
Development of an Australian Bread Wheat Nested Association Mapping Population, a New Genetic Diversity Resource for Breeding under Dry and Hot Climates

 ,
, 
 ,
,
Abstract
1. Introduction
2. Results
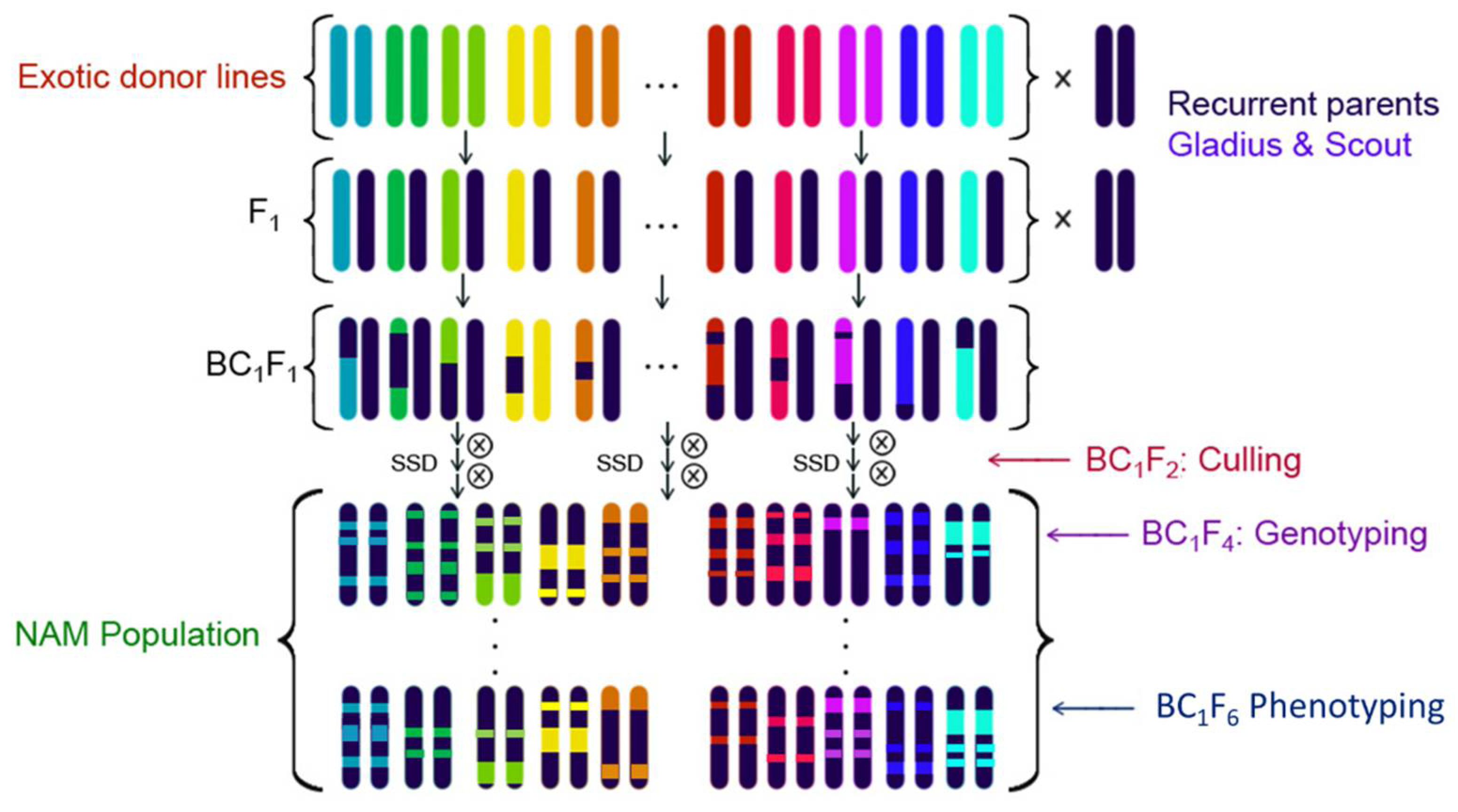
2.1. Development of the OzNAM Population
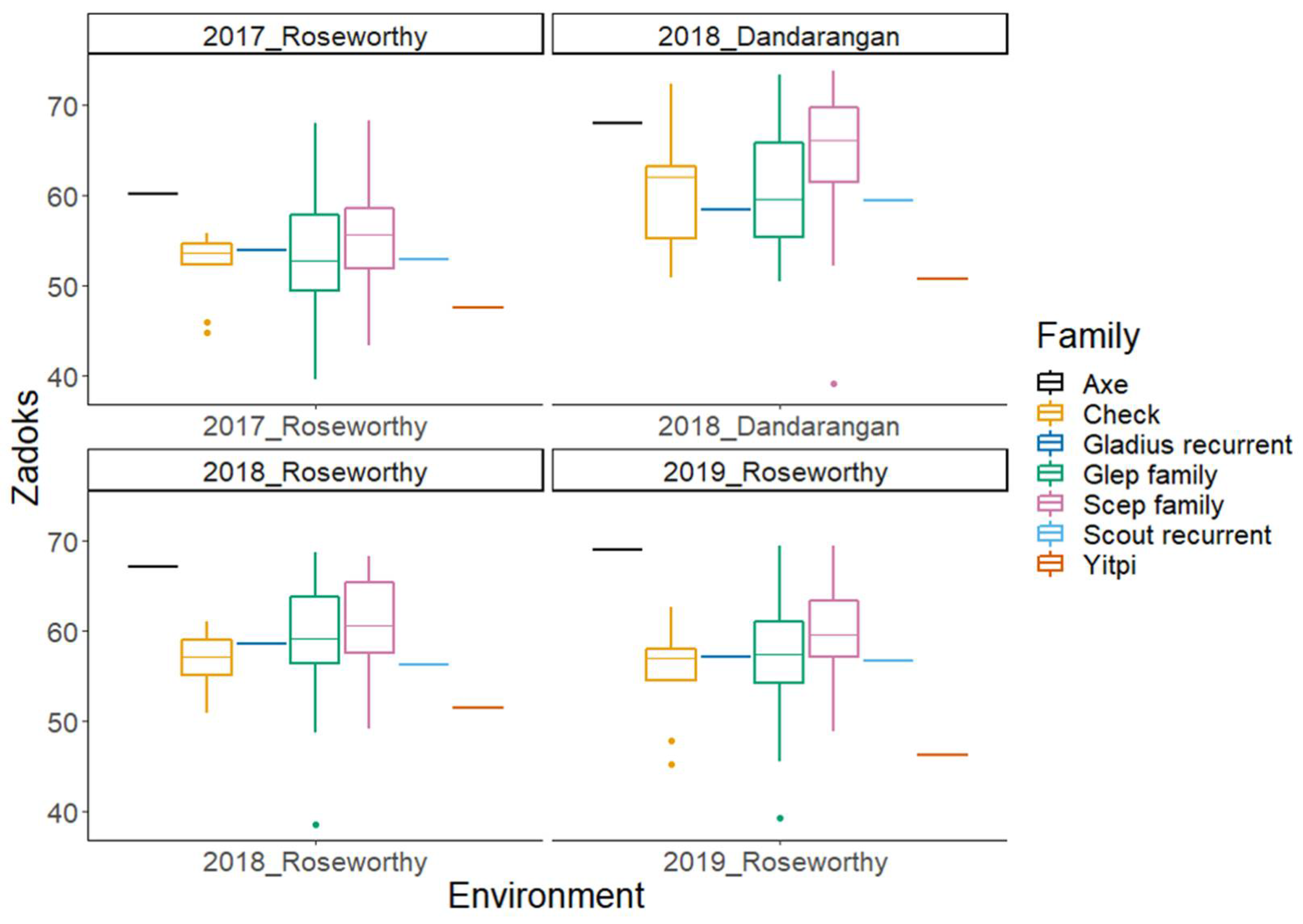
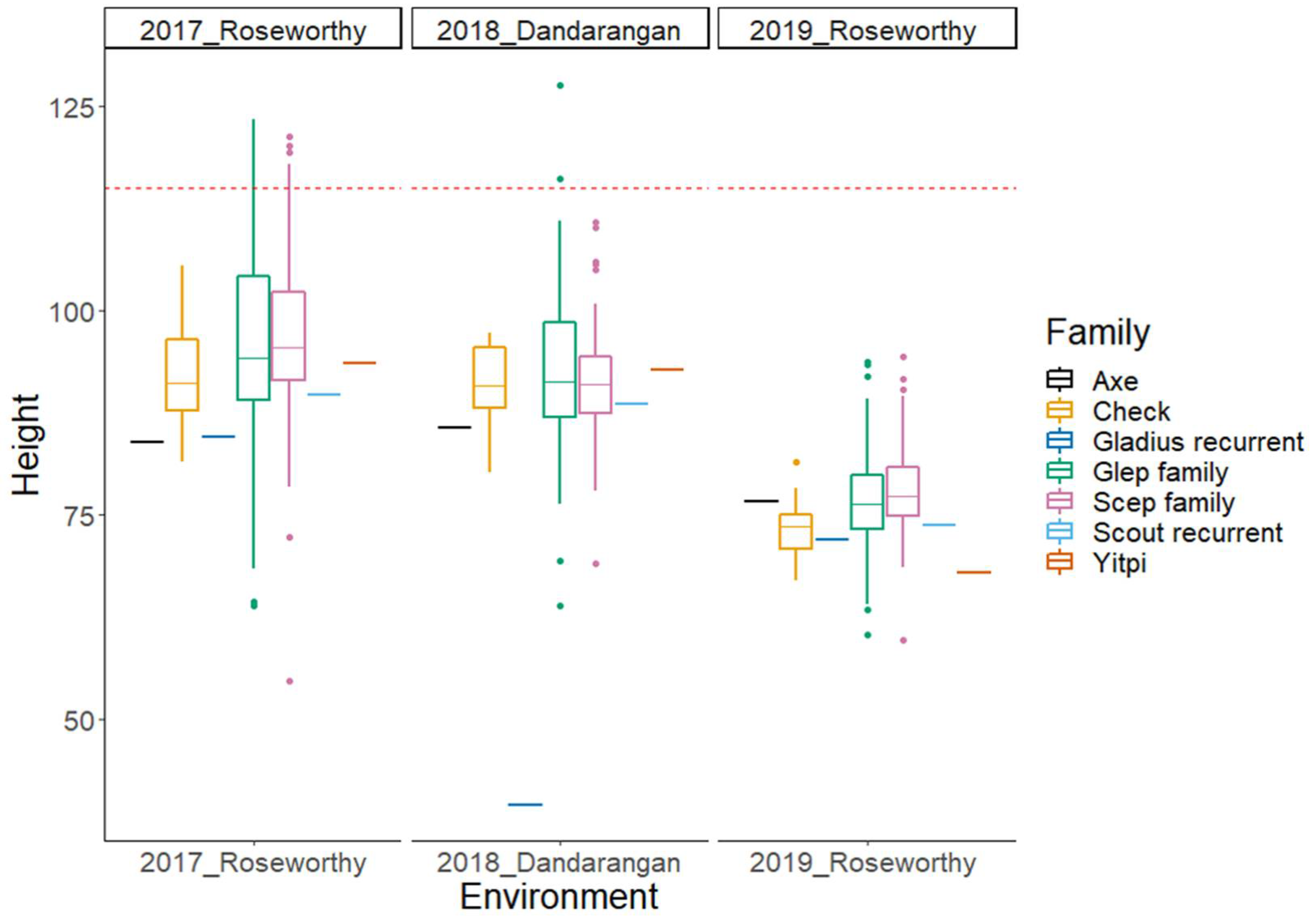
2.2. Phenotypic Data Analysis
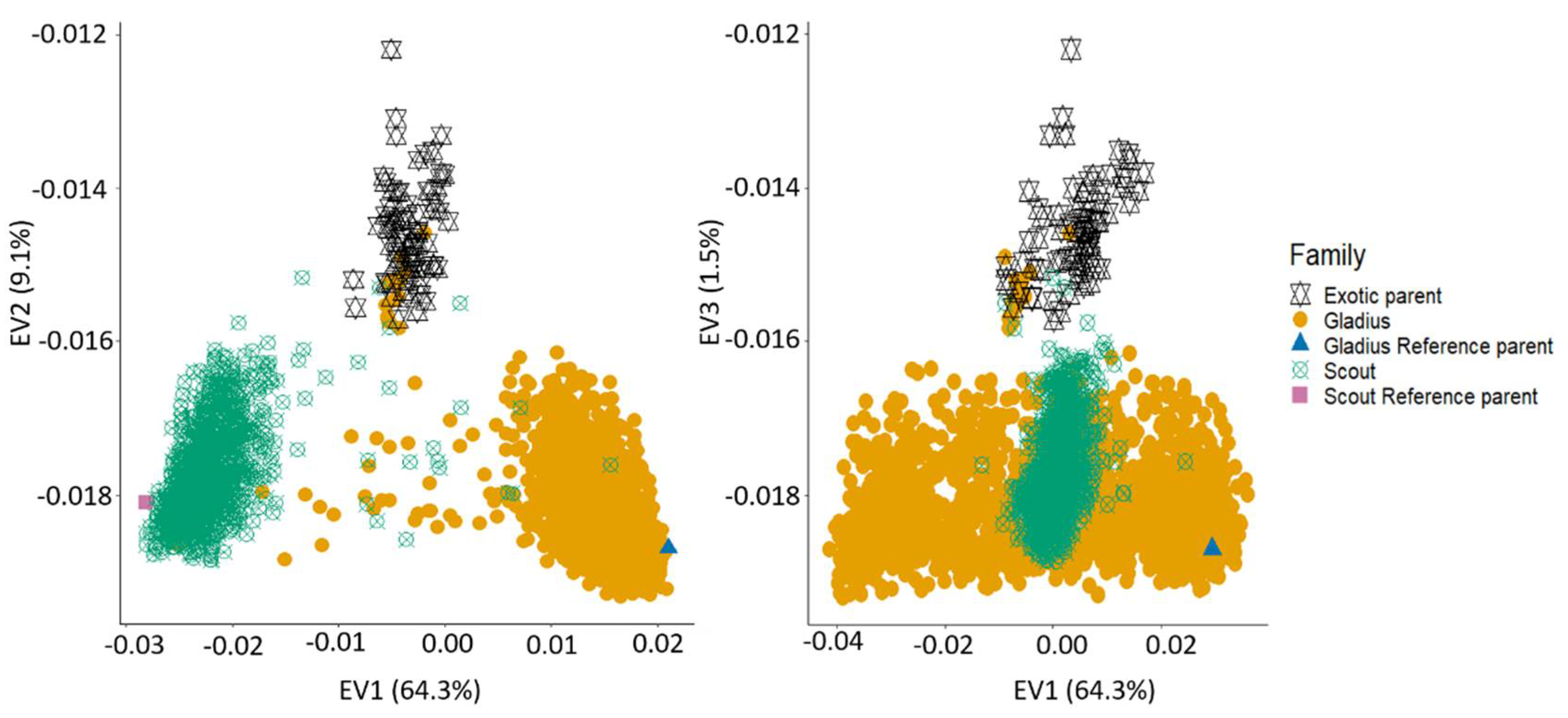
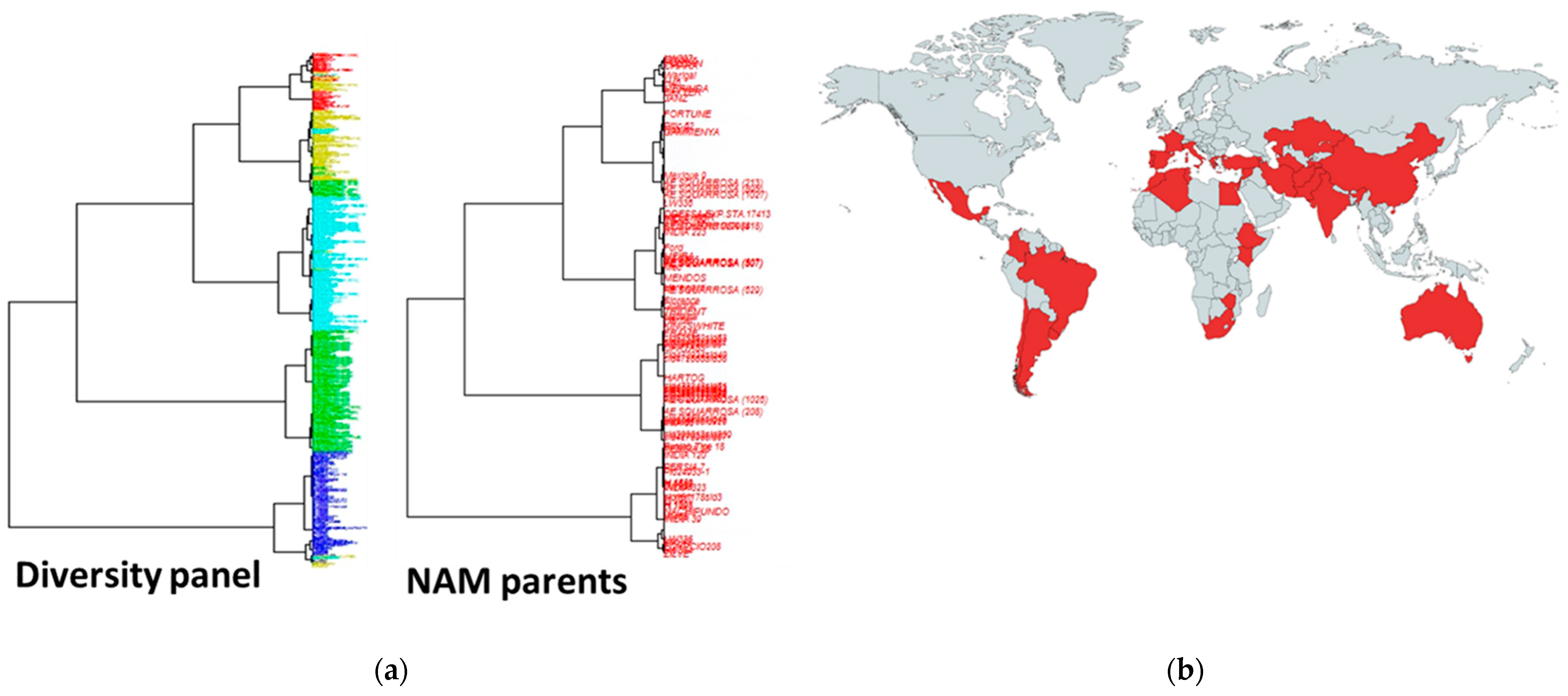
2.3. Genotypic Data and Population Structures
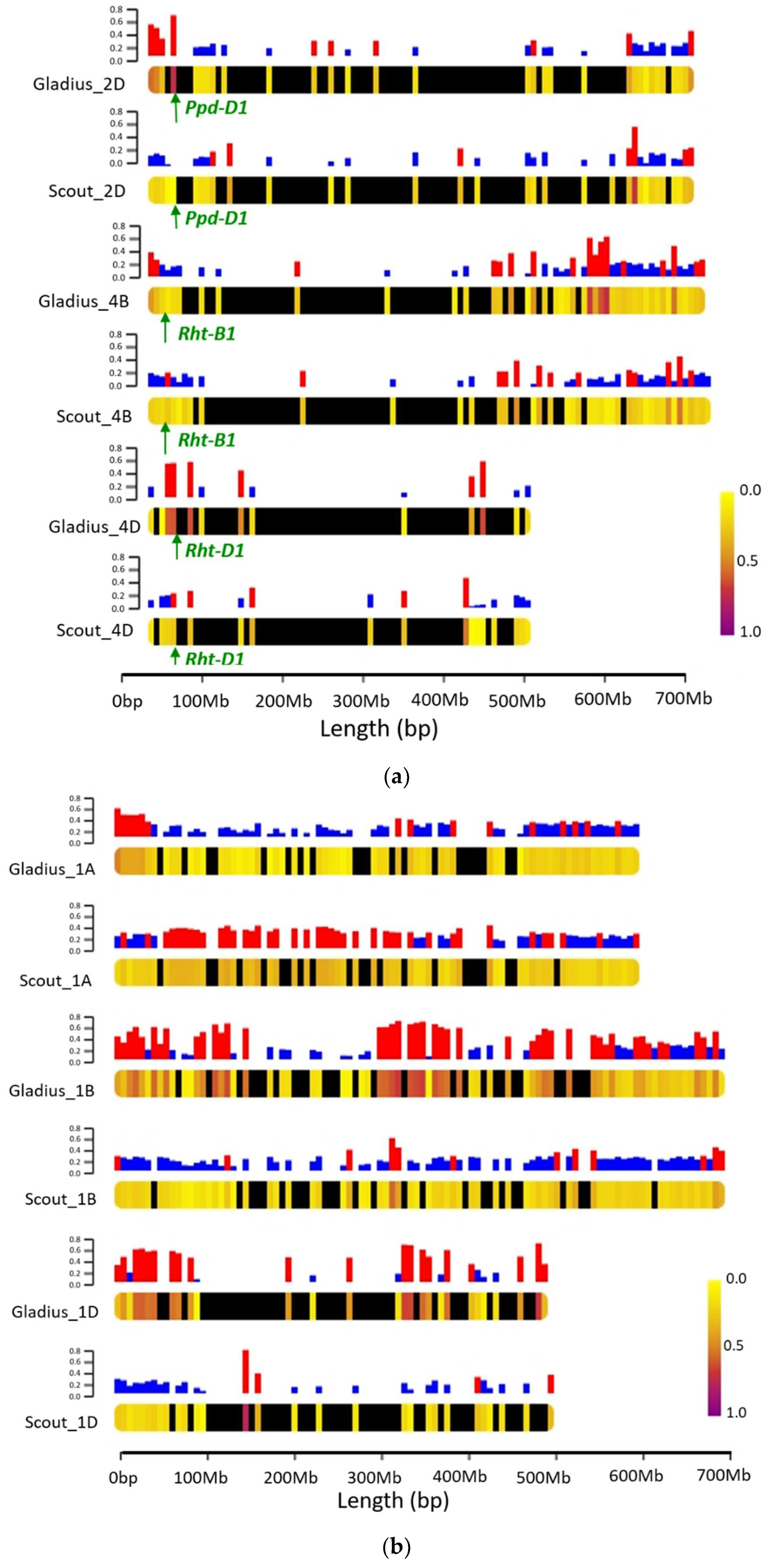
2.4. Introgression and Population Structure
2.5. QTL Mapping in the NAM Population
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Population Development
4.3. Genotyping of the NAM Population
4.3.1. Library Preparation
4.3.2. tGBS Allele Calling and Data Analysis
4.3.3. Linkage Disequilibrium Analysis and Data Imputation
4.4. Phenotyping and Statistical Analysis of the NAM Population
4.5. Introgression and Population Structure
4.6. QTL Mapping in the NAM Population
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackay, T.F. The genetic architecture of quantitative traits. Ann. Rev. Genet. 2001, 35, 303–339. [Google Scholar] [CrossRef]
- Mackay, T.F.C.; Stone, E.A.; Ayroles, J.F. The genetics of quantitative traits: Challenges and prospects. Nat. Rev. Genet. 2009, 10, 565–577. [Google Scholar] [CrossRef]
- Collins, N.C.; Tardieu, F.; Tuberosa, R. Quantitative trait loci and crop performance under abiotic stress: Where do we stand? Plant Physiol. 2008, 147, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Botstein, D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 1989, 121, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Myles, S.; Peiffer, J.; Brown, P.J.; Ersoz, E.S.; Zhang, Z.; Costich, D.E.; Buckler, E.S. Association mapping: Critical considerations shift from genotyping to experimental design. Plant Cell 2009, 21, 2194–2202. [Google Scholar] [CrossRef]
- Veldboom, L.; Lee, M.; Woodman, W. Molecular marker-facilitated studies in an elite maize population: I. linkage analysis and determination of QTL for morphological traits. Theor. Appl. Genet. 1994, 88, 7–16. [Google Scholar] [CrossRef]
