
ATPase Inhibitory Factor-1 Disrupts Mitochondrial Ca2+ Handling and Promotes Pathological Cardiac Hypertrophy through CaMKIIδ

 , ,
, ,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
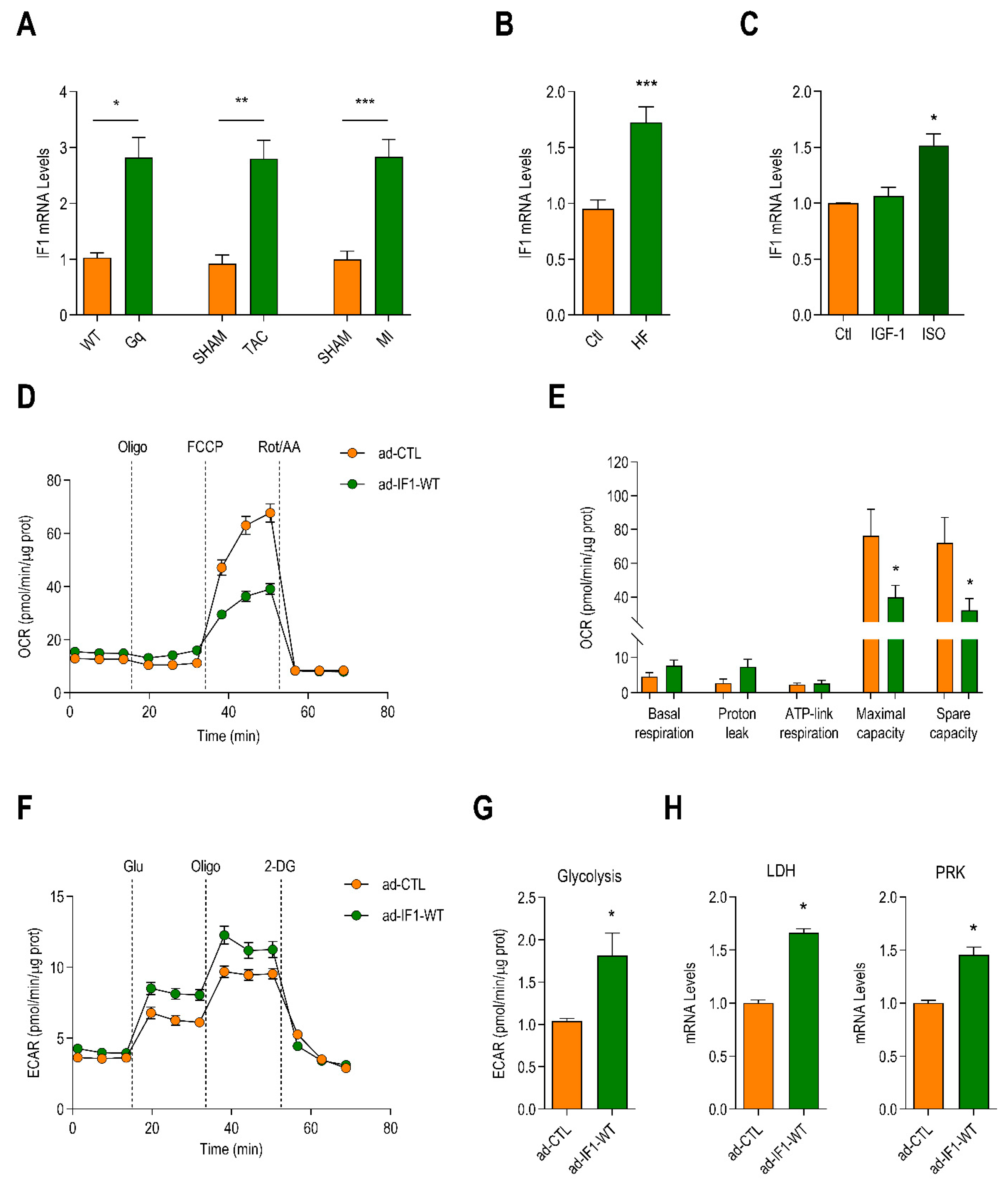
2.1. Myocardial Expression of IF1 Is Increased in Heart Failure and Induces Metabolic Reprogramming in Neonatal Rat Ventricular Myocytes (NRVMs)
2.2. IF1 Induces Mitochondrial Oxidative Stress and Stimulates Cardiomyocyte Hypertrophy
2.3. IF1-Induced Cardiomyocyte Reprograming Occurs Independent of Binding to ATP Synthase
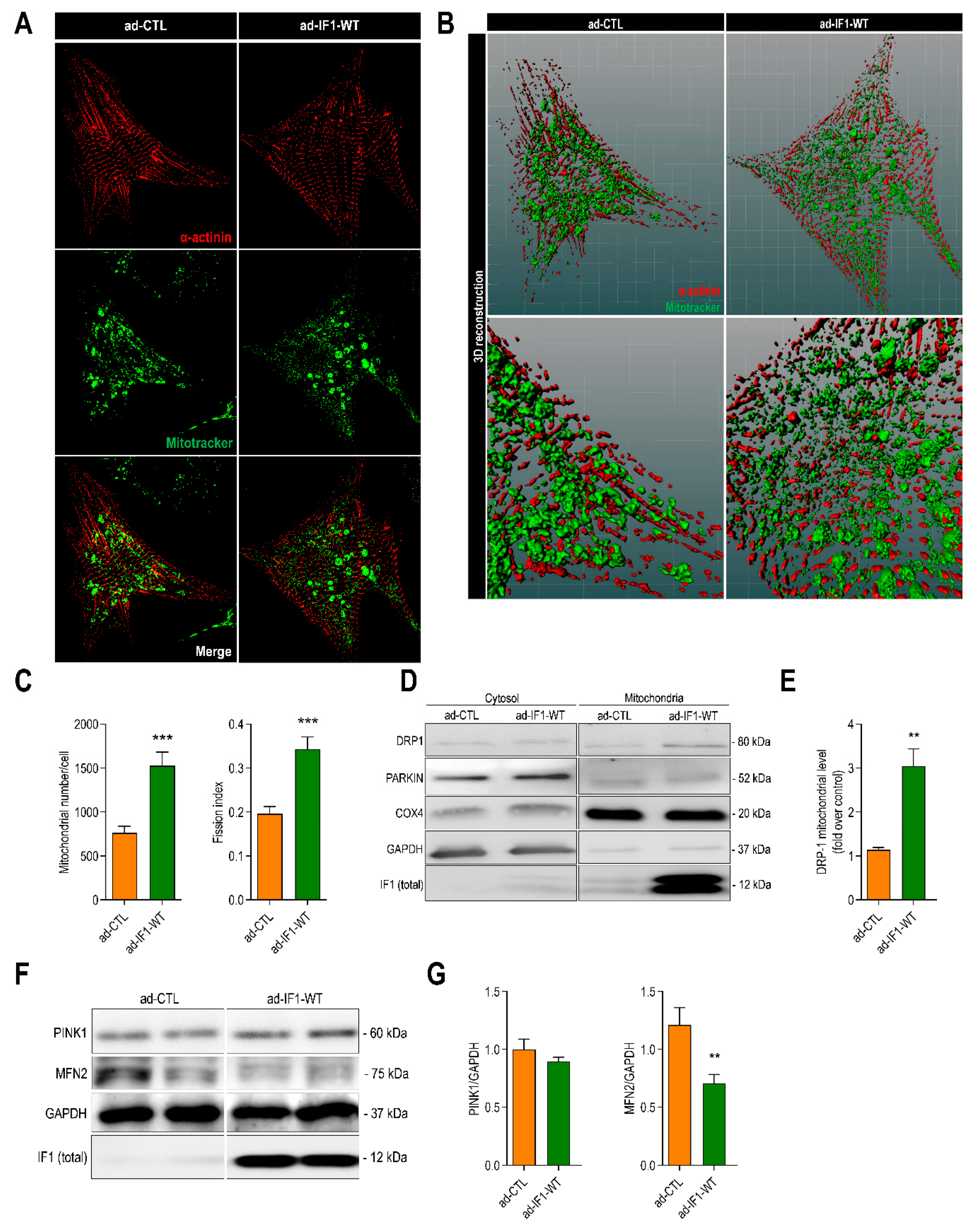
2.4. IF1 Induces Mitochondrial Fission through Dynamin-Related Protein 1 (DRP1)
2.5. IF1 Reduces Mitochondrial Ca2+ and Induces Sarcoplasmic Reticulum Ca2+ Overload
2.6. IF1-Induced Cardiomyocyte Hypertrophy Is Dependent upon CaMKIIδ
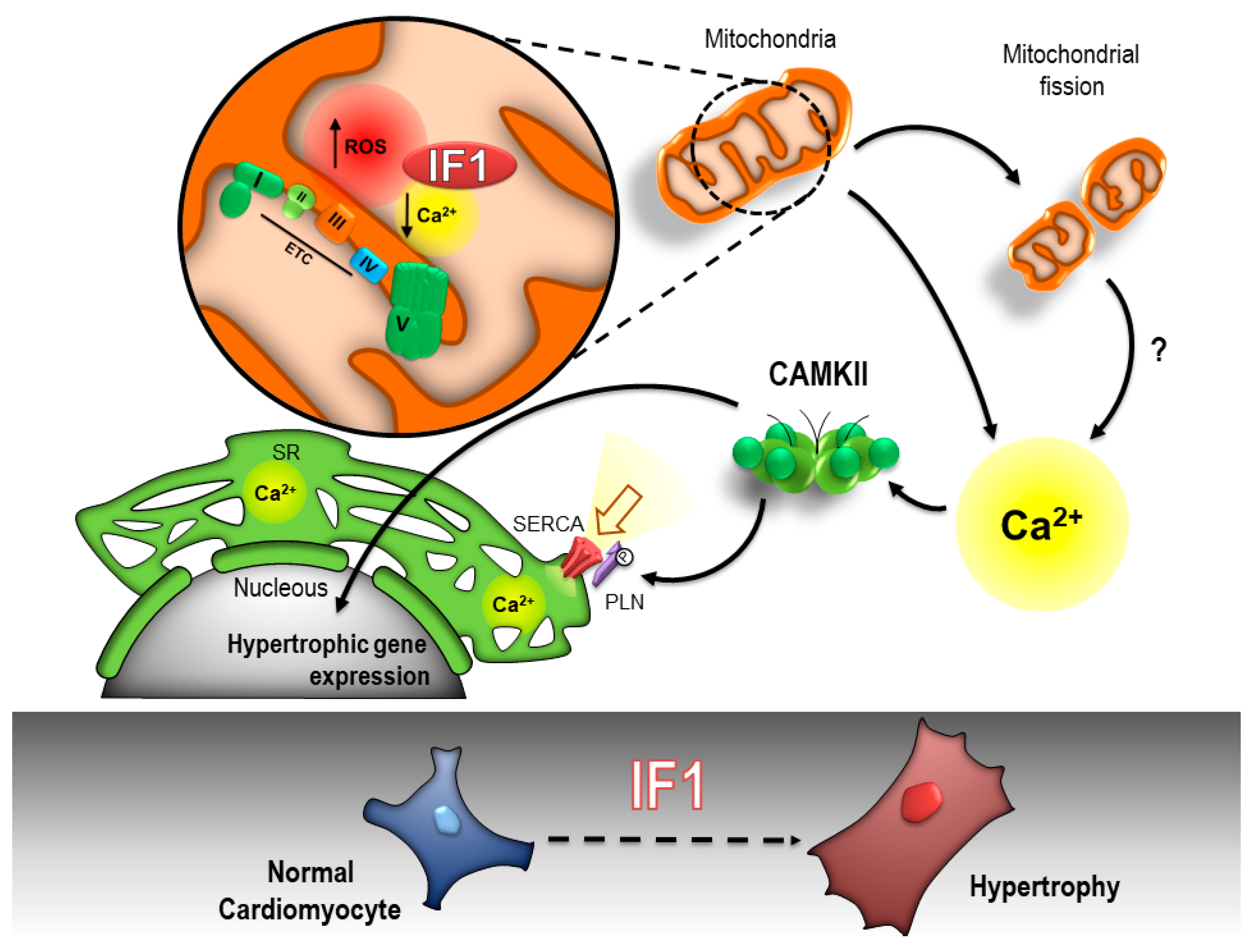
3. Discussion
4. Material and Methods
4.1. Heart Failure Samples
4.2. Neonatal Rat Cardiomyocyte Isolation
4.3. Western Blot
4.4. Real-Time PCR
4.5. Mitochondrial Membrane Potential
4.6. ATP Measurements
4.7. Seahorse Assays
4.8. Mitochondrial Morphology
4.9. Mitochondrial Isolation
4.10. Mitochondrial and Cytosolic Ca2+ Levels
4.11. Cell Size
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown:, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Tian, R.; Colucci, W.S.; Arany, Z.; Bachschmid, M.M.; Ballinger, S.W.; Boudina, S.; Bruce, J.E.; Busija, D.W.; Dikalov, S.; Dorn, G.W., II; et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System: Path to a Cure in Heart Failure—A Report from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation 2019, 140, 1205–1216. [Google Scholar] [CrossRef]
- Feniouk, B.A.; Yoshida, M. Regulatory mechanisms of proton-translocating F(O)F (1)-ATP synthase. Results Probl. Cell Differ. 2008, 45, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Cabezon, E.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. The structure of bovine F1-ATPase in complex with its regulatory protein IF1. Nat. Struct. Biol. 2003, 10, 744–750. [Google Scholar] [CrossRef]
