating CD34 + cells. Predisposition (30%) to acquired lymphoid, myeloid, and myeloproliferative syndromes. Absent to mild bleeding tendency. | [18,22,23] |
| IKZF5-related thrombocytopenia | AD | IKZF5 | Tcp | Mild to moderate thrombocytopenia. Platelets with fewer α and δ granules. No bleeding tendency. | [24] |
| CYCS-related thrombocytopenia (thrombocytopenia type 4) (612004) | AD | CYCS | Tcp | Mild to moderate thrombocytopenia. May also present with small platelets. No bleeding tendency. | [25,26] |
| KDSR-related thrombocytopenia | AR | KDSR | S | Early severe thrombocytopenia (although the platelet count may be normal at birth). Platelet secretion and activation defect. Variable hyperkeratosis from palmoplantar and anogenital hyperkeratosis/erythema to a Harlequin ichthyosis. Moderate to severe bleeding tendency. | [27,28] |
| Stormorken syndrome (185070) | AD | STIM1 ORAI1 | S | Moderate thrombocytopenia. High systemic platelet binding of annexin V. Abnormal clot formation and retraction; asplenia, mild anemia, myopathy with tubular aggregates, congenital miosis, ichthyosis, short stature, migraine and mild cognitive impairment. Mild bleeding tendency. | [26,29] |
| York platelet syndrome | AD | STIM1 | S | More rare than Stormorken syndrome. Moderate to severe thrombocytopenia. Myopathy with rimmed vacuoles; Platelets with fewer α and δ granules; giant dense bodies. Mild bleeding tendency. | [30] |
| INHERITED THROMBOCYTOPENIAS WITH LARGE PLATELETS | |||||
| Bernard-Soulier syndrome (BSS) (231200] | AR | GP1BA, GP1BB, GP9 | Tcp | Moderate to severe thrombocytopenia and giant platelets. Absence of platelet aggregation with ristocetin, not restored with plasma, due to absence of severe dysfunction of the main vWF platelet receptor Ibα/IX. Normal/reduced response to other agonists. Defect of adhesion to FVW. Severe bleeding tendency. | [31,32] |
| Bernard-Soulier syndrome, monoallelic form (mBSS) (153670) (previously referred to as Mediterranean thrombocytopenia) | AD | GP1BA, GP1BB | Tcp | Mild to moderate thrombocytopenia. Absent to mild bleeding tendency. | [33,34,35] |
| Platelet-type von Willebrand disease (PTvWD) (177820) | AD | GP1BA | Tcp | Mild to severe thrombocytopenia. Platelet count can be normal but decrease under stress conditions. Platelet aggregates in blood smear. Abnormally high platelet VWF binding and hyperaggregation with low-dose ristocetin due to gain-of–function variants in GPIbα. Aggregation of washed platelets inducible with cryoprecipitate (not in VWD type 2B). Absence of high molecular weight multimers (as in VWD type 2B). Absent to mild bleeding tendency. | [36] |
| ITGA2B/ITGB3-related thrombocytopenia (187800) | AD | ITGA2B, ITGB3 | Tcp | Mild to moderate thrombocytopenia. Defective expression and αIIbβ3 activation. Moderate bleeding tendency. Some monoallelic variants in ITGA2B/ ITGB3 mutations cause constitutive activation of integrin αIIbβ3 with a phenotype similar to variant Glanzmann’s thrombasthenia. | [37,38,39] |
| DiGeorge/velocardiofacial syndrome | AD | Deletions in 22q11.2 (TBX1/GP1BB, deletions) | Yes | Moderate thrombocytopenia. Heart abnormalities, parathyroid, and thymus insufficiency, cognitive delay, facial dysmorphia. Most patients have developmental differences. Mild to severe bleeding tendency. Bleeding diathesis can be contributed not only by platelet dysfunction (absent in some cases) but also by other alterations such as velopharyngeal insufficiency, chronic nasal irritation, nasal allergies, or abnormal vWF multimers. | [40] |
| Jacobsen syndrome (147791), Paris-Trousseau thrombocytopenia (188025) | AD | Deletions in 11q23 | S | Moderate to severe thrombocytopenia; may improve over time. Cardiac, facial, urinary tract, kidney, or central nervous system malformations; mental retardation; giant platelet granules. Mild to severe bleeding tendency. | [41,42] |
