
A New Mouse Model for Complete Congenital Stationary Night Blindness Due to Gpr179 Deficiency

 , and
, and 
Abstract
1. Introduction
2. Results
2.1. Design and Genotyping of the Gpr179 KO First Mouse Model
2.2. Structural Characterization by SD-OCT
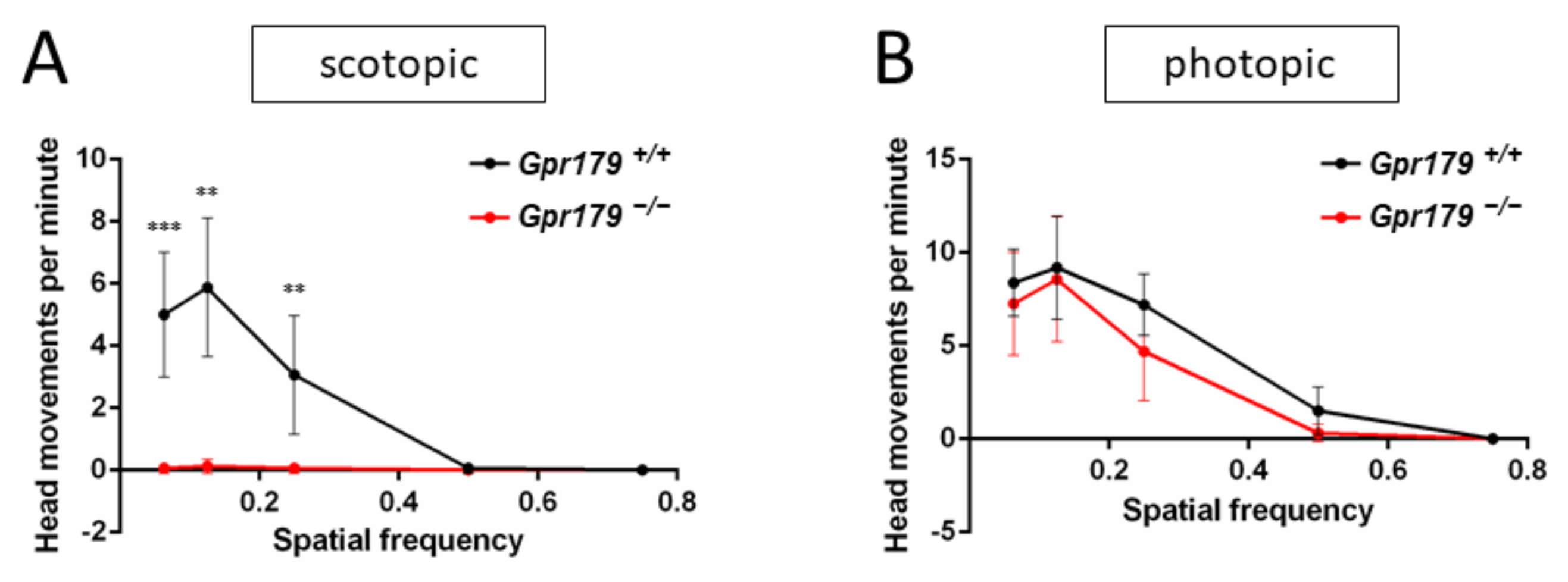
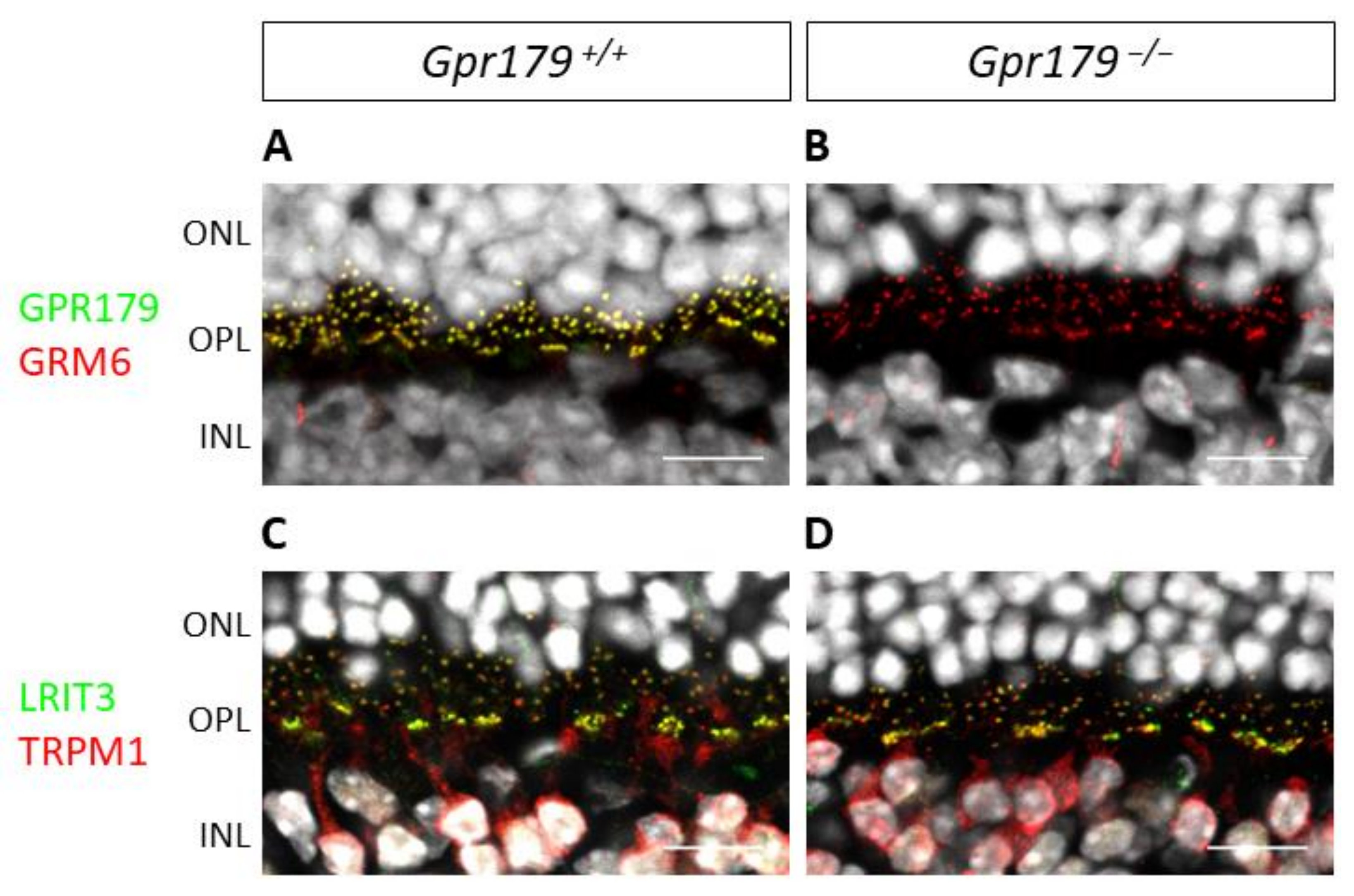
2.3. Functional Characterization
3. Discussion
4. Materials and Methods
4.1. Animal Care
4.2. Genotyping
4.2.1. Polymerase Chain Reaction (PCR) Genotyping for Gpr179
4.2.2. Genotyping for Common Mutations Found in Laboratory Mouse Strains
4.2.3. Genotyping for Genes with Mutations Underlying cCSNB
4.3. Spectral Domain Optical Coherence Tomography
4.4. Full Field Electroretinogram
4.5. Optomotor Response
4.6. Immunohistochemistry
4.6.1. Preparation of Retinal Sections for Immunohistochemistry
4.6.2. Immunostaining of Retinal Cryosections
4.6.3. Image Acquisition
4.7. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vardi, N.; Morigiwa, K. ON cone bipolar cells in rat express the metabotropic receptor mGluR6. Vis. Neurosci. 1997, 14, 789–794. [Google Scholar] [CrossRef]
- Nawy, S. The metabotropic receptor mGluR6 may signal through G(o), but not phosphodiesterase, in retinal bipolar cells. J. Neurosci. 1999, 19, 2938–2944. [Google Scholar] [CrossRef]
- Koike, C.; Obara, T.; Uriu, Y.; Numata, T.; Sanuki, R.; Miyata, K.; Koyasu, T.; Ueno, S.; Funabiki, K.; Tani, A.; et al. TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc. Natl. Acad. Sci. USA 2010, 107, 332–337. [Google Scholar] [CrossRef]
- Shen, Y.; Heimel, J.A.; Kamermans, M.; Peachey, N.S.; Gregg, R.G.; Nawy, S. A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J. Neurosci. 2009, 29, 6088–6093. [Google Scholar] [CrossRef]
- Morgans, C.W.; Zhang, J.; Jeffrey, B.G.; Nelson, S.M.; Burke, N.S.; Duvoisin, R.M.; Brown, R.L. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19174–19178. [Google Scholar] [CrossRef]
- Pearring, J.N.; Bojang, P., Jr.; Shen, Y.; Koike, C.; Furukawa, T.; Nawy, S.; Gregg, R.G. A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J. Neurosci. 2011, 31, 10060–10066. [Google Scholar] [CrossRef]
- Zeitz, C.; Jacobson, S.G.; Hamel, C.P.; Bujakowska, K.; Neuille, M.; Orhan, E.; Zanlonghi, X.; Lancelot, M.E.; Michiels, C.; Schwartz, S.B.; et al. Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2013, 92, 67–75. [Google Scholar] [CrossRef]