- Liu, K.; Sun, X.; Ning, T.; Duan, X.; Wang, Q.; Liu, T.; An, Y.; Guan, X.; Tian, J.; Chen, J. Genetic dissection of wheat panicle traits using linkage analysis and a genome-wide association study. Theor. Appl. Genet. 2018, 131, 1073–1090. [Google Scholar] [CrossRef]
- Ezenwaka, L.; Rabbi, I.; Onyeka, J.; Kulakow, P.; Egesi, C. Identification of additional /novel QTL associated with resistance to cassava green mite in a biparental mapping population. PLoS ONE 2020, 15, e0231008. [Google Scholar] [CrossRef]
- Briggs, W.H.; McMullen, M.D.; Gaut, B.S.; Doebley, J. Linkage mapping of domestication loci in a large maize teosinte backcross resource. Genetics 2007, 177, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Flint-Garcia, S.A.; Thuillet, A.C.; Yu, J.; Pressoir, G.; Romero, S.M.; Mitchell, S.E.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Maize association population: A high-resolution platform for quantitative trait locus dissection. Plant J. Plant Mol. Biol. 2005, 44, 1054–1064. [Google Scholar] [CrossRef]
- Remington, D.L.; Thornsberry, J.M.; Matsuoka, Y.; Wilson, L.M.; Whitt, S.R.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 2001, 98, 11479–11484. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S., IV. Structure of linkage disequilibrium in plants. Ann. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef]
- Platt, A.; Vilhjálmsson, B.J.; Nordborg, M. Conditions under which genome-wide association studies will be positively misleading. Genetics 2010, 186, 1045–1052. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.; Roux, F. Towards identifying genes underlying ecologically relevant traits in Arabidopsis thaliana. Nat. Rev. Genet. 2010, 11, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. Genetic design and statistical power of nested association mapping in maize. Genetics 2008, 178, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Gage, J.L.; Monier, B.; Giri, A.; Buckler, E.S. Ten years of the maize nested association mapping population: Impact, limitations, and future directions. Plant Cell 2020, 32, 2083. [Google Scholar] [CrossRef] [PubMed]