- Formentini, L.; Sanchez-Arago, M.; Sanchez-Cenizo, L.; Cuezva, J.M. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol. Cell 2012, 45, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aguilar, A.; Cuezva, J.M. A Review of the Inhibition of the Mitochondrial ATP Synthase by IF1 in vivo: Reprogramming Energy Metabolism and Inducing Mitohormesis. Front. Physiol. 2018, 9, 1322. [Google Scholar] [CrossRef]
- Faccenda, D.; Gorini, G.; Jones, A.; Thornton, C.; Baracca, A.; Solaini, G.; Campanella, M. The ATPase Inhibitory Factor 1 (IF1) regulates the expression of the mitochondrial Ca(2+) uniporter (MCU) via the AMPK/CREB pathway. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118860. [Google Scholar] [CrossRef]
- Campanella, M.; Casswell, E.; Chong, S.; Farah, Z.; Wieckowski, M.R.; Abramov, A.Y.; Tinker, A.; Duchen, M.R. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008, 8, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Faccenda, D.; Nakamura, J.; Gorini, G.; Dhoot, G.K.; Piacentini, M.; Yoshida, M.; Campanella, M. Control of Mitochondrial Remodeling by the ATPase Inhibitory Factor 1 Unveils a Pro-survival Relay via OPA1. Cell Rep. 2017, 18, 1869–1883. [Google Scholar] [CrossRef]
- Formentini, L.; Pereira, M.P.; Sanchez-Cenizo, L.; Santacatterina, F.; Lucas, J.J.; Navarro, C.; Martinez-Serrano, A.; Cuezva, J.M. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J. 2014, 33, 762–778. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Moon, J.; Chung, I.; Chung, J.H.; Park, C.; Lee, J.O.; Han, J.A.; Kang, M.J.; Yoo, E.H.; Kwak, S.Y.; et al. ATP synthase inhibitory factor 1 (IF1), a novel myokine, regulates glucose metabolism by AMPK and Akt dual pathways. FASEB J. 2019, 33, 14825–14840. [Google Scholar] [CrossRef]