- Neuille, M.; Morgans, C.W.; Cao, Y.; Orhan, E.; Michiels, C.; Sahel, J.A.; Audo, I.; Duvoisin, R.M.; Martemyanov, K.A.; Zeitz, C. LRIT3 is essential to localize TRPM1 to the dendritic tips of depolarizing bipolar cells and may play a role in cone synapse formation. Eur. J. Neurosci. 2015, 42, 1966–1975. [Google Scholar] [CrossRef]
- Hasan, N.; Pangeni, G.; Ray, T.A.; Fransen, K.M.; Noel, J.; Borghuis, B.G.; McCall, M.A.; Gregg, R.G. LRIT3 is Required for Nyctalopin Expression and Normal ON and OFF Pathway Signaling in the Retina. eNeuro 2020, 7. [Google Scholar] [CrossRef]
- Ross, E.M.; Wilkie, T.M. GTPase-activating proteins for heterotrimeric G proteins: Regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 2000, 69, 795–827. [Google Scholar] [CrossRef]
- Rao, A.; Dallman, R.; Henderson, S.; Chen, C.K. Gbeta5 is required for normal light responses and morphology of retinal ON-bipolar cells. J. Neurosci. 2007, 27, 14199–14204. [Google Scholar] [CrossRef]
- Morgans, C.W.; Wensel, T.G.; Brown, R.L.; Perez-Leon, J.A.; Bearnot, B.; Duvoisin, R.M. Gbeta5-RGS complexes co-localize with mGluR6 in retinal ON-bipolar cells. Eur. J. Neurosci. 2007, 26, 2899–2905. [Google Scholar] [CrossRef]
- Song, J.H.; Song, H.; Wensel, T.G.; Sokolov, M.; Martemyanov, K.A. Localization and differential interaction of R7 RGS proteins with their membrane anchors R7BP and R9AP in neurons of vertebrate retina. Mol. Cell. Neurosci. 2007, 35, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, M.; Zhou, H.; Jia, L.; Cain, M.D.; Blumer, K.J. R9AP and R7BP: Traffic cops for the RGS7 family in phototransduction and neuronal GPCR signaling. Trends Pharmacol. Sci. 2009, 30, 17–24. [Google Scholar] [CrossRef]
- Cao, Y.; Masuho, I.; Okawa, H.; Xie, K.; Asami, J.; Kammermeier, P.J.; Maddox, D.M.; Furukawa, T.; Inoue, T.; Sampath, A.P.; et al. Retina-specific GTPase accelerator RGS11/G beta 5S/R9AP is a constitutive heterotrimer selectively targeted to mGluR6 in ON-bipolar neurons. J. Neurosci. 2009, 29, 9301–9313. [Google Scholar] [CrossRef]
- Martemyanov, K.A.; Yoo, P.J.; Skiba, N.P.; Arshavsky, V.Y. R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. J. Biol. Chem. 2005, 280, 5133–5136. [Google Scholar] [CrossRef]
- Drenan, R.M.; Doupnik, C.A.; Jayaraman, M.; Buchwalter, A.L.; Kaltenbronn, K.M.; Huettner, J.E.; Linder, M.E.; Blumer, K.J. R7BP augments the function of RGS7*Gbeta5 complexes by a plasma membrane-targeting mechanism. J. Biol. Chem. 2006, 281, 28222–28231. [Google Scholar] [CrossRef]
- Zeitz, C. Molecular genetics and protein function involved in nocturnal vision. Expert. Rev. Ophthalmol. 2007, 2, 467–485. [Google Scholar] [CrossRef]