- Vatter, T.; Maurer, A.; Kopahnke, D.; Perovic, D.; Ordon, F.; Pillen, K. A nested association mapping population identifies multiple small effect QTL conferring resistance against net blotch (Pyrenophora teres f. teres) in wild barley. PLoS ONE 2017, 12, e0186803. [Google Scholar] [CrossRef]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef]
- Cook, J.P.; McMullen, M.D.; Holland, J.B.; Tian, F.; Bradbury, P.; Ross-Ibarra, J.; Buckler, E.S.; Flint-Garcia, S.A. Genetic Architecture of maize kernel composition in the nested association mapping and inbred association panels. Plant Physiol. 2012, 158, 824–834. [Google Scholar] [CrossRef]
- Bajgain, P.; Rouse, M.N.; Tsilo, T.J.; Macharia, G.K.; Bhavani, S.; Jin, Y.; Anderson, J.A. Nested association mapping of stem rust resistance in wheat using genotyping by sequencing. PLoS ONE 2016, 11, e0155760. [Google Scholar] [CrossRef]
- Carroll, N.; Frijters, P.; Shields, M.A. Quantifying the costs of drought: New evidence from life satisfaction data. J. Popul. Econ. 2009, 22, 445–461. [Google Scholar] [CrossRef]
- Farooq, M.; Hussain, M.; Siddique, K.H.M. Drought stress in wheat during flowering and grain-filling periods. CRC Crit. Rev. Plant Sci. 2014, 33, 331–349. [Google Scholar] [CrossRef]
- Moose, S.P.; Mumm, R.H. Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol. 2008, 147, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Tricker, P.J.; Eckermann, P.; Kalambettu, P.; Garcia, M.; Fleury, D. Novel alleles for combined drought and heat stress tolerance in wheat. Front. Plant Sci. 2019, 10, 1800. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Eckermann, P.; Haefele, S.; Satija, S.; Sznajder, B.; Timmins, A.; Baumann, U.; Wolters, P.; Mather, D.E.; Fleury, D. Genome-wide association mapping of grain yield in a diverse collection of spring wheat (Triticum aestivum L.) evaluated in southern Australia. PLoS ONE 2019, 14, e0211730. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kulwal, P.L.; Balyan, H.S.; Gupta, P.K. QTL mapping for yield and yield contributing traits in two mapping populations of bread wheat. Mol. Breed. 2007, 19, 163–177. [Google Scholar] [CrossRef]