| FLI1-related thrombocytopenia, (61744) | AD/AR | FLI1 | Tcp | Mild to moderate thrombocytopenia. Large platelet granules. Absent to moderate bleeding tendency (monoallelic and biallelic forms, respectively). | [43,44] |
| GATA1-related disorders (314050; 300367) | XL | GATA1 | S | Mild to severe thrombocytopenia. Can associate with dyserythropoiesis with or without anemia, β-thalassemia, neutropenia, splenomegaly or congenital erythropoietic porphyria); Dysplastic megakaryocytes. Platelets granule deficiency and functional defect. A very rare form associates with blood group Lu a- b- (Lu null). Mild to severe bleeding tendency. | [45,46] |
| GFI1b-related thrombocytopenia (187900) | AD/AR | GFI1B | Tcp | Mild to severe thrombocytopenia (monoallelic & biallelic forms). Red blood cells with anisopoikilocytosis, dysplastic megakaryocytes, emperipolesis. Platelets with α/β granule deficiency and aggregation defect. CD34+ abnormal expression in platelets. Absent to severe bleeding tendency (monoallelic & biallelic forms). | [47,48,49] |
| Gray platelet syndrome (GPS) (139090) | AR | NBEAL2 | S | Moderate to severe thrombocytopenia with typically grayish platelets. Could aggravate over time. Selective absence of α-granules demonstrable by electron microscopy; defect of α-granular proteins by biochemical assays (β-TG, PDGF, vWF, etc.). Impaired platelet function with weak agonists. In some patients, selective defects of GPVI and activation by collagen. Elevated vitamin B12 serum level. Early myelofibrosis; occasional splenomegaly; predisposition to autoimmune diseases. Moderate to severe bleeding tendency. | [50,51,52,53] |
| G6b-B-related Thrombocytopenia (617441) | AR | MPIG6B | S | Mild to severe thrombocytopenia; atypical megakaryocytes. Microcytic anemia, leukocytosis, myelofibrosis. Mild to moderate bleeding tendency. | [54] |
| MYH9-related disease, (MYH9-RD) (155100) | AD | MYH9 | S | Mild to severe thrombocytopenia; In some cases, basophilic neutrophilic inclusions (Döhle bodies), hearing loss, kidney disease, liver disease, cataracts; Strong genotype–phenotype relationship. Absent to mild bleeding tendency. | [10,41,42,55,56] |
| Thrombocytopenia associated with sitosterolemia | AR | ABCG5, ABCG8 | S | Mild to moderate thrombocytopenia. Elevated plasma levels of plant sterols. Xanthomas in tendons and xanthelasmas. Premature atherosclerosis; hemolytic anemia with stomatocytosis. Also non-syndromic picture. Mild to moderate bleeding tendency. | [57,58] |
| FLNA-related thrombocytopenia | X-linked | FLNA | S | Moderate thrombocytopenia. Nodular periventricular heterotopia; skeletal malformations, mental retardation, heart valve dystrophy, intestinal obstruction, bone dysplasia. May associate with Ehlers-Danlos syndrome; May also be nonsyndromic thrombocytopenia. Mild bleeding tendency. | [59,60,61] |
| TUBB1-related thrombocytopenia (613112) | AD | TUBB1 | Tcp | Mild to moderate thrombocytopenia. In some patients, normal platelet count with increased platelet size. Absent to mild bleeding tendency. | [26,62,63] |
| ACTN1-related thrombocytopenia (615193) | AD | ACTN1 | Tcp | Thrombocytopenia and moderate platelet dysfunction. Mild to moderate thrombocytopenia. Some giant platelets can be seen. Absent to mild bleeding tendency. | [26,64,65] |
| DIAPH1-related thrombocytopenia | AD | DIAPH1 | S | Mild thrombocytopenia (occasionally normal platelet counts); moderate fluctuating neutropenia with low to moderate infectious risk; early sensorineural deafness. Absent to mild bleeding tendency. | [66] |
| SLFN14-related thrombocytopenia (616913) | AD | SLFN14 | Tcp | Mild to moderate thrombocytopenia. δ granule secretion defect. Increased number of immature platelets. Moderate to severe bleeding tendency. | [67,68] |
