
Structural and Functional Characterization of the ABA-Water Deficit Stress Domain from Wheat and Barley: An Intrinsically Disordered Domain behind the Versatile Functions of the Plant Abscissic Acid, Stress and Ripening Protein Family



Abstract
1. Introduction
2. Results
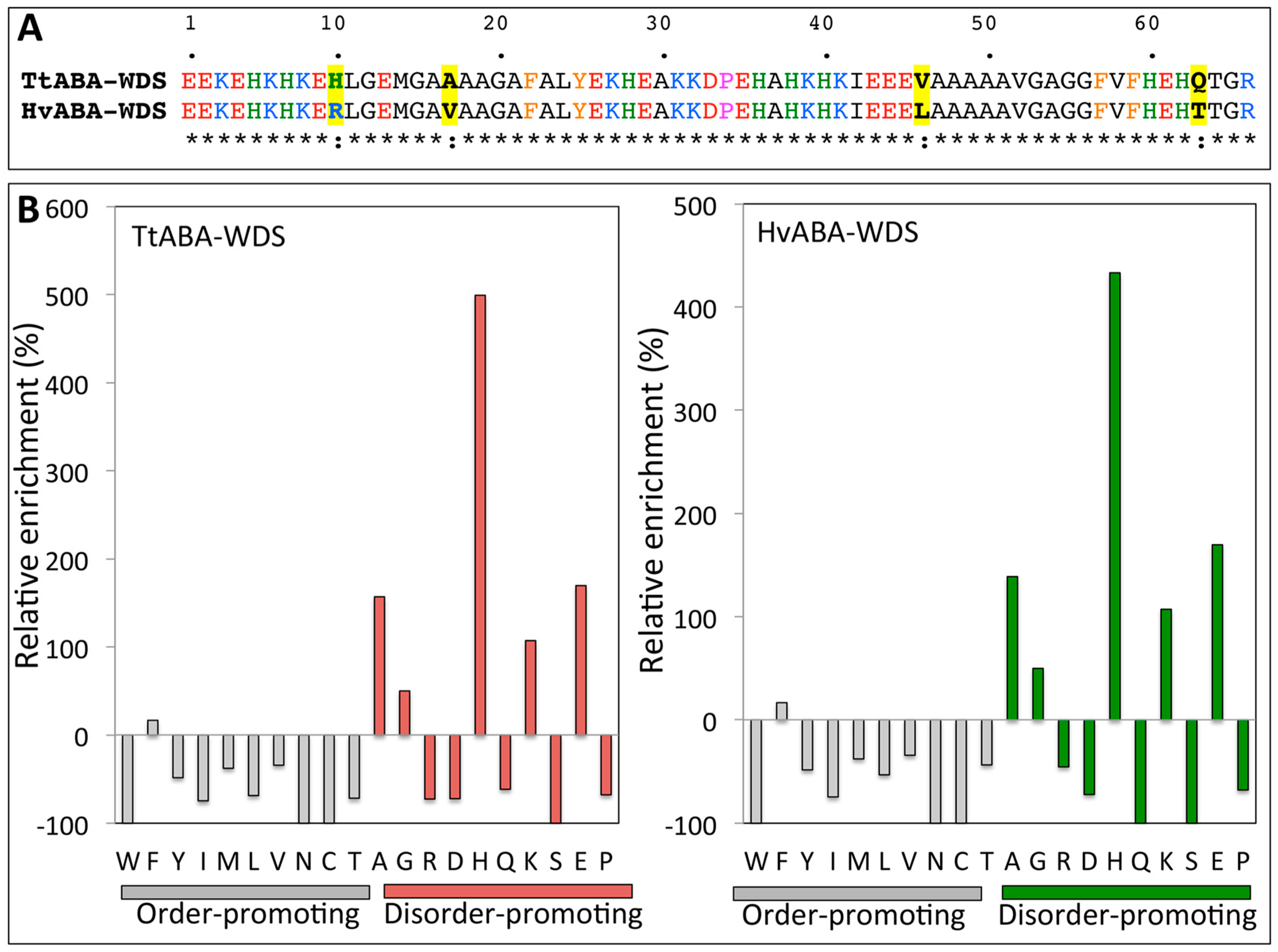
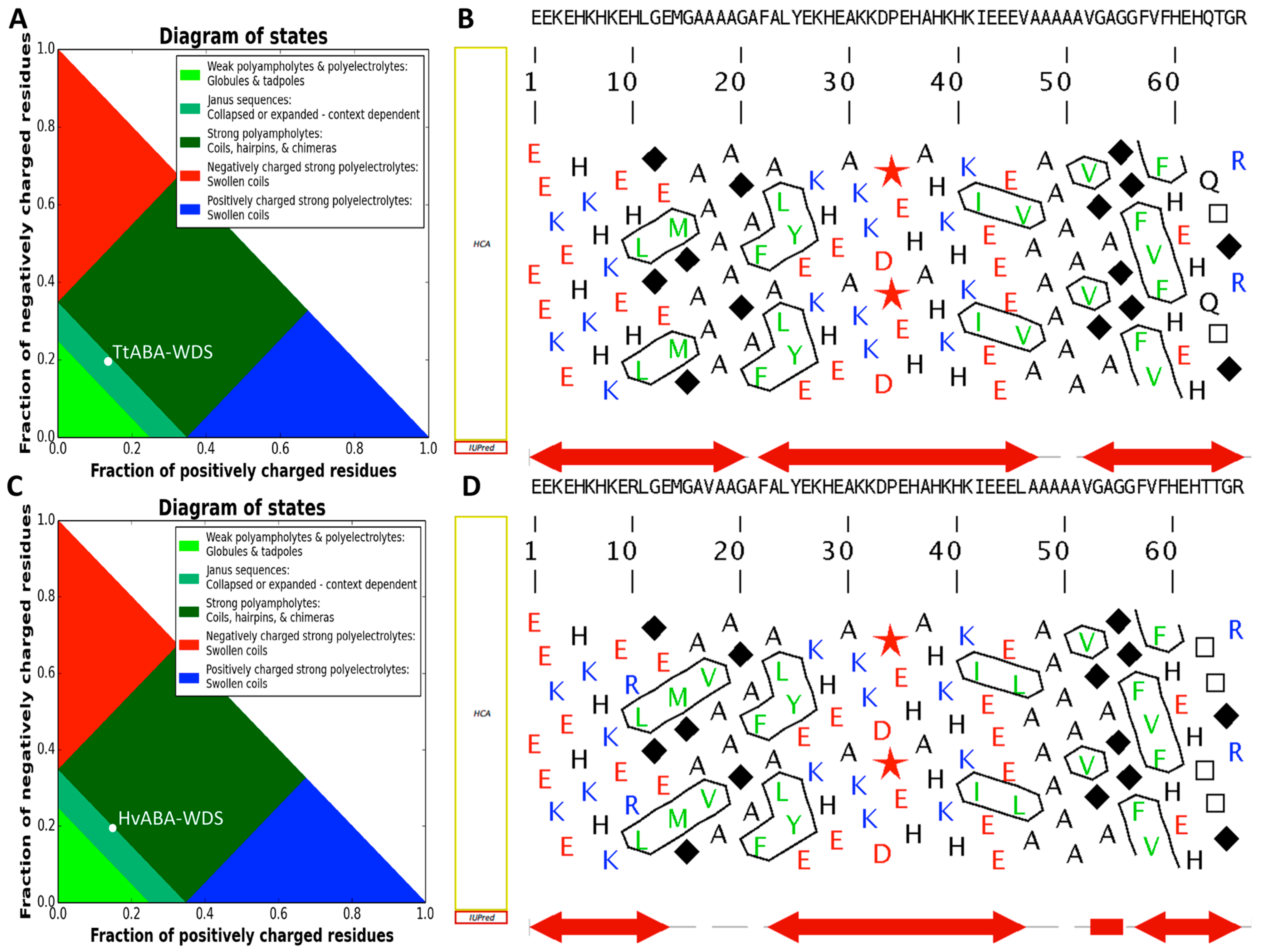
2.1. Cloning of TtABA-WDS and HvABA-WDS cDNA and Disorder Prediction of the Encoded Protein Domains
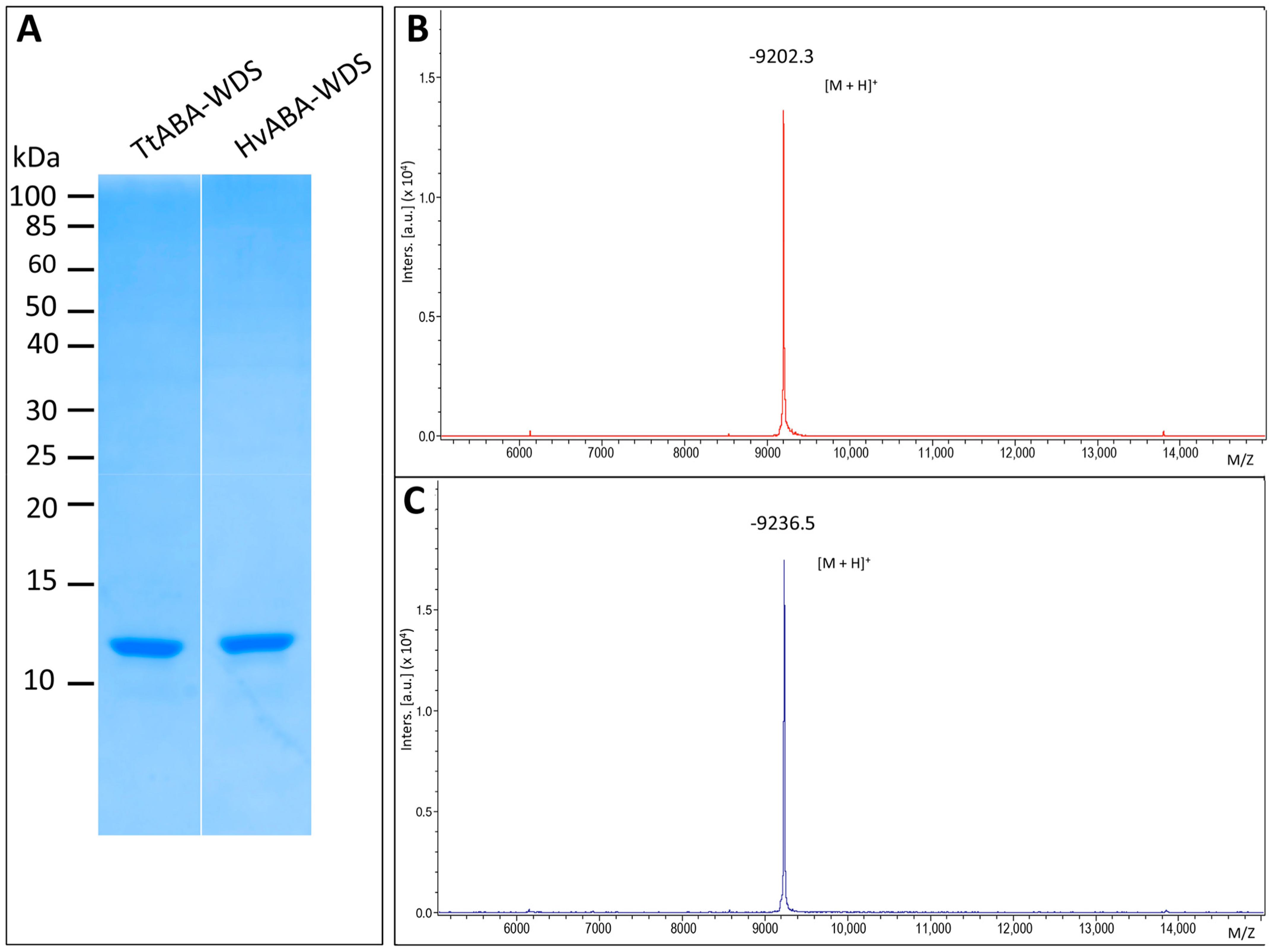
2.2. Expression and Purification of HvABA-WDS and TtABA-WDS Domains
2.3. Hydrodynamiques Properties of HvABA-WDS and TtABA-WDS Domains from Size Exclusion Chromatography (SEC)
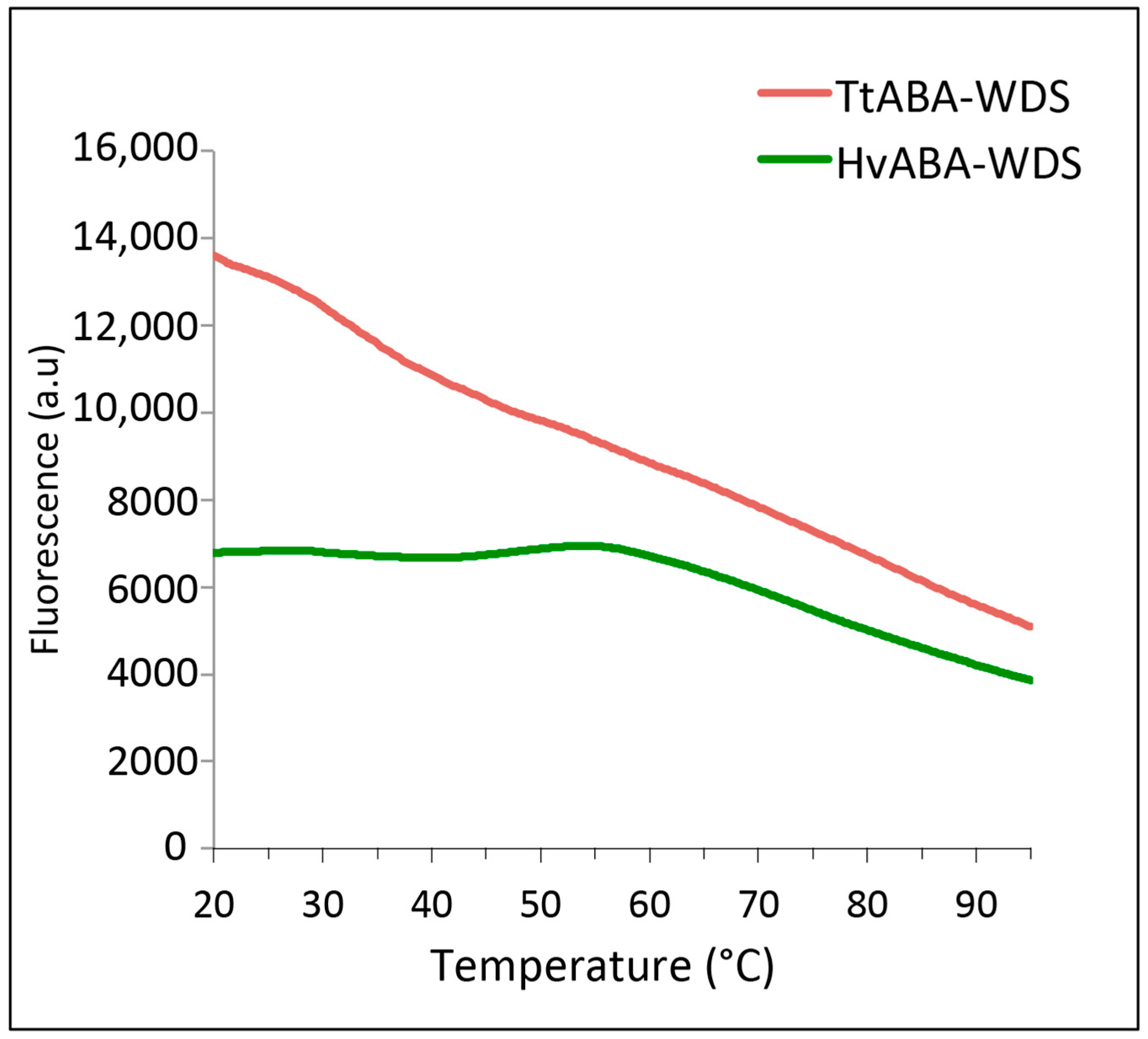
2.4. Differential Scanning Fluorimetry of HvABA-WDS and TtABA-WDS Domains