- Zeitz, C.; Varin, J.; Audo, I. Congenital Stationary Night Blindness Pediatric Retina, 3rd ed.; Hartnett, M., Ed.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2021; pp. 400–410. [Google Scholar]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef]
- Schubert, G.; Bornschein, H. Analysis of the human electroretinogram. Ophthalmologica 1952, 123, 396–413. [Google Scholar] [CrossRef]
- Miyake, Y.; Yagasaki, K.; Horiguchi, M.; Kawase, Y.; Kanda, T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986, 104, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Robson, A.G.; Holder, G.E.; Moore, A.T. The negative ERG: Clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv. Ophthalmol. 2008, 53, 16–40. [Google Scholar] [CrossRef] [PubMed]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Sparkes, R.L.; Koop, B.; Birch, D.G.; Bergen, A.A.; Prinsen, C.F.; Polomeno, R.C.; Gal, A.; et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000, 26, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Pusch, C.M.; Zeitz, C.; Brandau, O.; Pesch, K.; Achatz, H.; Feil, S.; Scharfe, C.; Maurer, J.; Jacobi, F.K.; Pinckers, A.; et al. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat. Genet. 2000, 26, 324–327. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Berson, E.L.; Fishman, G.A.; Sandberg, M.A.; Alexander, K.R.; Derlacki, D.J.; Rajagopalan, A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA 2005, 102, 4884–4889. [Google Scholar] [CrossRef]
- Zeitz, C.; van Genderen, M.; Neidhardt, J.; Luhmann, U.F.; Hoeben, F.; Forster, U.; Wycisk, K.; Matyas, G.; Hoyng, C.B.; Riemslag, F.; et al. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4328–4335. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sergouniotis, P.I.; Michaelides, M.; Mackay, D.S.; Wright, G.A.; Devery, S.; Moore, A.T.; Holder, G.E.; Robson, A.G.; Webster, A.R. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am. J. Hum. Genet. 2009, 85, 711–719. [Google Scholar] [CrossRef]
- Audo, I.; Kohl, S.; Leroy, B.P.; Munier, F.L.; Guillonneau, X.; Mohand-Said, S.; Bujakowska, K.; Nandrot, E.F.; Lorenz, B.; Preising, M.; et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 720–729. [Google Scholar] [CrossRef]
- Van Genderen, M.M.; Bijveld, M.M.; Claassen, Y.B.; Florijn, R.J.; Pearring, J.N.; Meire, F.M.; McCall, M.A.; Riemslag, F.C.; Gregg, R.G.; Bergen, A.A.; et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 730–736. [Google Scholar] [CrossRef]