| SRC-related thrombocytopenia (SRC-RT) (thrombocytopenia type-6) (616937) | AD | SRC | S | Mild to severe thrombocytopenia. Platelet granule defect, myelofibrosis and early edentulism, facial dysmorphia. High number of megakaryocytes, with immature features such as hypolobulated nuclei. Mild to severe bleeding tendency. | [69,70,71] |
| TPM4-related thrombocytopenia | AD | TPM4 | Tcp | Mild thrombocytopenia. Absent to mild bleeding tendency. | [72] |
| TRPM7-related thrombocytopenia (TRPM7-RT) | AD | TRPM7 | Tcp | Moderate thrombocytopenia. Absent to mild bleeding tendency | [73] |
| PRKACG-related thrombocytopenia (PRKACG-RT) (616176) | AR | PRKACG | Tcp | Severe macrothrombocytopenia. PKA-dependent protein phosphorylation defect (filamin A, GPIbβ). Severe bleeding tendency. | [74] |
| Takenouchi-Kosaki syndrome with macrothrombocytopenia (616737) | AD | CDC42 | S | Moderate thrombocytopenia. Defective intellectual, growth, and psychomotor development. Possible brain, facial/ muscle/skeletal abnormalities. Also, immunodeficiency, eczema, hearing/visual disability, lymphedema, and cardiac or genitourinary malformations. No bleeding tendency. | [75,76] |
| GNE-related thrombocytopenia | AR | GNE | S | Severe thrombocytopenia. Myopathy with rimmed vacuoles since early adulthood. Isolated thrombocytopenia some patients. Increased reticulated platelets, suggesting accelerated platelet clearance. Mild to severe bleeding tendency. | [77,78,79] |
| GALE-related thrombocytopenia | AR | GALE | S | Severe thrombocytopenia. Galactosemia, anemia and febrile neutropenia. Severe bleeding tendency. | [78,80] |
| SLC35A1- related thrombocytopenia | AR | SLC35A1 | S | Moderate thrombocytopenia. Impaired psychomotor development, epilepsy, ataxia, microcephaly, and choreiform movements. Mild bleeding tendency. | [26,81] |
| ACTB- related thrombocytopenia | AD | ACTB | S | Mild to moderate thrombocytopenia. Leucocytosis with eosinophilia or leukopenia. Other possible features are microcephaly, minor facial anomalies, developmental delay, and mild intellectual disability. No bleeding tendency. | [82] |
| INHERITED THROMBOCYTOPENIAS WITH SMALL PLATELETS | |||||
| Wiskott-Aldrich syndrome (WAS)(301000) | XL | WAS | S | Severe thrombocytopenia. Immunodeficiency, eczema, lymphoproliferative, and autoimmune disorders. Severe bleeding tendency. | [83,84] |
| X-linked thrombocytopenia (XLT or thrombocytopenia type 1) (313900) | XL | WAS | S | Moderate to severe thrombocytopenia. Mild form of Wiskott-Aldrich. Possible immunodeficiency and mild eczema. Increased risk of malignancy and autoimmune disorders. Absent to moderate bleeding tendency. | [83,84] |
| FYB-related thrombocytopenia (ADAP-related thrombocytopenia or (thrombocytopenia type 3) (273900) | AR | FYB | TCP | Moderate to severe thrombocytopenia. Baseline platelet hyperactivation. Impaired activation of αIIbβ3. Mild to moderate bleeding tendency. | [85,86] |
| ARCP1B-related thrombocytopenia (617718) | AR | ARCP1B | S | Moderate to severe thrombocytopenia. Normal platelet count in some cases. Eosinophilia, immune-mediated inflammatory disease, eczema, lymphadenopathy, hepato-splenomegaly. Impaired growth. Moderate bleeding tendency. | [87,88] |
| PTPRJ-related thrombocytopenia (CD148-related thrombocytopenia) (PTPRJ-RT, na) | AR | PTPRJ | Tcp | Moderate to severe thrombocytopenia. So far, a single pedigree with PTPRJ-RT has been identified. Impaired platelet reactivity with GPVI agonists (collagen, CRP, convulxin). SRC-type kinase activation defect. Megakaryocyte maturation defect. Mild to moderate bleeding tendency. | [89] |
| Receptor | Disease (OMIM Code) | Inh. | Genes | Clinical & Laboratory Phenotype in Most Reported Cases | Ref. |
