
An Update on Glioblastoma Biology, Genetics, and Current Therapies: Novel Inhibitors of the G Protein-Coupled Receptor CCR5


 ,
, 
Abstract

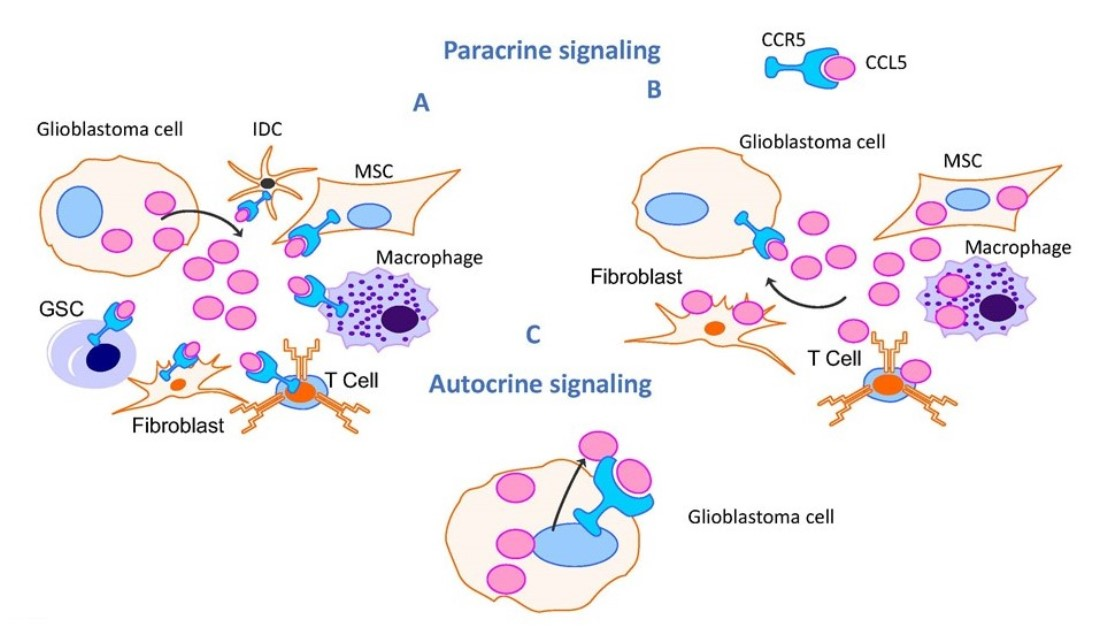{kind=link}
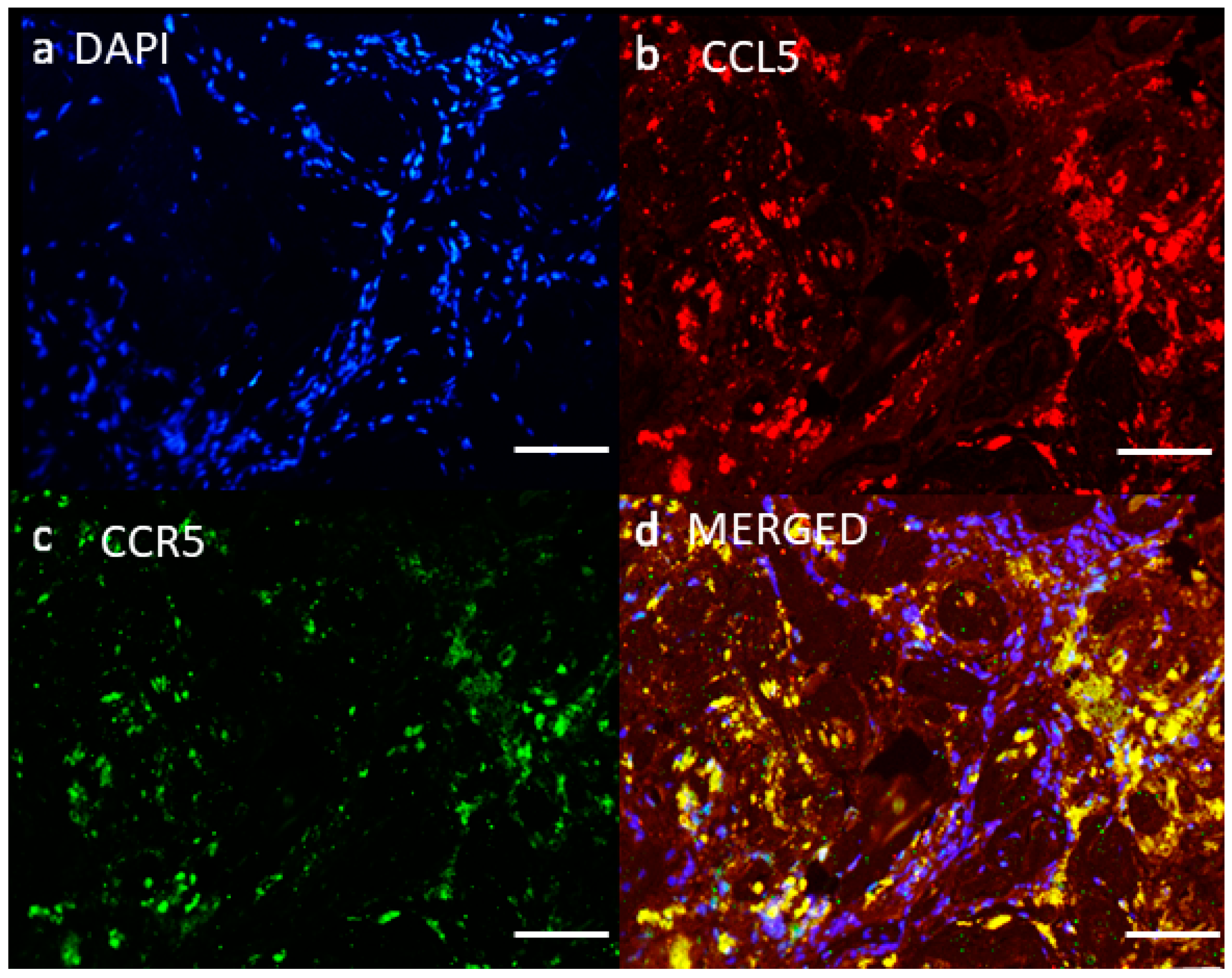{kind=link}
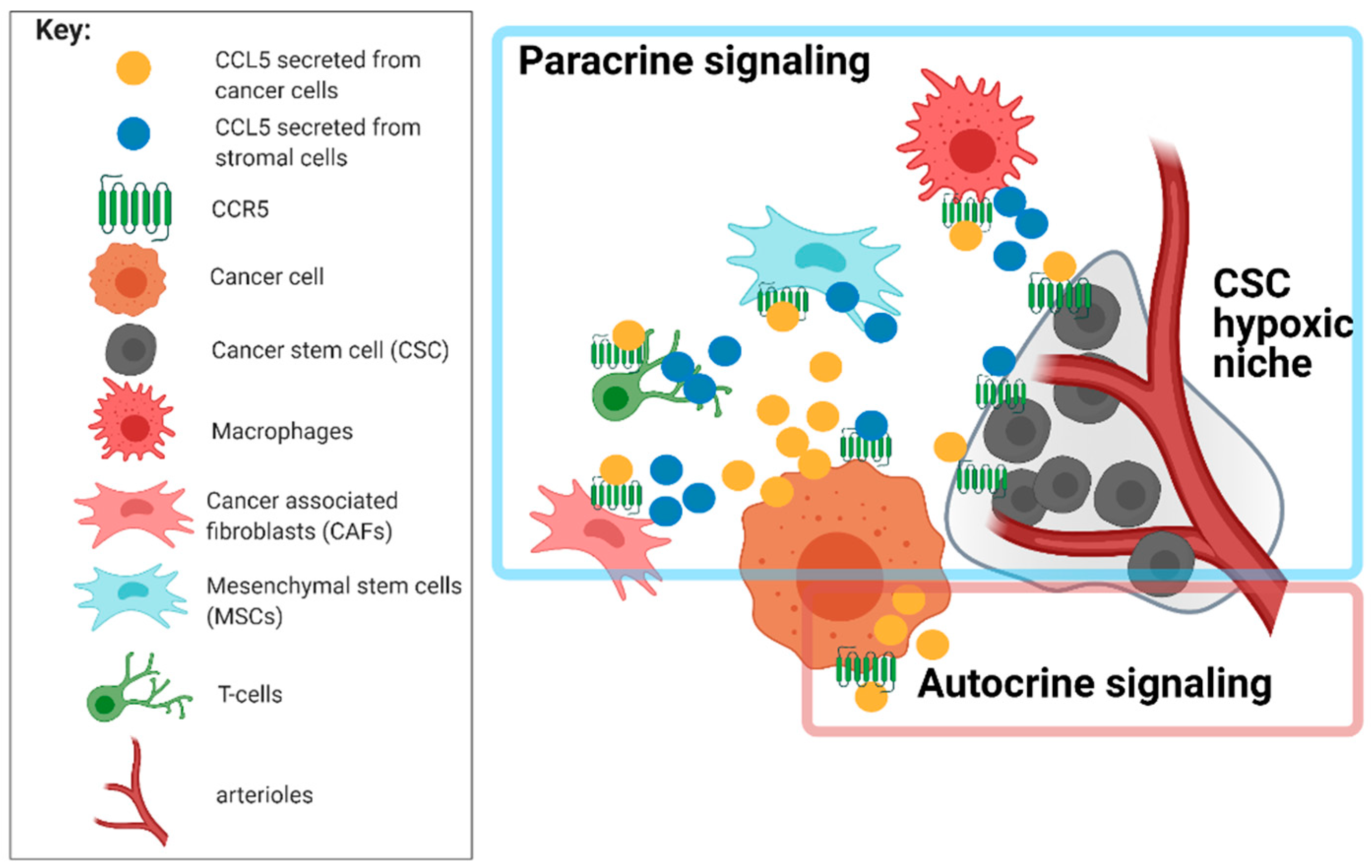{kind=link}
1. Introduction
2. Glioblastoma
2.1. Subtypes and Genetics and of Glioblastoma
2.2. Cancer Stem Cells in Glioblastoma
2.3. Glioblastoma Therapy
2.3.1. Targeting Mutations in Glioblastoma
2.3.2. Metabolic Targets in Glioblastoma
3. Glioblastoma Microenvironment (GME)
3.1. Glioblastoma Transitions Are Affected by Stromal Cells in Therapy
3.2. The Immune Glioblastoma Subtypes and Immunotherapy
4. Cellular Cross Talks by Chemokines and Their Receptors
4.1. Chemokine Receptor CCR5 and the LIGAND CCL5 in Cancers
4.2. Network of Receptor CCR5 and CCL5 Interactions in Glioblastoma
5. Targeting Chemokine–Receptor Axis
5.1. Targeting Glioblastoma Microenvironment and GSC Niches
5.2. CCR5 (CCR5i) Inhibitors as Adjuvant Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 1203–1217. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; Da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Antonio Chiocca, E.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro. Oncol. 2017, 19, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Stylli, S.S. Novel Treatment Strategies for Glioblastoma. Cancers (Basel) 2020, 12, 2883. [Google Scholar] [CrossRef]
- Koehler, A.; Karve, A.; Desai, P.; Arbiser, J.; Plas, D.R.; Qi, X.; Read, R.D.; Sasaki, A.T.; Gawali, V.S.; Toukam, D.K.; et al. Reuse of Molecules for Glioblastoma Therapy. Pharmaceuticals 2021, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent Advances targeting CCR5 for Cancer and its Role in Immuno-Oncology. Cancer Res. 2019, 79. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Koprivnikar Krajnc, M.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-Mediated Signaling Is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Xue, Y.; Lv, W.; Zhang, Y.; He, S. Critical roles of chemokine receptor CCR5 in regulating glioblastoma proliferation and invasion. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 890–898. [Google Scholar] [CrossRef]
- Kranjc, M.K.; Novak, M.; Pestell, R.G.; Lah, T.T. Cytokine CCL5 and receptor CCR5 axis in glioblastoma multiforme. Radiol. Oncol. 2019, 53, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.; Henshaw, D.L.; Lamburn, G.; O’Carroll, M.J. Brain tumours: Rise in glioblastoma multiforme incidence in England 1995-2015 Suggests an Adverse Environmental or Lifestyle Factor. J. Environ. Public Health 2018, 2018, 7910754. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. 2016, 529, 110–114. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Meir, E.G.V.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.; Wen, P.Y.; Olson, J.J. NIH Public Access. Cancer Journal, The 2010, 60, 166–193. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Shim, J.K.; Yoon, S.J.; Kim, S.H.; Chang, J.H.; Kang, S.G. Transcriptome profiling-based identification of prognostic subtypes and multi-omics signatures of glioblastoma. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (80-.) 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal Differentiation Mediated by NF-κB Promotes Radiation Resistance in Glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905.e7. [Google Scholar] [CrossRef]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma a randomized clinical trial. JAMA - J. Am. Med. Assoc. 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Verbovšek, U.; Van Noorden, C.J.F.; Lah, T.T. Complexity of cancer protease biology: Cathepsin K expression and function in cancer progression. Semin. Cancer Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Breznik, B.; Vittori, M.; Loncq de Jong, A.; Mlakar, J.; Oostra, R.J.; Khurshed, M.; Molenaar, R.J.; Lah, T.; Van Noorden, C.J.F. Similarities Between Stem Cell Niches in Glioblastoma and Bone Marrow: Rays of Hope for Novel Treatment Strategies. J. Histochem. Cytochem. 2019, 68. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Molenaar, R.J.; Breznik, B.; Lah, T.; Aronica, E.; Noorden, C.J.F. Van Immunohistochemical Detection of Neural Stem Cells and Glioblastoma Stem Cells in the Subventricular Zone of Glioblastoma Patients. J. Histochem. Cytochem. 2021. [Google Scholar] [CrossRef]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef] [PubMed]
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the Treatment of Glioblastoma: Multisystem Mechanisms of Therapeutic Resistance. World Neurosurg. 2018, 116, 505–517. [Google Scholar] [CrossRef]
- Motaln, H.; Koren, A.; Gruden, K.; Ramšak, Ž.; Schichor, C.; Lah, T.T. Heterogeneous glioblastoma cell cross-talk promotes phenotype alterations and enhanced drug resistance. Oncotarget 2015, 6, 40998–41017. [Google Scholar] [CrossRef]
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of glioblastoma (GBM) molecular classification methods. Semin. Cancer Biol. 2018, 53, 201–211. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Kazda, T.; Dziacky, A.; Burkon, P.; Pospisil, P.; Slavik, M.; Rehak, Z.; Jancalek, R.; Slampa, P.; Slaby, O.; Lakomy, R. Radiotherapy of glioblastoma 15 years after the landmark Stupp’s trial: More controversies than standards? Radiol. Oncol. 2018, 52, 121–128. [Google Scholar] [CrossRef]