- Audo, I.; Bujakowska, K.; Orhan, E.; Poloschek, C.M.; Defoort-Dhellemmes, S.; Drumare, I.; Kohl, S.; Luu, T.D.; Lecompte, O.; Zrenner, E.; et al. Whole-Exome Sequencing Identifies Mutations in GPR179 Leading to Autosomal-Recessive Complete Congenital Stationary Night Blindness. Am. J. Hum. Genet. 2012, 90, 321–330. [Google Scholar] [CrossRef]
- Peachey, N.S.; Ray, T.A.; Florijn, R.; Rowe, L.B.; Sjoerdsma, T.; Contreras-Alcantara, S.; Baba, K.; Tosini, G.; Pozdeyev, N.; Iuvone, P.M.; et al. GPR179 is required for depolarizing bipolar cell function and is mutated in autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2012, 90, 331–339. [Google Scholar] [CrossRef]
- Bahadori, R.; Biehlmaier, O.; Zeitz, C.; Labhart, T.; Makhankov, Y.V.; Forster, U.; Gesemann, M.; Berger, W.; Neuhauss, S.C. Nyctalopin is essential for synaptic transmission in the cone dominated zebrafish retina. Eur. J. Neurosci. 2006, 24, 1664–1674. [Google Scholar] [CrossRef]
- Orhan, E.; Prezeau, L.; El Shamieh, S.; Bujakowska, K.M.; Michiels, C.; Zagar, Y.; Vol, C.; Bhattacharya, S.S.; Sahel, J.A.; Sennlaub, F.; et al. Further insights into GPR179: Expression, localization, and associated pathogenic mechanisms leading to complete congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8041–8050. [Google Scholar] [CrossRef] [PubMed]
- Masu, M.; Iwakabe, H.; Tagawa, Y.; Miyoshi, T.; Yamashita, M.; Fukuda, Y.; Sasaki, H.; Hiroi, K.; Nakamura, Y.; Shigemoto, R.; et al. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell 1995, 80, 757–765. [Google Scholar] [CrossRef]
- Vardi, N.; Duvoisin, R.; Wu, G.; Sterling, P. Localization of mGluR6 to dendrites of ON bipolar cells in primate retina. J. Comp. Neurol. 2000, 423, 402–412. [Google Scholar] [CrossRef]
- Morgans, C.W.; Ren, G.; Akileswaran, L. Localization of nyctalopin in the mammalian retina. Eur. J. Neurosci. 2006, 23, 1163–1171. [Google Scholar] [CrossRef]
- Klooster, J.; Blokker, J.; Brink, J.B.T.; Unmehopa, U.; Fluiter, K.; Bergen, A.A.; Kamermans, M. Ultrastructural localization and expression of TRPM1 in the human retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8356–8362. [Google Scholar] [CrossRef]
- Orlandi, C.; Posokhova, E.; Masuho, I.; Ray, T.A.; Hasan, N.; Gregg, R.G.; Martemyanov, K.A. GPR158/179 regulate G protein signaling by controlling localization and activity of the RGS7 complexes. J. Cell Biol. 2012, 197, 711–719. [Google Scholar] [CrossRef]