- Fernandez-Cardenas, L.P.; Villanueva-Chimal, E.; Salinas, L.S.; Jose-Nunez, C.; Tuena de Gomez Puyou, M.; Navarro, R.E. Caenorhabditis elegans ATPase inhibitor factor 1 (IF1) MAI-2 preserves the mitochondrial membrane potential (Deltapsim) and is important to induce germ cell apoptosis. PLoS ONE 2017, 12, e0181984. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jimenez, R.; Faccenda, D.; Allen, E.; Reichel, H.B.; Arcos, L.; Ferraina, C.; Strobbe, D.; Russell, C.; Campanella, M. Reduction of the ATPase inhibitory factor 1 (IF1) leads to visual impairment in vertebrates. Cell Death Dis. 2018, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Long, Q.; Saja, K.; Huang, F.; Pogwizd, S.M.; Zhou, L.; Yoshida, M.; Yang, Q. Knockout of the ATPase inhibitory factor 1 protects the heart from pressure overload-induced cardiac hypertrophy. Sci. Rep. 2017, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Westenbrink, B.D.; Ling, H.; Divakaruni, A.S.; Gray, C.B.; Zambon, A.C.; Dalton, N.D.; Peterson, K.L.; Gu, Y.; Matkovich, S.J.; Murphy, A.N.; et al. Mitochondrial reprogramming induced by CaMKIIdelta mediates hypertrophy decompensation. Circ. Res. 2015, 116, e28–e39. [Google Scholar] [CrossRef]
- Booij, H.G.; Yu, H.; de Boer, R.A.; van de Kolk, C.W.; van de Sluis, B.; van Deursen, J.M.; van Gilst, W.H.; Sillje, H.H.; Westenbrink, B.D. Overexpression of A kinase interacting protein 1 attenuates myocardial ischaemia/reperfusion injury but does not influence heart failure development. Cardiovasc. Res. 2016, 111, 217–226. [Google Scholar] [CrossRef]
- Esparza-Molto, P.B.; Cuezva, J.M. The Role of Mitochondrial H(+)-ATP Synthase in Cancer. Front. Oncol. 2018, 8, 53. [Google Scholar] [CrossRef]