- Cabrera, A.R.; Kirkpatrick, J.P.; Fiveash, J.B.; Shih, H.A.; Koay, E.J.; Lutz, S.; Petit, J.; Chao, S.T.; Brown, P.D.; Vogelbaum, M.; et al. Radiation therapy for glioblastoma: Executive summary of an American Society for Radiation Oncology Evidence-Based Clinical Practice Guideline. Pract. Radiat. Oncol. 2016, 6, 217–225. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.; Erridge, S.; Vogelbaum, M.; Nowak, A.; Sanson, M.; Brandes, A.; Wick, W.; Clement, P.; Baurain, J.-F.; Mason, W.; et al. ACTR-11. second interim and 1st molecular analysis of the eortc randomized phase iii intergroup catnon trial on concurrent and adjuvant temozolomide in anaplastic glioma without 1p/19q codeletion. Neuro. Oncol. 2019, 21, vi14. [Google Scholar] [CrossRef]
- Balana, C.; Vaz, M.A.; Sepúlveda, J.M.; Mesia, C.; Del Barco, S.; Pineda, E.; Muñoz-Langa, J.; Estival, A.; De Las Peñas, R.; Fuster, J.; et al. A phase II randomized, multicenter, open-label trial of continuing adjuvant temozolomide beyond 6 cycles in patients with glioblastoma (GEINO 14-01). Neuro. Oncol. 2020, 22, 1851–1861. [Google Scholar] [CrossRef]
- Herbener, V.J.; Burster, T.; Goreth, A.; Pruss, M.; Bandemer, H.v.; Baisch, T.; Fitzel, R.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; et al. Considering the experimental use of Temozolomide in glioblastoma research. Biomedicines 2020, 8, 151. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Peñaranda-Fajardo, N.M.; Meijer, C.; Liang, Y.; Dijkstra, B.M.; Aguirre-Gamboa, R.; Den Dunnen, W.F.A.; Kruyt, F.A.E. ER stress and UPR activation in glioblastoma: Identification of a noncanonical PERK mechanism regulating GBM stem cells through SOX2 modulation. Cell Death Dis. 2019. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Chemotherapy resistance explained through endoplasmic reticulum stress-dependent signaling. Cancers (Basel) 2019, 11, 338. [Google Scholar] [CrossRef]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef]
- Neagu, M.R.; Reardon, D.A. An Update on the Role of Immunotherapy and Vaccine Strategies for Primary Brain Tumors. Curr. Treat. Options Oncol. 2015, 16. [Google Scholar] [CrossRef]
- Salinas, R.D.; Durgin, J.S.; O’Rourke, D.M. Potential of Glioblastoma-Targeted Chimeric Antigen Receptor (CAR) T-Cell Therapy. CNS Drugs 2020, 34, 127–145. [Google Scholar] [CrossRef]
- Majc, B.; Novak, M.; Jerala, N.K.; Jewett, A.; Breznik, B. Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development. Cells 2021, 10, 265. [Google Scholar] [CrossRef]
- Golán, I.; De La Fuente, L.R.; Costoya, J.A. NK cell-based glioblastoma immunotherapy. Cancers (Basel) 2018, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Borin, T.F.; Piranlioglu, R.; Ara, R.; Lebedyeva, I.; Angara, K.; Achyut, B.R.; Arbab, A.S.; Rashid, M.H. Changes in the tumor microenvironment and outcome for TME-targeting therapy in glioblastoma: A pilot study. PLoS ONE 2021, 16, e0246646. [Google Scholar] [CrossRef]