- Scott, M.F.; Ladejobi, O.; Amer, S.; Bentley, A.R.; Biernaskie, J.; Boden, S.A.; Clark, M.; Dell’Acqua, M.; Dixon, L.E.; Filippi, C.V.; et al. Multi-parent populations in crops: A toolbox integrating genomics and genetic mapping with breeding. Heredity 2020, 125, 396–416. [Google Scholar] [CrossRef]
- Huynh, B.L.; Ehlers, J.D.; Huang, B.E.; Muñoz-Amatriaín, M.; Lonardi, S.; Santos, J.R.P.; Ndeve, A.; Batieno, B.J.; Boukar, O.; Cisse, N.; et al. A multi-parent advanced generation inter-cross (MAGIC) population for genetic analysis and improvement of cowpea (Vigna unguiculata L. Walp.). Plant J. 2018, 93, 1129–1142. [Google Scholar] [CrossRef]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Beales, J.; Turner, A.; Griffiths, S.; Snape, J.W.; Laurie, D.A. A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor. Appl. Genet. 2007, 115, 721–733. [Google Scholar] [CrossRef]
- Ellis, M.; Spielmeyer, W.; Gale, K.; Rebetzke, G.; Richards, R. Perfect markers for the Rht-B1b and Rht-D1b dwarfing genes in wheat. Theor. Appl. Genet. 2002, 105, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hu, S.; Gardner, C.; Lübberstedt, T. Emerging avenues for utilization of exotic germplasm. Trends Plant Sci. 2017, 22, 624–637. [Google Scholar] [CrossRef] [PubMed]
- Zamir, D. Improving plant breeding with exotic genetic libraries. Nat. Rev. Genet. 2001, 2, 983–989. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, C.J.; York, A.M.; Xue, W.; Daskalska, L.L.; DeValk, C.A.; Krueger, K.W.; Lawton, S.B.; Spiegelberg, B.G.; Schnell, J.M.; et al. TeoNAM: A nested association mapping population for domestication and agronomic trait analysis in maize. Genetics 2019, 213, 1065–1078. [Google Scholar] [CrossRef]
- Fragoso, C.A.; Moreno, M.; Wang, Z.; Heffelfinger, C.; Arbelaez, L.J.; Aguirre, J.A.; Franco, N.; Romero, L.E.; Labadie, K.; Zhao, H.; et al. Genetic architecture of a rice nested association mapping population. G3 Genes Genom. Genet. 2017, 7, 1913–1926. [Google Scholar] [CrossRef]