- Chen, W.W.; Birsoy, K.; Mihaylova, M.M.; Snitkin, H.; Stasinski, I.; Yucel, B.; Bayraktar, E.C.; Carette, J.E.; Clish, C.B.; Brummelkamp, T.R.; et al. Inhibition of ATPIF1 ameliorates severe mitochondrial respiratory chain dysfunction in mammalian cells. Cell Rep. 2014, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., II. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pennanen, C.; Parra, V.; Lopez-Crisosto, C.; Morales, P.E.; del Campo, A.; Gutierrez, T.; Rivera-Mejias, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014, 127, 2659–2671. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef]
- Hoshino, A.; Wang, W.J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef]
- Jagasia, R.; Grote, P.; Westermann, B.; Conradt, B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature 2005, 433, 754–760. [Google Scholar] [CrossRef]
- Gutierrez, T.; Parra, V.; Troncoso, R.; Pennanen, C.; Contreras-Ferrat, A.; Vasquez-Trincado, C.; Morales, P.E.; Lopez-Crisosto, C.; Sotomayor-Flores, C.; Chiong, M.; et al. Alteration in mitochondrial Ca(2+) uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun. Signal 2014, 12, 68. [Google Scholar] [CrossRef][Green Version]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetic, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; de Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflugers Arch. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Westenbrink, B.D.; Edwards, A.G.; McCulloch, A.D.; Brown, J.H. The promise of CaMKII inhibition for heart disease: Preventing heart failure and arrhythmias. Expert Opin. Ther. Targets 2013, 17, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Braunwald, E. The war against heart failure: The Lancet lecture. Lancet 2015, 385, 812–824. [Google Scholar] [CrossRef]
- Boreikaite, V.; Wicky, B.I.M.; Watt, I.N.; Clarke, J.; Walker, J.E. Extrinsic conditions influence the self-association and structure of IF1, the regulatory protein of mitochondrial ATP synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 10354–10359. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat. Commun. 