- Kushnirsky, M.; Feun, L.G.; Gultekin, S.H.; de la Fuente, M.I. Prolonged Complete Response With Combined Dabrafenib and Trametinib After BRAF Inhibitor Failure in BRAF-Mutant Glioblastoma. JCO Precis. Oncol. 2020, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Jane, E.P.; Premkumar, D.R.; Thambireddy, S.; Golbourn, B.; Agnihotri, S.; Bertrand, K.C.; Mack, S.C.; Myers, M.I.; Chattopadhyay, A.; Lansing Taylor, D.; et al. Targeting NADþ biosynthesis overcomes panobinostat and bortezomib-induced malignant glioma resistance. Mol. Cancer Res. 2020, 18, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Garcia-Fabiani, M.B.; Carney, S.; Altshuler, D.; Núñez, F.J.; Méndez, F.M.; Núñez, F.; Lowenstein, P.R.; Castro, M.G. Mutant ATRX: Uncovering a new therapeutic target for glioma. Expert Opin. Ther. Targets 2018, 22, 599–613. [Google Scholar] [CrossRef]
- Li, J.; Wu, J.; Bao, X.; Honea, N.; Xie, Y.; Kim, S.; Sparreboom, A.; Sanai, N. Quantitative and mechanistic understanding of AZD1775 penetration across human blood-brain barrier in glioblastoma patients using an IVIVE-PBPK modeling approach. Clin. Cancer Res. 2017, 23, 7454–7466. [Google Scholar] [CrossRef]
- Sanai, N.; Li, J.; Boerner, J.; Stark, K.; Wu, J.; Kim, S.; Derogatis, A.; Mehta, S.; Dhruv, H.D.; Heilbrun, L.K.; et al. Phase 0 trial of azd1775 in first-recurrence glioblastoma patients. Clin. Cancer Res. 2018, 24, 3820–3828. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- van Noorden, C.J.F.; Hira, V.V.V.; van Dijck, A.J.; Novak, M.; Breznik, B.; Molenaar, R.J. Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy? Cells 2021, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Seliger, C.; Genbrugge, E.; Gorlia, T.; Chinot, O.; Stupp, R.; Nabors, B.; Weller, M.; Hau, P. Use of metformin and outcome of patients with newly diagnosed glioblastoma: Pooled analysis. Int. J. Cancer 2020, 146, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.P.; Hoffmeyer, E.; Chetty, S.L.; Cruz-Cruz, J.; Hamrick, F.; Youssef, O.; Cheshier, S.; Mitra, S.S. Microglia in the Brain Tumor Microenvironment. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2020; Volume 1273, pp. 197–208. [Google Scholar]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Majc, B.; Sever, T.; Zarić, M.; Breznik, B.; Turk, B. BBA - Molecular Cell Research Epithelial-to-mesenchymal transition as the driver of changing carcinoma. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118782. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Onuchic, J.N.; Levine, H.; Ben-Jacob, E. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Smigiel, J.M.; Taylor, S.E.; Bryson, B.L.; Tamagno, I.; Polak, K.; Jackson, M.W. Cellular plasticity and metastasis in breast cancer: A pre- and post-malignant problem. J. Cancer Metastasis Treat. 2019, 5, 47. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Perus, L.J.M.; Walsh, L.A. Microenvironmental Heterogeneity in Brain Malignancies. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Chen, P.; Hsu, W.H.; Han, J.; Xia, Y.; DePinho, R.A. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Cloughesy, T.F.; Maus, M.V.; Prins, R.M.; Reardon, D.A.; Sonabend, A.M. Emerging immunotherapies for malignant glioma: From immunogenomics to cell therapy. Neuro Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Motaln, H.; Turnšek, T.L. Proteases and cytokines as mediators of interactions between cancer and stromal cells in tumours. Biol. Chem. 2017, 398, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends in Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes–Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell. Neurosci. 2018, 12, 1–22. [Google Scholar] [CrossRef]
- Oliveira, M.N.; Pillat, M.M.; Motaln, H.; Ulrich, H.; Lah, T.T. Kinin-B1 Receptor Stimulation Promotes Invasion and is Involved in Cell-Cell Interaction of Co-Cultured Glioblastoma and Mesenchymal Stem Cells. Sci. Rep. 2018, 8, 1299. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; De Borja, R.; Aronson, M.; Durn, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Jiao, X.; Velasco-Vel Azquez, M.A.; Wang, M.; Li, Z.; Rui, H.; Peck, A.R.; Korkola, J.E.; Chen, X.; Xu, S.; Duhadaway, J.B.; et al. Tumor Biology and Immunology CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion. Cancer Res 2018, 78, 1657–1671. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Verbovšek, U.; Breznik, B.; Srdič, M.; Novinec, M.; Kakar, H.; Wormer, J.; der Swaan, B.V.; Lenarčič, B.; Juliano, L.; et al. Cathepsin K cleavage of SDF-1α inhibits its chemotactic activity towards glioblastoma stem-like cells. Biochim. Biophys. Acta - Mol. Cell Res. 2017, 1864, 594–603. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Van Noorden, C.J.F.; Molenaar, R.J. CXCR4 antagonists as stem cell mobilizers and therapy sensitizers for acute myeloid leukemia and glioblastoma? Biology (Basel) 2020, 9, 31. [Google Scholar] [CrossRef]
- Aldinucci, D.; Casagrande, N. Inhibition of the CCL5/CCR5 axis against the progression of gastric cancer. Int. J. Mol. Sci. 2018, 19, 1477. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers (Basel) 2020, 12, 1765. [Google Scholar] [CrossRef]
- Laudati, E.; Currò, D.; Navarra, P.; Lisi, L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 2017, 108, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017. [Google Scholar] [CrossRef]
- Morisse, M.C.; Jouannet, S.; Dominguez-Villar, M.; Sanson, M.; Idbaih, A. Interactions between tumor-associated macrophages and tumor cells in glioblastoma: Unraveling promising targeted therapies. Expert Rev. Neurother. 2018, 18, 729–737. [Google Scholar] [CrossRef]
- Sicoli, D.; Jiao, X.; Ju, X.; Velasco-Velazquez, M.; Ertel, A.; Addya, S.; Li, Z.; Andò, S.; Fatatis, A.; Paudyal, B.; et al. CCR5 receptor antagonists block metastasis to bone of v-Src oncogene-transformed metastatic prostate cancer cell lines. Cancer Res. 2014, 74, 7103–7114. [Google Scholar] [CrossRef]