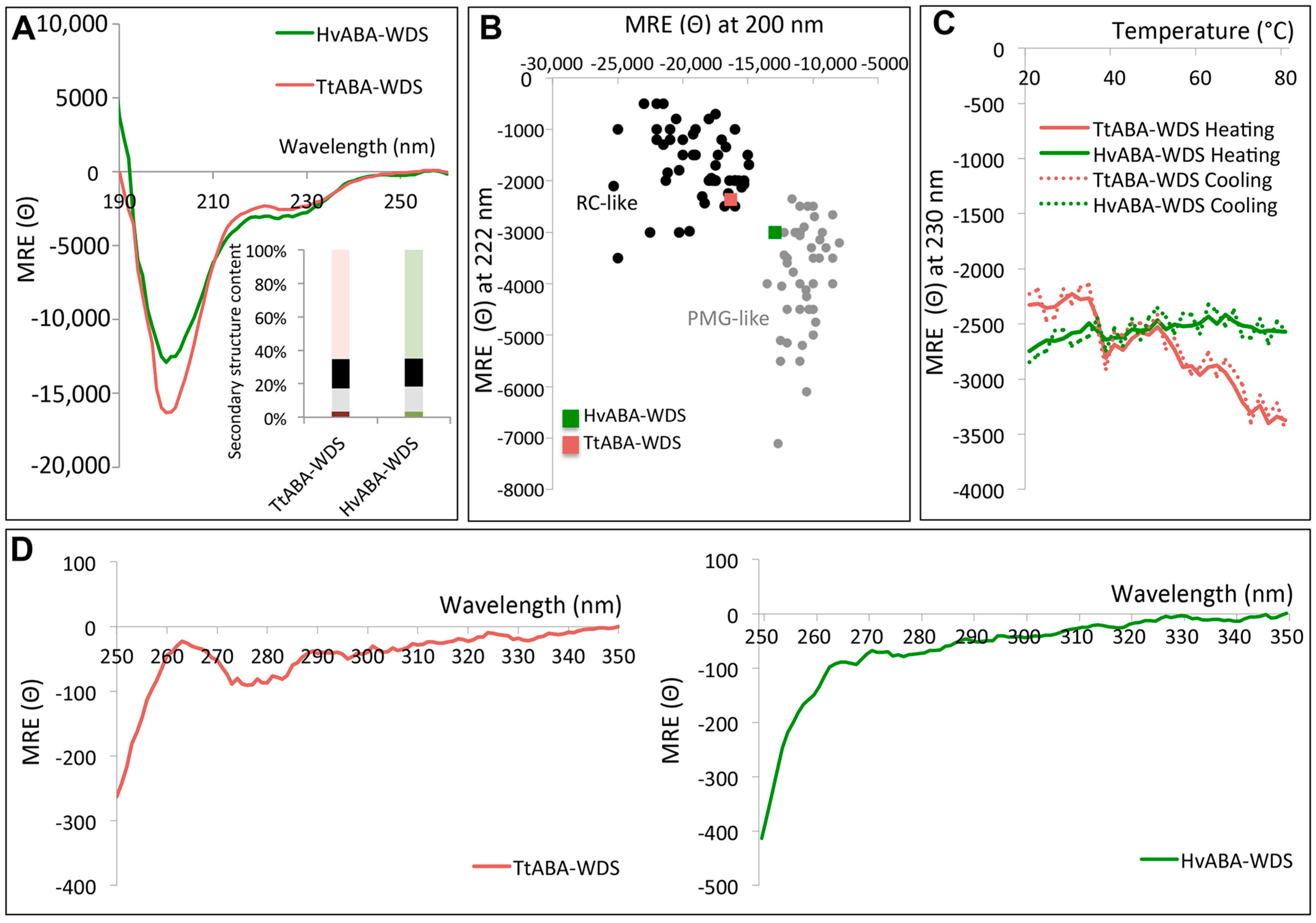
2.5. Circular Dichroism (CD) Studies of HvABA-WDS and TtABA-WDS
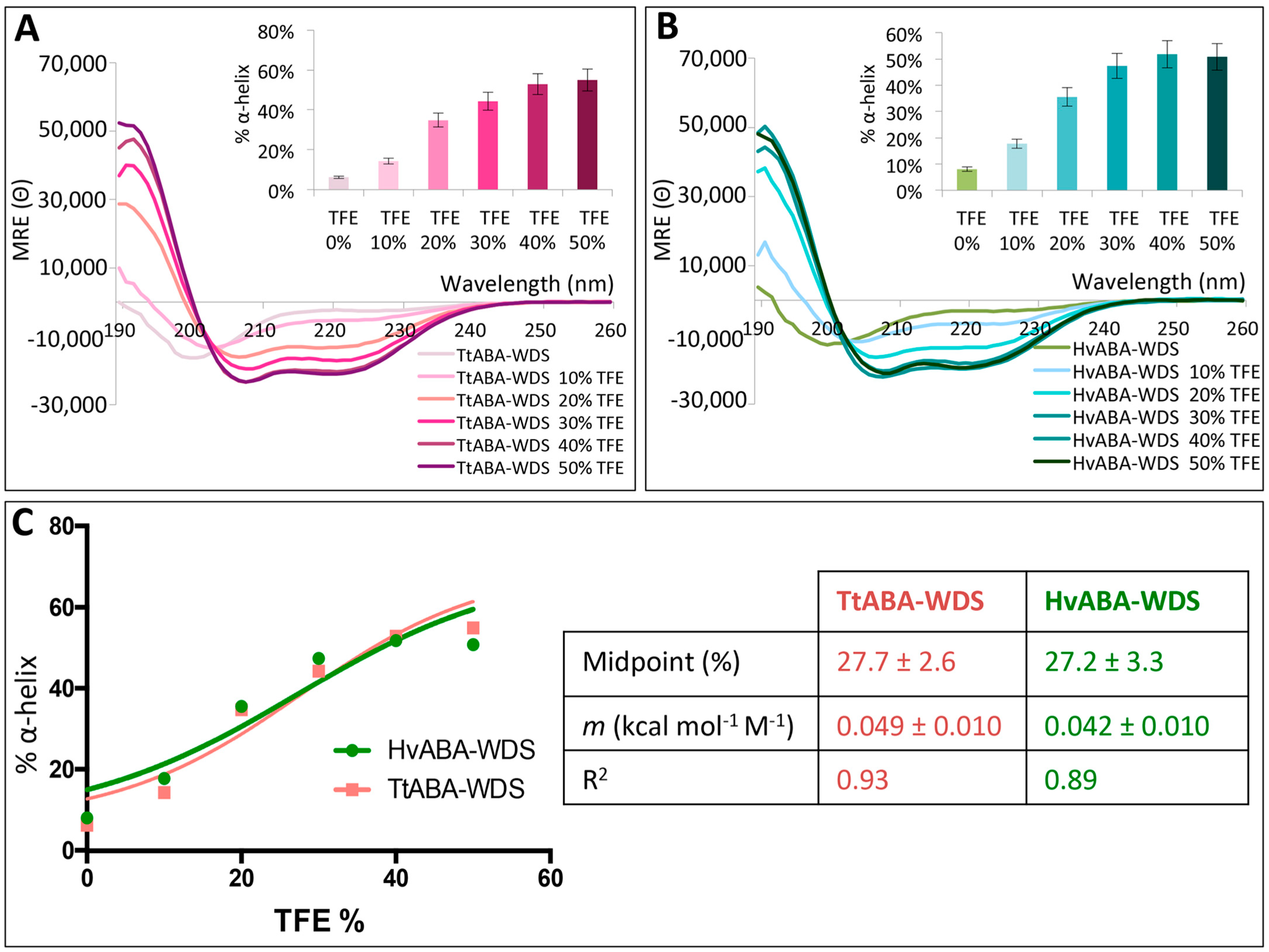
2.6. TtABA-WDS and HvABA-WDS Domains Have an Intrinsic Propensity to Fold into an α-Helical Conformation
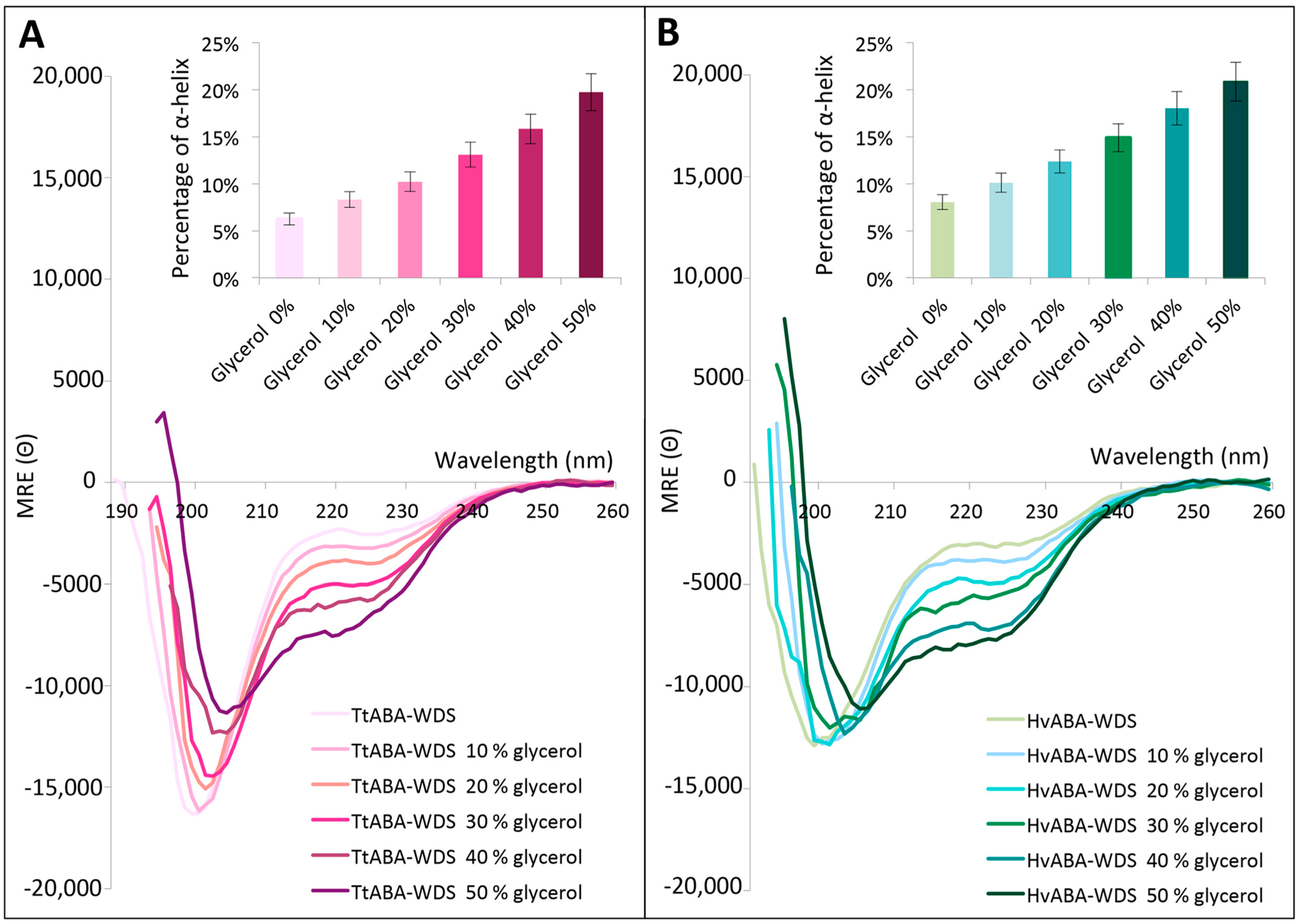
2.7. TtABA-WDS and HvABA-WDS Domains Gain α-Helicity under Conditions Mimicking Dehydration
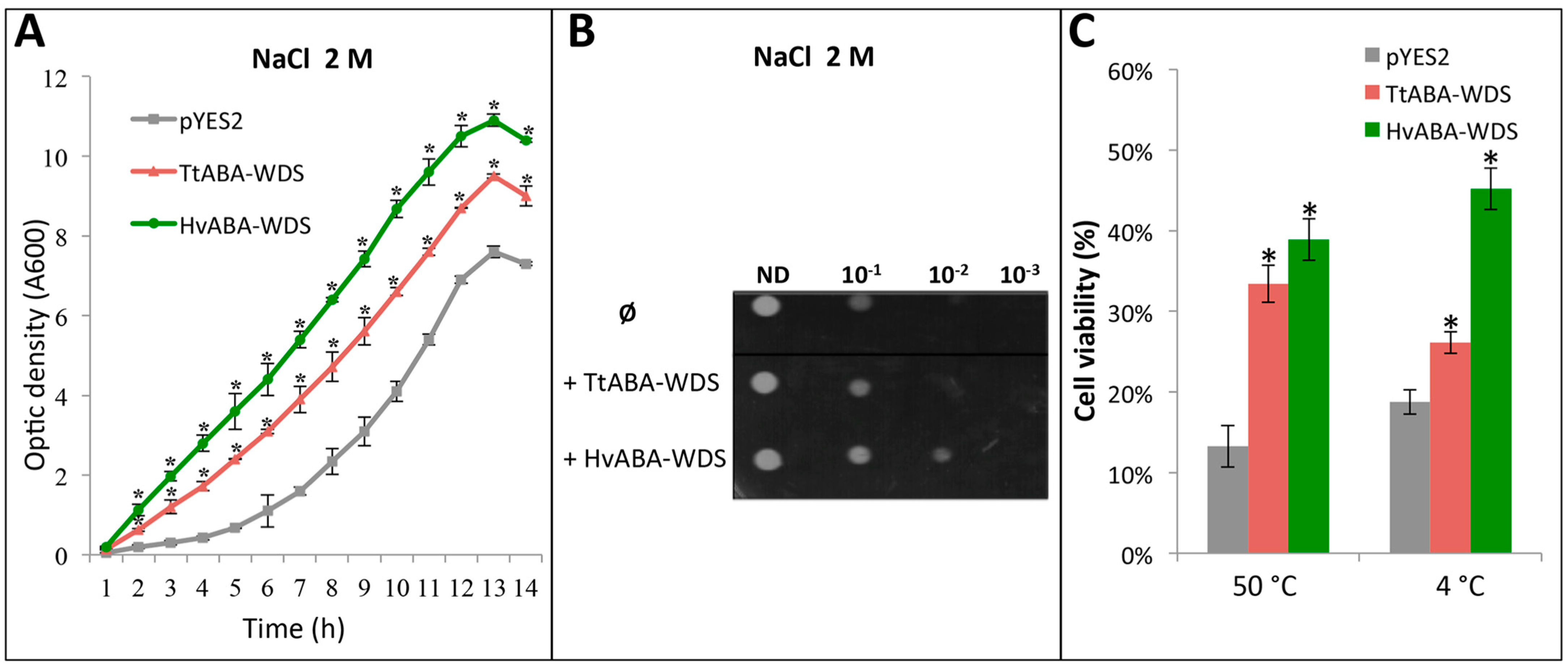
2.8. TtABA-WDS and HvABA-WDS Confer Salt, Heat and Cold Stress Tolerance to Yeast Cells
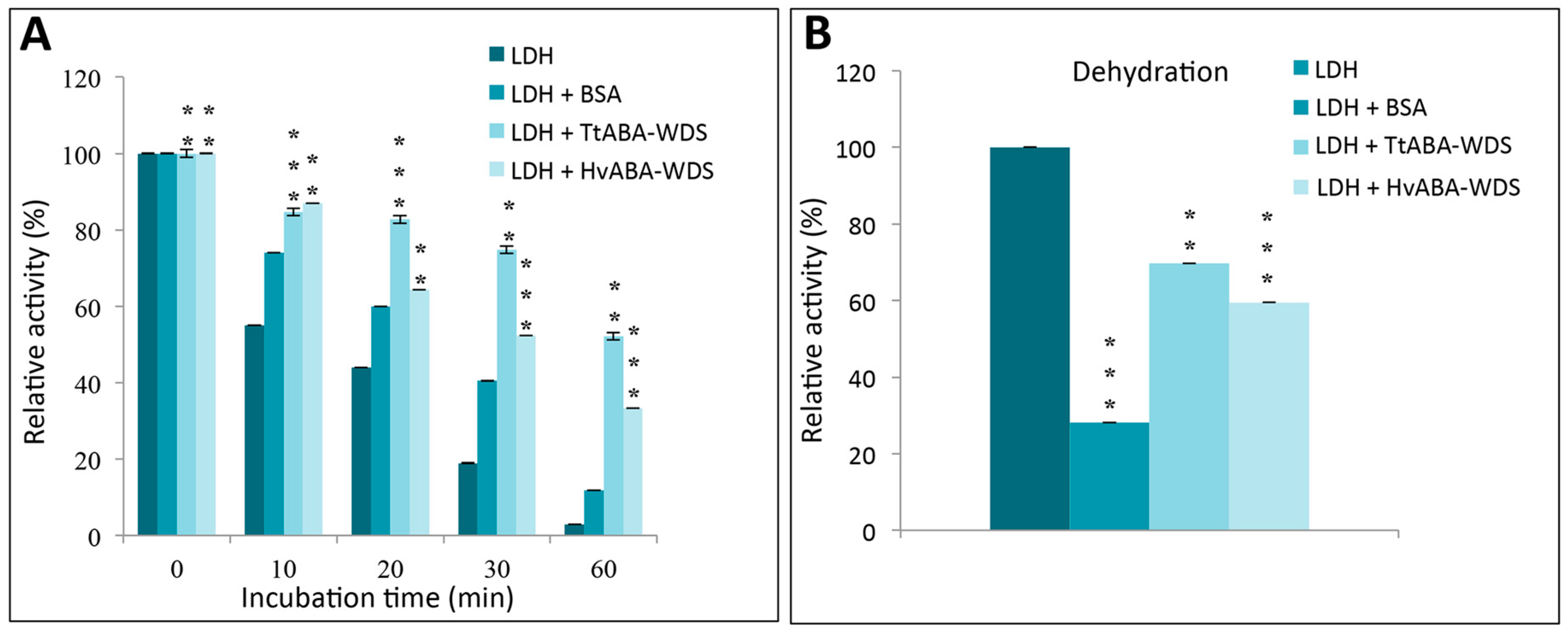
2.9. HvABA-WDS and TtABA-WDS Domains Stabilize the Enzyme LDH against Thermal and Dessication Stress
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Stress Treatments