2016, 7, 13189. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, A.; Bassani, R.A.; Escobar, A.L.; Palomeque, J.; Valverde, C.A.; Vila Petroff, M.; Bers, D.M. Chasing cardiac physiology and pathology down the CaMKII cascade. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, 1177–1191. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yurista, S.R.; Sillje, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.M.; Pavez Giani, M.G.; Hillebrands, J.L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail 2019, 21, 862–873. [Google Scholar] [CrossRef]
- Yurista, S.R.; Matsuura, T.R.; Sillje, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Heart Fail 2021, 14, e007684. [Google Scholar] [CrossRef]
- Yurista, S.R.; Chong, C.R.; Badimon, J.J.; Kelly, D.P.; de Boer, R.A.; Westenbrink, B.D. Therapeutic Potential of Ketone Bodies for Patients with Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 1660–1669. [Google Scholar] [CrossRef]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of Elamipretide on Left Ventricular Function in Patients with Heart Failure with Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail 2020, 26, 429–437. [Google Scholar] [CrossRef]
- Ivanes, F.; Faccenda, D.; Gatliff, J.; Ahmed, A.A.; Cocco, S.; Cheng, C.H.; Allan, E.; Russell, C.; Duchen, M.R.; Campanella, M. The compound BTB06584 is an IF1 -dependent selective inhibitor of the mitochondrial F1 Fo-ATPase. Br. J. Pharmacol. 2014, 171, 4193–4206. [Google Scholar] [CrossRef]
- Strobbe, D.; Pecorari, R.; Conte, O.; Minutolo, A.; Hendriks, C.M.M.; Wiezorek, S.; Faccenda, D.; Abeti, R.; Montesano, C.; Bolm, C.; et al. NH-sulfoximine: A novel pharmacological inhibitor of the mitochondrial F1 Fo-ATPase, which suppresses viability of cancerous cells. Br. J. Pharmacol. 