- Kidane, Y.G.; Gesesse, C.A.; Hailemariam, B.N.; Desta, E.A.; Mengistu, D.K.; Fadda, C.; Pè, M.E.; Dell’Acqua, M. A large nested association mapping population for breeding and quantitative trait locus mapping in Ethiopian durum wheat. Plant Biotechnol. J. 2019, 17, 1380–1393. [Google Scholar] [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP Markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Gai, J.; Chen, L.; Zhang, Y.; Zhao, T.; Xing, G.; Xing, H. Genome-wide genetic dissection of germplasm resources and implications for breeding by design in soybean. Breed Sci. 2012, 61, 495–510. [Google Scholar] [CrossRef]
- Li, S.; Cao, Y.; He, J.; Zhao, T.; Gai, J. Detecting the QTL-allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure. Theor. Appl. Genet. 2017, 130, 2297–2314. [Google Scholar] [CrossRef] [PubMed]
- Jobson, E.M.; Johnston, R.E.; Oiestad, A.J.; Martin, J.M.; Giroux, M.J. The impact of the wheat Rht-B1b semi-dwarfing allele on photosynthesis and seed development under field conditions. Front. Plant Sci. 2019, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Iwata, H. Bayesian QTL mapping for multiple families derived from crossing a set of inbred lines to a reference line. Heredity 2009, 102, 497. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Bradbury, P.; Ersoz, E.; Buckler, E.S.; Wang, J. Joint QTL linkage mapping for multiple-cross mating design sharing one common parent. PLoS ONE 2011, 6, e17573. [Google Scholar] [CrossRef]
- Liu, W.; Gowda, M.; Steinhoff, J.; Maurer, H.P.; Würschum, T.; Longin, C.F.H.; Cossic, F.; Reif, J.C. Association mapping in an elite maize breeding population. Theor. Appl. Genet. 2011, 123, 847. [Google Scholar] [CrossRef]
- Mace, E.S.; Hunt, C.H.; Jordan, D.R. Supermodels: Sorghum and maize provide mutual insight into the genetics of flowering time. Theor. Appl. Genet. 2013, 126, 1377–1395. [Google Scholar] [CrossRef]
- Maurer, A.; Draba, V.; Jiang, Y.; Schnaithmann, F.; Sharma, R.; Schumann, E.; Kilian, B.; Reif, J.C.; Pillen, K. Modelling the genetic architecture of flowering time control in barley through nested association mapping. BMC Genom. 2015, 16, 290. [Google Scholar] [CrossRef]