- Hasan, N.; Pangeni, G.; Cobb, C.A.; Ray, T.A.; Nettesheim, E.R.; Ertel, K.J.; Lipinski, D.M.; McCall, M.A.; Gregg, R.G. Presynaptic Expression of LRIT3 Transsynaptically Organizes the Postsynaptic Glutamate Signaling Complex Containing TRPM1. Cell Rep. 2019, 27, 3107–3116. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef]
- Di Scipio, M.; Tavares, E.; Deshmukh, S.; Audo, I.; Green-Sanderson, K.; Zubak, Y.; Zine-Eddine, F.; Pearson, A.; Vig, A.; Tang, C.Y.; et al. Phenotype Driven Analysis of Whole Genome Sequencing Identifies Deep Intronic Variants that Cause Retinal Dystrophies by Aberrant Exonization. Investig. Ophthalmol. Vis. Sci. 2020, 61, 36. [Google Scholar] [CrossRef]
- Neuille, M.; El Shamieh, S.; Orhan, E.; Michiels, C.; Antonio, A.; Lancelot, M.E.; Condroyer, C.; Bujakowska, K.; Poch, O.; Sahel, J.A.; et al. Lrit3 deficient mouse (nob6): A novel model of complete congenital stationary night blindness (cCSNB). PLoS ONE 2014, 9, e90342. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Iwakabe, H.; Akazawa, C.; Nawa, H.; Shigemoto, R.; Mizuno, N.; Nakanishi, S. Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for L-2-amino-4-phosphonobutyrate. J. Biol. Chem. 1993, 268, 11868–11873. [Google Scholar] [CrossRef]
- Gregg, R.G.; Mukhopadhyay, S.; Candille, S.I.; Ball, S.L.; Pardue, M.T.; McCall, M.A.; Peachey, N.S. Identification of the gene and the mutation responsible for the mouse nob phenotype. Investig. Ophthalmol. Vis. Sci. 2003, 44, 378–384. [Google Scholar] [CrossRef]
- Pardue, M.T.; McCall, M.A.; LaVail, M.M.; Gregg, R.G.; Peachey, N.S. A naturally occurring mouse model of X-linked congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2443–2449. [Google Scholar]
- Pinto, L.H.; Vitaterna, M.H.; Shimomura, K.; Siepka, S.M.; Balannik, V.; McDearmon, E.L.; Omura, C.; Lumayag, S.; Invergo, B.M.; Glawe, B.; et al. Generation, identification and functional characterization of the nob4 mutation of Grm6 in the mouse. Vis. Neurosci. 2007, 24, 111–123. [Google Scholar] [CrossRef]
- Maddox, D.M.; Vessey, K.A.; Yarbrough, G.L.; Invergo, B.M.; Cantrell, D.R.; Inayat, S.; Balannik, V.; Hicks, W.L.; Hawes, N.L.; Byers, S.; et al. Allelic variance between GRM6 mutants, Grm6nob3 and Grm6nob4 results in differences in retinal ganglion cell visual responses. J. Physiol. 2008, 586, 4409–4424. [Google Scholar] [CrossRef]
- Peachey, N.S.; Pearring, J.N.; Bojang, P., Jr.; Hirschtritt, M.E.; Sturgill-Short, G.; Ray, T.A.; Furukawa, T.; Koike, C.; Goldberg, A.F.; Shen, Y.; et al. Depolarizing bipolar cell dysfunction due to a Trpm1 point mutation. J. Neurophysiol. 2012, 108, 2442–2451. [Google Scholar] [CrossRef][Green Version]