4.2. RNA Extraction and Amplification of TtABA-WDS and HvABA-WDS cDNA Domains from Durum Wheat and Barely
4.3. Cloning, Expression, and Production of Recombinant TtABA-WDS and HvABA-WDS cDNA in E. coli
4.4. Mass Spectrometry
4.4.1. Intact Protein Mass Analysis
4.4.2. Peptide Mass Fingerprinting
4.5. Size Exclusion Chromatography and Calculation of Hydrodynamic Radii
4.6. Differential Scanning Fluorimetry (DSF)
4.7. Circular Dichroism (CD) Measurements
4.8. Salt Tolerance and Growth Assays of Yeast Cells
4.9. LDH Protective Assay
4.10. Bioinformatics Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABA | Abscissic acid |
| ASR | Abscissic acid stress ripening protein |
| ABA_WDS | Abscissic acid water deficit stress domain |
| IDPs | Intrinsically disordered proteins |
| TF | Transcription factor |
| LEA | Late embryogenesis abundant protein |
| IDRs | Intrinsically disordered regions |
| IDDs | Intrinsically disordered domains |
| TFE | Trifluoroethanol |
| LDH | Lactate dehydrogenase |
| NaCl | Sodium Chloride |
| GST | Glutathione S-transferase |
| RS | Stokes radii |
| SEC | Size Exclusion Chromatography |
| DSF | Differential scanning fluorimetry |
| CD | Circular dichroism |
| BSA | Bovine serum albumin |
| CDD | Conserved Domain Database |
References
- Doupis, G.; Chartzoulakis, K.; Beis, A.; Patakas, A. Allometric and biochemical responses of grapevines subjected to drought and enhanced ultraviolet-B radiation. Aust. J. Grape Wine 2011, 17, 36–42. [Google Scholar] [CrossRef]