- Upadhyaya, C.; Jiao, X.; Ashton, A.; Patel, K.; Kossenkov, A.V.; Pestell, R.G. The G protein coupled receptor CCR5 in cancer. In Advances in Cancer Research, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 145. [Google Scholar]
- Velasco-Velázquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef]
- Pervaiz, A.; Ansari, S.; Berger, M.R.; Adwan, H. CCR5 blockage by maraviroc induces cytotoxic and apoptotic effects in colorectal cancer cells. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [PubMed]
- González-Arriagada, W.A.; Lozano-Burgos, C.; Zúñiga-Moreta, R.; González-Díaz, P.; Coletta, R.D. Clinicopathological significance of chemokine receptor (CCR1, CCR3, CCR4, CCR5, CCR7 and CXCR4) expression in head and neck squamous cell carcinomas. J. Oral Pathol. Med. 2018, 47, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Shen, Y.C.; Chang, J.W.C.; Hsieh, J.J.; Chu, Y.; Wang, C.H. Autocrine CCL5 promotes tumor progression in esophageal squamous cell carcinoma in vitro. Cytokine 2018, 110, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Zi, J.; Yuan, S.; Qiao, J.; Zhao, K.; Xu, L.; Qi, K.; Xu, K.; Zeng, L. Treatment with the C-C chemokine receptor type 5 (CCR5)-inhibitor maraviroc suppresses growth and induces apoptosis of acute lymphoblastic leukemia cells. Am. J. Cancer Res. 2017, 7, 869–880. [Google Scholar]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science (80-.) 1996, 273, 1856–1862. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–726. [Google Scholar] [CrossRef]
- Olson, W.C.; Rabut, G.E.E.; Nagashima, K.A.; Tran, D.N.H.; Anselma, D.J.; Monard, S.P.; Segal, J.P.; Thompson, D.A.D.; Kajumo, F.; Guo, Y.; et al. Differential Inhibition of Human Immunodeficiency Virus Type 1 Fusion, gp120 Binding, and CC-Chemokine Activity by Monoclonal Antibodies to CCR5. J. Virol. 1999, 73, 4145–4155. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Joy, M.T.; Assayag, E.B.; Shabashov-stone, D.; Liraz-zaltsman, S.; Mazzitelli, J.; Arenas, M.; Abduljawad, N.; Kliper, E.; Korczyn, A.D.; Thareja, N.S.; et al. CCR5 Is a Therapeutic Target for Recovery after Stroke and Traumatic Brain Injury. Cell 2020, 176, 1143–1157. [Google Scholar] [CrossRef]
- Jiao, X.; Wang, M.; Zhang, Z.; Li, Z.; Ni, D.; Ashton, A.W.; Tang, H.Y.; Speicher, D.W.; Pestell, R.G. Leronlimab, a humanized monoclonal antibody to CCR5, blocks breast cancer cellular metastasis and enhances cell death induced by DNA damaging chemotherapy. Breast Cancer Res. 2021, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, H.; Ichikura, T.; Kinoshita, M.; Ono, S.; Majima, T.; Tsujimoto, H.; Chochi, K.; Hiroi, S.; Takayama, E.; Saitoh, D.; et al. Gastric cancer cells exploit CD4+ cell-derived CCL5 for their growth and prevention of CD8+ cell-involved tumor elimination. Int. J. Cancer 2008. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.A.; Bae, S. CCR5 / CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Cambien, B.; Richard-Fiardo, P.; Karimdjee, B.F.; Martini, V.; Ferrua, B.; Pitard, B.; Schmid-Antomarchi, H.; Schmid-Alliana, A. CCL5 Neutralization Restricts Cancer Growth and Potentiates the Targeting of PDGFRβ in Colorectal Carcinoma. PLoS ONE 2011, 6, e28842. [Google Scholar] [CrossRef]