- Bellone, R.R.; Forsyth, G.; Leeb, T.; Archer, S.; Sigurdsson, S.; Imsland, F.; Mauceli, E.; Engensteiner, M.; Bailey, E.; Sandmeyer, L.; et al. Fine-mapping and mutation analysis of TRPM1: A candidate gene for leopard complex (LP) spotting and congenital stationary night blindness in horses. Brief. Funct. Genom. 2010, 9, 193–207. [Google Scholar] [CrossRef][Green Version]
- Qian, H.; Ji, R.; Gregg, R.G.; Peachey, N.S. Identification of a new mutant allele, Grm6(nob7), for complete congenital stationary night blindness. Vis. Neurosci. 2015, 32, E004. [Google Scholar] [CrossRef]
- Das, R.G.; Becker, D.; Jagannathan, V.; Goldstein, O.; Santana, E.; Carlin, K.; Sudharsan, R.; Leeb, T.; Nishizawa, Y.; Kondo, M.; et al. Genome-wide association study and whole-genome sequencing identify a deletion in LRIT3 associated with canine congenital stationary night blindness. Sci. Rep. 2019, 9, 14166. [Google Scholar] [CrossRef]
- Hack, Y.L.; Crabtree, E.E.; Avila, F.; Sutton, R.B.; Grahn, R.; Oh, A.; Gilger, B.; Bellone, R.R. Whole-genome sequencing identifies missense mutation in GRM6 as the likely cause of congenital stationary night blindness in a Tennessee Walking Horse. Equine Vet. J. 2021, 53, 316–323. [Google Scholar] [CrossRef]
- Charette, J.R.; Samuels, I.S.; Yu, M.; Stone, L.; Hicks, W.; Shi, L.Y.; Krebs, M.P.; Naggert, J.K.; Nishina, P.M.; Peachey, N.S. A Chemical Mutagenesis Screen Identifies Mouse Models with ERG Defects. Adv. Exp. Med. Biol. 2016, 854, 177–183. [Google Scholar] [CrossRef]
- Scalabrino, M.L.; Boye, S.L.; Fransen, K.M.; Noel, J.M.; Dyka, F.M.; Min, S.H.; Ruan, Q.; De Leeuw, C.N.; Simpson, E.M.; Gregg, R.G.; et al. Intravitreal delivery of a novel AAV vector targets ON bipolar cells and restores visual function in a mouse model of complete congenital stationary night blindness. Hum. Mol. Genet. 2015, 24, 6229–6239. [Google Scholar] [CrossRef]
- Varin, J.; Bouzidi, N.; Dias, M.M.S.; Pugliese, T.; Michiels, C.; Robert, C.; Desrosiers, M.; Sahel, J.A.; Audo, I.; Dalkara, D.; et al. Restoration of mGluR6 Localization Following AAV-Mediated Delivery in a Mouse Model of Congenital Stationary Night Blindness. Investig. Ophthalmol. Vis. Sci. 2021, 62, 24. [Google Scholar] [CrossRef]
- Mattapallil, M.J.; Wawrousek, E.F.; Chan, C.C.; Zhao, H.; Roychoudhury, J.; Ferguson, T.A.; Caspi, R.R. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2921–2927. [Google Scholar] [CrossRef]