- Iusem, N.D.; Bartholomew, D.M.; Hitz, W.D.; Scolnik, P.A. Tomato (Lycopersicon esculentum) transcript induced by water deficit and ripening. Plant Physiol. 1993, 102, 1353–1354. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.M.; Iusem, N.D. Twenty years of research on Asr (ABA-stress-ripening) genes and proteins. Planta 2014, 239, 941–949. [Google Scholar] [CrossRef]
- Saumonneau, A.; Laloi, M.; Lallemand, M.; Rabot, A.; Atanassova, R. Dissection of the transcriptional regulation of grape ASR and response to glucose and abscisic acid. J. Exp. Bot. 2012, 63, 1495–1510. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Singh, P.C.; Chaudhry, V.; Shirke, P.A.; Chakrabarty, D.; Farooqui, A.; Nautiyal, C.S.; Sane, A.P.; Sane, V.A. PGPR-induced OsASR6 improves plant growth and yield by altering root auxin sensitivity and the xylem structure in transgenic Arabidopsis thaliana. J. Plant Physiol. 2019, 240, 153010. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, W.; Feng, J.; Yang, X.; Huang, Q.; Xiao, J.; Liu, Y.; Yang, G.; He, G. Identification of the ASR gene family from Brachypodium distachyon and functional characterization of BdASR1 in response to drought stress. Plant Cell Rep. 2016, 35, 1221–1234. [Google Scholar] [CrossRef]
- Goldgur, Y.; Rom, S.; Ghirlando, R.; Shkolnik, D.; Shadrin, N.; Konrad, Z.; Bar-Zvi, D. Desiccation and zinc binding induce transition of tomato abscisic acid stress ripening 1, a water stress- and salt stress-regulated plant-specific protein, from unfolded to folded state. Plant Physiol. 2007, 143, 617–628. [Google Scholar] [CrossRef]