- Kouno, J.; Nagai, H.; Nagahata, T.; Onda, M.; Yamaguchi, H.; Adachi, K.; Takahashi, H.; Teramoto, A.; Emi, M. Up-regulation of CC chemokine, CCL3L1, and receptors, CCR3, CCR5 in human glioblastoma that promotes cell growth. J. Neurooncol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Moogooei, M.; Shamaei, M.; Khorramdelazad, H.; Fattahpour, S.; Seyedmehdi, S.M.; Moogooei, M.; Hassanshahi, G.; Kalantari Khandani, B. The Intricate Expression of CC Chemokines in Glial Tumors: Evidence for Involvement of CCL2 and CCL5 but Not CCL11. Acta Med. Iran. 2015, 53, 770–777. [Google Scholar]
- Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X.; Wang, D. Hypoxia and macrophages promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef]
- Chen, L.; Liu, X.; Zhang, H.Y.; Du, W.; Qin, Z.; Yao, Y.; Mao, Y.; Zhou, L. Upregulation of chemokine receptor CCR10 is essential for glioma proliferation, invasion and patient survival. Oncotarget 2014, 5, 6576–6583. [Google Scholar] [CrossRef][Green Version]
- Pan, Y.; Lu, F.; Fei, Q.; Yu, X.; Xiong, P.; Yu, X.; Dang, Y.; Hou, Z.; Lin, W.; Lin, X.; et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef]
- Pham, K.; Luo, D.; Liu, C.; Harrison, J.K. CCL5, CCR1 and CCR5 in murine glioblastoma: Immune cell infiltration and survival rates are not dependent on individual expression of either CCR1 or CCR5. J. Neuroimmunol. 2012, 246, 10–17. [Google Scholar] [CrossRef]
- Kast, R.E. Glioblastoma: Synergy of growth promotion between CCL5 and NK-1R can be thwarted by blocking CCL5 with miraviroc, an FDA approved anti-HIV drug and blocking NK-1R with aprepitant, an FDA approved anti-nausea drug. J. Clin. Pharm. Ther. 2010, 35, 657–663. [Google Scholar] [CrossRef]
- Wu, C.Y.-J.; Chen, C.-H.; Lin, C.-Y.; Feng, L.-Y.; Lin, Y.-C.; Wei, K.-C.; Huang, C.-Y.; Fang, J.-Y.; Chen, P.-Y. CCL5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase-2. Neuro. Oncol. 2019, 1–14. [Google Scholar] [CrossRef]
- Solga, A.C.; Pong, W.W.; Kim, K.Y.; Cimino, P.J.; Toonen, J.A.; Walker, J.; Wylie, T.; Magrini, V.; Griffith, M.; Griffith, O.L.; et al. RNA Sequencing of Tumor-Associated Microglia Reveals Ccl5 as a Stromal Chemokine Critical for Neurofibromatosis-1 Glioma Growth. Neoplasia (United States) 2015, 17, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef]