- Berger, A.; Cavallero, S.; Dominguez, E.; Barbe, P.; Simonutti, M.; Sahel, J.A.; Sennlaub, F.; Raoul, W.; Paques, M.; Bemelmans, A.P. Spectral-domain optical coherence tomography of the rodent eye: Highlighting layers of the outer retina using signal averaging and comparison with histology. PLoS ONE 2014, 9, e96494. [Google Scholar] [CrossRef]
- Orhan, E.; Dalkara, D.; Neuille, M.; Lechauve, C.; Michiels, C.; Picaud, S.; Leveillard, T.; Sahel, J.A.; Naash, M.I.; Lavail, M.M.; et al. Genotypic and phenotypic characterization of P23H line 1 rat model. PLoS ONE 2015, 10, e0127319. [Google Scholar] [CrossRef]
- Neuille, M.; Cao, Y.; Caplette, R.; Guerrero-Given, D.; Thomas, C.; Kamasawa, N.; Sahel, J.A.; Hamel, C.P.; Audo, I.; Picaud, S.; et al. LRIT3 Differentially Affects Connectivity and Synaptic Transmission of Cones to ON- and OFF-Bipolar Cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1768–1778. [Google Scholar] [CrossRef]
- Ray, T.A.; Heath, K.M.; Hasan, N.; Noel, J.M.; Samuels, I.S.; Martemyanov, K.A.; Peachey, N.S.; McCall, M.A.; Gregg, R.G. GPR179 is required for high sensitivity of the mGluR6 signaling cascade in depolarizing bipolar cells. J. Neurosci. 2014, 34, 6334–6343. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, C.; Cao, Y.; Martemyanov, K.A. Orphan receptor GPR179 forms macromolecular complexes with components of metabotropic signaling cascade in retina ON-bipolar neurons. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7153–7161. [Google Scholar] [CrossRef] [PubMed]
- Klooster, J.; van Genderen, M.M.; Yu, M.; Florijn, R.J.; Riemslag, F.C.; Bergen, A.A.; Gregg, R.G.; Peachey, N.S.; Kamermans, M. Ultrastructural localization of GPR179 and the impact of mutant forms on retinal function in CSNB1 patients and a mouse model. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6973–6981. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Pahlberg, J.; Sarria, I.; Kamasawa, N.; Sampath, A.P.; Martemyanov, K.A. Regulators of G protein signaling RGS7 and RGS11 determine the onset of the light response in ON bipolar neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 7905–7910. [Google Scholar] [CrossRef]
- Trapani, I.; Banfi, S.; Simonelli, F.; Surace, E.M.; Auricchio, A. Gene therapy of inherited retinal degenerations: Prospects and challenges. Hum. Gene Ther. 2015, 26, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Posokhova, E.; Martemyanov, K.A. TRPM1 forms complexes with nyctalopin in vivo and accumulates in postsynaptic compartment of ON-bipolar neurons in mGluR6-dependent manner. J. Neurosci. 2011, 31, 11521–11526. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Species | Dilution | Reference | Retinal Sections Preparation | Immunohistochemistry |