- He, J.B.; Gai, J.Y. QTL-allele matrix detected from RTM-GWAS is a powerful tool for studies in genetics, evolution, and breeding by design of crops. J. Integr. Agric. 2020, 19, 1407–1410. [Google Scholar] [CrossRef]
- He, J.; Meng, S.; Zhao, T.; Xing, G.; Yang, S.; Li, Y.; Guan, R.; Lu, J.; Wang, Y.; Xia, Q.; et al. An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding. Theor. Appl. Genet. 2017, 130, 2327–2343. [Google Scholar] [CrossRef]
- Zikhali, M.; Griffiths, S. The Effect of Earliness per se (Eps) Genes on Flowering Time in Bread Wheat; Springer: Tokyo, Japan, 2015; pp. 339–345. [Google Scholar]
- Guo, Z.; Song, Y.; Zhou, R.; Ren, Z.; Jia, J. Discovery, evaluation and distribution of haplotypes of the wheat Ppd-D1 gene. New Phytol. 2010, 185, 841–851. [Google Scholar] [CrossRef]
- Yan, L.; Loukoianov, A.; Tranquilli, G.; Helguera, M.; Fahima, T.; Dubcovsky, J. Positional cloning of the wheat vernalization gene VRN1. Proc. Natl. Acad. Sci. USA 2003, 100, 6263–6268. [Google Scholar] [CrossRef]
- Miura, H.; Nakagawa, M.; Worland, A.J. Control of ear emergence time by chromosome 3A of wheat. Plant Breed. 1999, 118, 85–87. [Google Scholar] [CrossRef]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. Green revolution genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef]
- Sallam, A.H.; Manan, F.; Bajgain, P.; Martin, M.; Szinyei, T.; Conley, E.; Brown-Guedira, G.; Muehlbauer, G.J.; Anderson, J.A.; Steffenson, B.J. Genetic architecture of agronomic and quality traits in a nested association mapping population of spring wheat. TPG 2020, 13, e20051. [Google Scholar]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. Pear: A fast and accurate illumina paired-end reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Money, D.; Gardner, K.; Migicovsky, Z.; Schwaninger, H.; Zhong, G.-Y.; Myles, S. LinkImpute: Fast and accurate genotype imputation for nonmodel organisms. G3 Genes. Genom. Genet. 2015, 5, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Cullis, B.R.; Smith, A.B.; Coombes, N.E. On the design of early generation variety trials with correlated data. J. Agric. Biol. Environ. Stat. 2006, 11, 381. [Google Scholar] [CrossRef]