- Casagrande, N.; Borghese, C.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 antagonism by maraviroc inhibits hodgkin lymphoma microenvironment interactions and xenograft growth. Haematologica 2019, 104, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, C.; Liu, X.; Wang, Z.; Sun, L.; Li, G.; Liang, J.; Hu, H.; Liu, Y.; Zhang, W.; et al. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology 2016, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Fish, E.N. Chemokines in breast cancer: Regulating metabolism. Cytokine 2018, 109, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Emma, J.; Taylor, M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; et al. Immune Resistance. Nature 2015, 515, 568–571. [Google Scholar] [CrossRef]
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Jewett, A.; Kos, J.; Kaur, K.; Turnsek, T.L.; Breznik, B.; Senjor, E.; Wong, P.; Nguyen, K.Y.; Ko, M.W. Multiple defects of natural killer cells in cancer patients: Anarchy, dysregulated systemic immunity, and immunosuppression in metastatic cancer. Crit. Rev. Immunol. 2020, 40, 93–133. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Albright, A.; Kaufman, D.R.; Cheng, J.D.; Shankaran, V.; Ribas, A.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- Jamal, R.; Lapointe, R.; Cocolakis, E.; Thébault, P.; Kazemi, S.; Friedmann, J.E.; Dionne, J.; Cailhier, J.F.; Bélanger, K.; Ayoub, J.P.; et al. Peripheral and local predictive immune signatures identified in a phase II trial of ipilimumab with carboplatin/paclitaxel in unresectable stage III or stage IV melanoma. J. Immunother. Cancer 2017, 5, 1–13. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lah Turnšek, T.; Jiao, X.; Novak, M.; Jammula, S.; Cicero, G.; Ashton, A.W.; Joyce, D.; Pestell, R.G. An Update on Glioblastoma Biology, Genetics, and Current Therapies: Novel Inhibitors of the G Protein-Coupled Receptor CCR5. Int. J. Mol. Sci. 2021, 22, 4464. https://doi.org/10.3390/ijms22094464
Lah Turnšek T, Jiao X, Novak M, Jammula S, Cicero G, Ashton AW, Joyce D, Pestell RG. An Update on Glioblastoma Biology, Genetics, and Current Therapies: Novel Inhibitors of the G Protein-Coupled Receptor CCR5. International Journal of Molecular Sciences. 2021; 22(9):4464. https://doi.org/10.3390/ijms22094464
Chicago/Turabian StyleLah Turnšek, Tamara, Xuanmao Jiao, Metka Novak, Sriharsha Jammula, Gina Cicero, Anthony W. Ashton, David Joyce, and Richard G. Pestell. 2021. "An Update on Glioblastoma Biology, Genetics, and Current Therapies: Novel Inhibitors of the G Protein-Coupled Receptor CCR5" International Journal of Molecular Sciences 22, no. 9: 4464. https://doi.org/10.3390/ijms22094464
APA StyleLah Turnšek, T., Jiao, X., Novak, M., Jammula, S., Cicero, G., Ashton, A. W., Joyce, D., & Pestell, R. G. (2021). An Update on Glioblastoma Biology, Genetics, and Current Therapies: Novel Inhibitors of the G Protein-Coupled Receptor CCR5. International Journal of Molecular Sciences, 22(9), 4464. https://doi.org/10.3390/ijms22094464