|---|---|---|---|---|---|
| Ib/IX/V | Bernard-Soulier Syndrome (SBS) (231200) | AR | GP1BA, GP1BB, GP9 | See Table 1. | - |
| Platelet Type von Willebrand Disease (PT-vWD) (177820) | AD | GP1BA | See Table 1. | - | |
| αIIbβ3 | Glanzmann Thrombasthenia (GT) (273800) | AR | ITGA2B, ITGB3 | Normal platelet count and morphology. Absent or severely reduced LTA with all agonists (ADP, TxA2, collagen, thrombin). LTA with ristocetin normal or 2nd wave reduced. Absent or severely reduced clot retraction. Absence or decreased αIIbβ3 expression demonstrable by flow cytometry: Type I <5%; Type II 10–20%; Variant GT with even >50% non-functional αIIbβ3. Severe bleeding tendency. | [26,90,91,92] |
| P2Y12 | ADP receptor deficiency (609821) | AD/AR | P2RY12 | Normal platelet count and morphology. LTA greatly decreased with ADP. With other agonists normal or reduced second wave. Absent ADP inhibition of PGE1-induced cAMP synthesis. Defective VASP de-phosphorylation with ADP. Mild to moderate bleeding tendency. Bleeding mostly associated with antiplatelet intake, dental interventions, or other hemostatic challenges. | [8,93,94] |
| TxA2 –R (TPα) | TxA2 receptor deficiency (614009) | AD/AR | TBXA2R | Normal platelet count and morphology. Absent or severely reduced LTA with arachidonic acid or TxA2 analogs such as U46619 or STA2. Normal with other agonists or reduced second wave. Mild to moderate bleeding tendency. Bleeding mostly associated with antiplatelet intake, dental interventions, or other hemostatic challenges. Post- surgery bleeding. | [8,95] |
| GPVI | GPVI collagen receptor defect (614201) | AR | GP6 | Normal platelet count and morphology. Absent or severely reduced LTA with collagen and GPVI agonists such a as collagen related-peptide or convulxin. Also defective platelet secretion and protein tyrosine phosphorylation with GPVI agonists Mild to moderate bleeding tendency. Bleeding mostly associated with antiplatelet intake, dental interventions, or other hemostatic challenges. | [8,96,97] |
| EPHB2 | Ephrin type-B receptor 2 defect (618262) | AR | EPHB2 | Normal platelet count and morphology. Impaired LTA and secretion with multiple agonists. Impaired GPVI pathway signaling and inside-out αIIbβ3 signaling. Moderate to severe bleeding tendency. | [98] |
| Granule | Disease (OMIM code) | Inh. | Genes | Clinical & Laboratory Phenotype in Most Reported Cases | Ref. |
|---|---|---|---|---|---|
| α and δ | Idiopathic granule deficiency | AR/AD | Nc | Platelet count normal or slightly decreased. Normal platelet morphology δ and α -granule defect by electron microscopy. Reduced LTA and/or absence of second aggregation wave with weak/low dose agonists (ADP, epinephrine, collagen). Defect of granular protein release by flow cytometry. Absent or very moderate hemorrhagic symptoms, associated with situations of high hemorrhagic risk. | [50,99] |
| α | Grey Platelet Syndrome (GPS) (139090) | AR AD | NBEAL2 GFI1B | See Table 1. | |
| Quebec Syndrome (QS) (601709) | AD | PLAU | Moderate thrombocytopenia and normal morphology. LTA absent or reduced with epinephrine and normal with other agonists. Defect of procoagulant platelet activity and increased fibrinolytic activity α-granule protein defect by flow cytometry. (P-selectin, factor V). Mucocutaneous, visceral and/or post-surgery bleeding. Response to anti-fibrinolytics but lack of response to platelet transfusions. | [100,101] | |