- Rodríguez-Álvarez, M.X.; Boer, M.P.; van Eeuwijk, F.A.; Eilers, P.H.C. Correcting for spatial heterogeneity in plant breeding experiments with P-splines. Spat. Stat. 2018, 23, 52–71. [Google Scholar] [CrossRef]
- Oakey, H.; Verbyla, A.P.; Cullis, B.R.; Wei, X.; Pitchford, W.S. Joint modeling of additive and non-additive (genetic line) effects in multi-environment trials. Theor. Appl. Genet. 2007, 114, 1319–1332. [Google Scholar] [CrossRef]
- Anand, L. chromoMap: An R package for interactive visualization and annotation of chromosomes. bioRxiv 2020, 605600. [Google Scholar] [CrossRef]
- Li, S.; Xu, H.; Yang, J.; Zhao, T. Dissecting the genetic architecture of seed protein and oil content in soybean from the Yangtze and Huaihe River valleys using multi-locus genome-wide association studies. Int. J. Mol. Sci. 2019, 20, 3041. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Uauy, C.; Distelfeld, A.; Fahima, T.; Blechl, A.; Dubcovsky, J. A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 2006, 314, 1298–1301. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Trial | Min | Max | Mean | N | SD | H2 |
|---|---|---|---|---|---|---|---|
| Zadoks’ score | Roseworthy 2017 | 40 | 68 | 54.0 | 546 | 5.7 | 0.93 |
| Roseworthy 2018 | 39 | 69 | 60 | 252 | 5.2 | 0.93 | |
| Dandaragan 2018 | 39 | 74 | 62 | 252 | 6.7 | 0.8 | |
| Roseworthy 2019 | 39 | 70 | 59 | 252 | 5.9 | 0.91 | |
| Height (cm) | Roseworthy 2017 | 55 | 123 | 97 | 511 | 10.4 | 0.95 |
| Dandaragan 2018 | 40 | 128 | 92 | 252 | 8.6 | 0.81 | |
| Roseworthy 2019 | 60 | 94 | 77 | 252 | 5.8 | 0.9 |
| Trait | Environment | Marker | Chromosome | Number of Alleles | Corresponding Gene | p-Value | R2 (%) |
|---|---|---|---|---|---|---|---|
| Zadoks’ Score (maturity) | 2017_Roseworthy | LDB_2D_1 | 2D | 2 | Ppd-D1 | 1.56283 × 10−46 | 34.3 |
| LDB_3A_624535417 | 3A | 5 | - | 1.01741 × 10−5 | 3.5 | ||
| LDB_2B_58324935 | 2B | 3 | - | 3.80867 × 10−5 | 3.2 | ||
| 2018_Dandarangan | LDB_2D_1 | 2D | 2 | Ppd-D1 | 4.09762 × 10−16 | 26.9 | |
| LDB_3A_510690367 | 3A | 3 | - | 1.57639 × 10−5 | 7.1 | ||
| 2018 Roseworthy | LDB_2D_1 | 2D | 2 | Ppd-D1 | 1.26164 × 10−15 | 28.5 | |