|---|---|---|---|---|---|
| GNB5 | rabbit | 1:300 | ABIN1451282 (Antibodies Online, Aachen, Germany) | Method 2 | Method 2 |
| GPR179 | mouse | 1:200 | Ab-887-YOM (Primm, Milan, Italy) | Either Method 1 or Method 3 | Method 1 |
| GRM6 | guinea pig | 1:200 | AP20134SU-N (Acris, Herford, Germany) | Method 1 | Method 1 |
| Lectin PNA 488 conjugate | Arachis hypogaea | 1:1000 | L21409 (Life Technologies, ThermoFisher Scientific, Waltham, MA, USA) | Method 1 | Method 1 |
| LRIT3 | rabbit | 1:500 | Neuillé et al., 2015 [8] | Method 1 | Method 2 |
| PKCα | mouse | 1:200 | P5704 (Sigma-Aldrich, Saint Louis, MI, USA) | Method 3 | Method 1 |
| RGS7 | rabbit | 1:250 | sc-28836 (Santa-Cruz) | Method 2 | Method 2 |
| RGS11 | goat | 1:200 | sc-9725 (Santa-Cruz, Dallas, TX, USA) | Method 2 | Method 2 |
| TRPM1 | sheep | 1:500 | Cao et al., 2011 [66] | Method 1 | Method 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orhan, E.; Neuillé, M.; de Sousa Dias, M.; Pugliese, T.; Michiels, C.; Condroyer, C.; Antonio, A.; Sahel, J.-A.; Audo, I.; Zeitz, C. A New Mouse Model for Complete Congenital Stationary Night Blindness Due to Gpr179 Deficiency. Int. J. Mol. Sci. 2021, 22, 4424. https://doi.org/10.3390/ijms22094424
Orhan E, Neuillé M, de Sousa Dias M, Pugliese T, Michiels C, Condroyer C, Antonio A, Sahel J-A, Audo I, Zeitz C. A New Mouse Model for Complete Congenital Stationary Night Blindness Due to Gpr179 Deficiency. International Journal of Molecular Sciences. 2021; 22(9):4424. https://doi.org/10.3390/ijms22094424
Chicago/Turabian StyleOrhan, Elise, Marion Neuillé, Miguel de Sousa Dias, Thomas Pugliese, Christelle Michiels, Christel Condroyer, Aline Antonio, José-Alain Sahel, Isabelle Audo, and Christina Zeitz. 2021. "A New Mouse Model for Complete Congenital Stationary Night Blindness Due to Gpr179 Deficiency" International Journal of Molecular Sciences 22, no. 9: 4424. https://doi.org/10.3390/ijms22094424
APA StyleOrhan, E., Neuillé, M., de Sousa Dias, M., Pugliese, T., Michiels, C., Condroyer, C., Antonio, A., Sahel, J.-A., Audo, I., & Zeitz, C. (2021). A New Mouse Model for Complete Congenital Stationary Night Blindness Due to Gpr179 Deficiency. International Journal of Molecular Sciences, 22(9), 4424. https://doi.org/10.3390/ijms22094424