| δ | Hermansky-Pudlak Syndrome (HPS) (203300, 608233, 614072, 614073, 614074, 614075, 614076, 614077, 614171, 617050, 619172) | AR | HPS1, HPS2-[ AP3B1], HPS3, HPS4, HPS5, HPS6, HPS7-[ DTNBP1] HPS8-[ BLOC1S3] HPS9-[ BLOC1S] HPS10-[ AP3D1], HPS11 [BLOC1S5] | Normal platelet count and morphology. Selective δ-granule defect by electron microscopy. Reduced LTA and/or absence of second wave with weak/low dose agonists (ADP, epinephrine, collagen). Radio-labelled serotonin uptake, mepacrine uptake, and CD63 release defects by flow cytometry. Oculocutaneous albinism; accumulation of lipofuscin-like ceroid material in cells of the phagocytic mononuclear system; neutropenia, immunodeficiency, pulmonary fibrosis, granulomatous colitis depending on the subtype. Genotype–phenotype relationship. Mild to moderate bleeding tendency. | [50,99,102,103,104,105] |
| Chediak-Higashi Syndrome (CHS) (214500) | AR | LYST | Normal platelet count and morphology. δ-granule defect by electron microscopy. Reduced LTA and/or absence of second aggregation wave with weak/low dose agonists (ADP, epinephrine, collagen). Impaired uptake of radioactively labeled serotonin or mepacrine, and defect in CD63 release by flow cytometry. Oculocutaneous albinism; Immunodeficiency with predisposition to recurrent infections. In 85% of cases evolution to hemophagocytic lymphohistiocytosis with an accelerated phase. A juvenile milder form can be caused by missense variants maintaining Lyst expression/function. Genotype-Phenotype relationship. Mild to moderate bleeding tendency. | [50,99,102,106] | |
| Griscelli Syndromes (214450, 607624, 609227) | AR | RAB27 MYO5A MLPH | Normal platelet count and morphology. δ-granule defect as in HPS and CHS. Variable neutropenia. Albinism, silver hair, neurological defects, lymphohistiocytosis, decreased cytotoxic function of NK cells and T-lymphocytes, according to subtype. | [50,99,107] | |
| Delta-storage pool disease | AD/AR | - | Normal platelet numbers. Reduced second phase of aggregation with weak/low dose agonists (ADP, epinephrine, collagen). Absence of δ-granules in electron microscopy. Absent or mild bleeding tendency. | [50,99] |
| Affected Element | Disease (OMIM Code) | Inh. | Genes | Clinical & Laboratory Phenotype in Most Reported Cases | Ref. |
|---|---|---|---|---|---|
| Enzyme activity/signaling pathways | Defect in TxA2 pathways (176805, 231095, 600522) | AR/AD | PTGS1 TBXAS1 PLA2G4A | Normal platelet count and morphology. Occasionally moderate thrombocytopenia. Selectively reduced LTA with arachidonic acid and normal with TxA2 analogs such as U46619. Reduced with weak/low dose agonists (ADP, epinephrine, collagen). Serum TxA2 generation defect. Heterozygous variant in PTGS1 affecting N-glycosylation may have dominant negative effect. Osteoporosis is a feature in Ghosal syndrome (TBXAS1 defect); recurrent gastrointestinal ulceration may associate to PLA2G4A defect. Moderate or absent bleeding symptoms; usually associated with high bleeding risk situations. | [8,108,109,110] |
| G protein defects (Gi, Gq, Gs, Gz) | - | GNAS GNAQ GNAZ GNAI1 | Normal platelet count and morphology. Gs hyperfunction may be associated with discrete macrothrombocytopenia and neurological disorders. Reduced LTA and secretion in response to multiple soluble agonists. Moderate hemorrhagic symptoms; mainly associated with hemostatic challenges. | [8,111,112] | |
| CalDAG-GEF1 defect (615888) | AR | RASGRP2 | Normal platelet count and morphology. LTA and secretion defects with weak agonists. αIIbβ3 integrin activation defects with weak agonists (fibrinogen binding deficit, PAC-1). Normal or moderately reduced response to PMA. | [113,114,115] | |
| LAD-III Syndrome (Kindlin-3 deficiency) (612840) | AR | FERMT3 | Leukocyte adhesion deficiency syndrome type 3 (LADIII): infections, poor healing, osteopetrosis. Severe bleeding tendency, even more severe than Glanzmann thrombasthenia. | [116,117] | |
| Procoagulant Activity | Scott Syndrome (262890) | AR | ANO6 [TREM16F] | Normal platelet count and morphology. Decreased agonist-induced generation of microparticles and annexin V platelet binding, demonstrable with flow cytometry. Abnormal clot formation and retraction. Moderate to severe bleeding; mainly in risk situations (childbirth, dental interventions). | [8,118] |