| LDB_5A_1 | 5A | 3 | - | 1.29529 × 10−5 | 9.4 | ||
| 2019_Roseworthy | LDB_2D_1 | 2D | 2 | Ppd-D1 | 7.46238 × 10−20 | 34.1 | |
| Plant Height | Roseworthy 2017 | LDB_4D_1 | 4D | 2 | Rht-D1 | 2.23596 × 10−49 | 39.6 |
| LDB_4B_1 | 4B | 2 | Rht-B1 | 2.80 × 10−25 | 13.6 | ||
| Dandaragan 2018 | LDB_4D_1 | 4D | 2 | Rht-D1 | 1.3935 × 10−14 | 27.1 | |
| LDB_6B_679622060 | 6B | 11 | - | 7.20103 × 10−13 | 18.6 | ||
| Roseworthy 2019 | LDB_4D_1 | 4D | 2 | Rht-D1 | 1.58475 × 10−11 | 21.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chidzanga, C.; Fleury, D.; Baumann, U.; Mullan, D.; Watanabe, S.; Kalambettu, P.; Pontre, R.; Edwards, J.; Forrest, K.; Wong, D.; et al. Development of an Australian Bread Wheat Nested Association Mapping Population, a New Genetic Diversity Resource for Breeding under Dry and Hot Climates. Int. J. Mol. Sci. 2021, 22, 4348. https://doi.org/10.3390/ijms22094348
Chidzanga C, Fleury D, Baumann U, Mullan D, Watanabe S, Kalambettu P, Pontre R, Edwards J, Forrest K, Wong D, et al. Development of an Australian Bread Wheat Nested Association Mapping Population, a New Genetic Diversity Resource for Breeding under Dry and Hot Climates. International Journal of Molecular Sciences. 2021; 22(9):4348. https://doi.org/10.3390/ijms22094348
Chicago/Turabian StyleChidzanga, Charity, Delphine Fleury, Ute Baumann, Dan Mullan, Sayuri Watanabe, Priyanka Kalambettu, Robert Pontre, James Edwards, Kerrie Forrest, Debbie Wong, and et al. 2021. "Development of an Australian Bread Wheat Nested Association Mapping Population, a New Genetic Diversity Resource for Breeding under Dry and Hot Climates" International Journal of Molecular Sciences 22, no. 9: 4348. https://doi.org/10.3390/ijms22094348
APA StyleChidzanga, C., Fleury, D., Baumann, U., Mullan, D., Watanabe, S., Kalambettu, P., Pontre, R., Edwards, J., Forrest, K., Wong, D., Langridge, P., Chalmers, K., & Garcia, M. (2021). Development of an Australian Bread Wheat Nested Association Mapping Population, a New Genetic Diversity Resource for Breeding under Dry and Hot Climates. International Journal of Molecular Sciences, 22(9), 4348. https://doi.org/10.3390/ijms22094348