2021, 178, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Tigchelaar, W.; de Jong, A.M.; van Gilst, W.H.; de Boer, R.A.; Sillje, H.H. In EXOG-depleted cardiomyocytes cell death is marked by a decreased mitochondrial reserve capacity of the electron transport chain. Bioessays 2016, 38, 136–145. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavez-Giani, M.G.; Sánchez-Aguilera, P.I.; Bomer, N.; Miyamoto, S.; Booij, H.G.; Giraldo, P.; Oberdorf-Maass, S.U.; Nijholt, K.T.; Yurista, S.R.; Milting, H.; et al. ATPase Inhibitory Factor-1 Disrupts Mitochondrial Ca2+ Handling and Promotes Pathological Cardiac Hypertrophy through CaMKIIδ. Int. J. Mol. Sci. 2021, 22, 4427. https://doi.org/10.3390/ijms22094427
Pavez-Giani MG, Sánchez-Aguilera PI, Bomer N, Miyamoto S, Booij HG, Giraldo P, Oberdorf-Maass SU, Nijholt KT, Yurista SR, Milting H, et al. ATPase Inhibitory Factor-1 Disrupts Mitochondrial Ca2+ Handling and Promotes Pathological Cardiac Hypertrophy through CaMKIIδ. International Journal of Molecular Sciences. 2021; 22(9):4427. https://doi.org/10.3390/ijms22094427
Chicago/Turabian StylePavez-Giani, Mario G., Pablo I. Sánchez-Aguilera, Nils Bomer, Shigeki Miyamoto, Harmen G. Booij, Paula Giraldo, Silke U. Oberdorf-Maass, Kirsten T. Nijholt, Salva R. Yurista, Hendrik Milting, and et al. 2021. "ATPase Inhibitory Factor-1 Disrupts Mitochondrial Ca2+ Handling and Promotes Pathological Cardiac Hypertrophy through CaMKIIδ" International Journal of Molecular Sciences 22, no. 9: 4427. https://doi.org/10.3390/ijms22094427
APA StylePavez-Giani, M. G., Sánchez-Aguilera, P. I., Bomer, N., Miyamoto, S., Booij, H. G., Giraldo, P., Oberdorf-Maass, S. U., Nijholt, K. T., Yurista, S. R., Milting, H., van der Meer, P., Boer, R. A. d., Heller Brown, J., Sillje, H. W. H., & Westenbrink, B. D. (2021). ATPase Inhibitory Factor-1 Disrupts Mitochondrial Ca2+ Handling and Promotes Pathological Cardiac Hypertrophy through CaMKIIδ. International Journal of Molecular Sciences, 22(9), 4427. https://doi.org/10.3390/ijms22094427