| Stormorken Syndrome (185070) | AD | STIM1 ORAI1 | See Table 1. |
| Value of Commonly Used Assays in the Diagnosis of IPD (0: Low; 1: Reasonable; 2: High)# | Basic Panel | Extended Panel (Confirmation/Characterization) |
|---|---|---|
| Complete blood counts and blood smear: 2 Bleeding time: 0 PFA-100: 1 Thromboelastography: 0 Platelet aggregation: 2 Lumiaggregometry: 2 Flow cytometry: 2 Electron microscopy: 2 Molecular analysis: 2 | Complete blood counts (automatic) Blood smear (platelet count and morphology, inclusion bodies in neutrophils, abnormalities in other blood cells) Basic coagulation and VWF study Platelet aggregation: basic panel of agonists: PAR-1 peptide (TRAP), ADP, arachidonic acid, epinephrine, collagen, ristocetin. Flow cytometric analysis of major platelet adhesive glycoproteins (GP Ib/IX; GP IIb/IIIa; GP Ia/IIa, GPVI) | Immunostaining of platelet proteins in blood smears. Platelet secretion: ATP release by lumiaggregometry; HPLC; uptake/release of serotonin (HPLC/ radiolabelled); release of CD62/CD63 by flow cytometry; release of granular proteins (PF4. Thrombospondin-1, VWF, P-selectin,) by ELISA. Agonist-induced activation of αIIbβ3 by flow cytometry (Binding of PAC-1 or fibrinogen labeled with fluorochromes). Clot retraction assay. Platelet aggregation: basic panel of agonists: PAR-4, collagen related peptide [CRP], convulxin, PMA, U46619, A23188; aggregation inhibition test with PGE1 or PGI2. Extended panel of platelet receptor expression (ADP receptors, TxA2, thrombin; CD31) by flow cytometry with selective antibody or ligand binding assays. Procoagulant activity by cytometry (annexin V binding). Measurement of second messengers (Ca2 +, IP3, cAMP, TxA2) by cytometry or biochemical assays. Assessment of signaling cascades by measuring phosphorylation of specific proteins by western blot or flow cytometry (VASP test). Platelet adhesion and spreading on adhesive surfaces (static and/or low flow). Platelet morphology/ultrastructure (electron microscopy, immunofluorescence microscopy, confocal). Molecular analysis (first-line HTS with a panel of selected genes; negative cases in HTS-gene panel can be analyzed by complete whole exome sequencing [WES) and/or complete genome sequencing [WGS]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma-Barqueros, V.; Revilla, N.; Sánchez, A.; Zamora Cánovas, A.; Rodriguez-Alén, A.; Marín-Quílez, A.; González-Porras, J.R.; Vicente, V.; Lozano, M.L.; Bastida, J.M.; et al. Inherited Platelet Disorders: An Updated Overview. Int. J. Mol. Sci. 2021, 22, 4521. https://doi.org/10.3390/ijms22094521
Palma-Barqueros V, Revilla N, Sánchez A, Zamora Cánovas A, Rodriguez-Alén A, Marín-Quílez A, González-Porras JR, Vicente V, Lozano ML, Bastida JM, et al. Inherited Platelet Disorders: An Updated Overview. International Journal of Molecular Sciences. 2021; 22(9):4521. https://doi.org/10.3390/ijms22094521
Chicago/Turabian StylePalma-Barqueros, Verónica, Nuria Revilla, Ana Sánchez, Ana Zamora Cánovas, Agustín Rodriguez-Alén, Ana Marín-Quílez, José Ramón González-Porras, Vicente Vicente, María Luisa Lozano, José María Bastida, and et al. 2021. "Inherited Platelet Disorders: An Updated Overview" International Journal of Molecular Sciences 22, no. 9: 4521. https://doi.org/10.3390/ijms22094521
APA StylePalma-Barqueros, V., Revilla, N., Sánchez, A., Zamora Cánovas, A., Rodriguez-Alén, A., Marín-Quílez, A., González-Porras, J. R., Vicente, V., Lozano, M. L., Bastida, J. M., & Rivera, J. (2021). Inherited Platelet Disorders: An Updated Overview. International Journal of Molecular Sciences, 22(9), 4521. https://doi.org/10.3390/ijms22094521