- Ricardi, M.M.; González, R.M.; Zhong, S.; Domínguez, P.G.; Duffy, T.; Turjanski, P.G.; Salter, J.D.S.; Alleva, K.; Carrari, F.; Giovannoni, J.J. Genome-wide data (ChIP-seq) enabled identification of cell wall-related and aquaporin genes as targets of tomato ASR1, a drought stress-responsive transcription factor. BMC Plant Biol. 2014, 14, 29. [Google Scholar] [CrossRef]
- Hamdi, K.; Brini, F.; Kharrat, N.; Masmoudi, K.; Yakoubi, I. Abscisic Acid, Stress, and Ripening (TtASR1) Gene as a Functional Marker for Salt Tolerance in Durum Wheat. BioMed. Res. Int. 2020, 2020, 7876357. [Google Scholar] [CrossRef]
- Arenhart, R.A.; Schunemann, M.; Bucker Neto, L.; Margis, R.; Wang, Z.Y.; Margis-Pinheiro, M. Rice ASR1 and ASR5 are complementary transcription factors regulating aluminium responsive genes. Plant Cell Environ. 2016, 39, 645–651. [Google Scholar] [CrossRef]
- Virlouvet, L.; Jacquemot, M.P.; Gerentes, D.; Corti, H.; Bouton, S.; Gilard, F.; Valot, B.; Trouverie, J.; Tcherkez, G.; Falque, M.; et al. The ZmASR1 protein influences branched-chain amino acid biosynthesis and maintains kernel yield in maize under water-limited conditions. Plant Physiol. 2011, 157, 917–936. [Google Scholar] [CrossRef]
- Hu, W.; Huang, C.; Deng, X.; Zhou, S.; Chen, L.; Li, Y.; Wang, C.; Ma, Z.; Yuan, Q.; Wang, Y.; et al. TaASR1, a transcription factor gene in wheat, confers drought stress tolerance in transgenic tobacco. Plant Cell. Environ. 2013, 36, 1449–1464. [Google Scholar] [CrossRef]
- Dai, J.R.; Liu, B.; Feng, D.R.; Liu, H.Y.; He, Y.M.; Qi, K.B.; Wang, H.B.; Wang, J.F. MpAsr encodes an intrinsically unstructured protein and enhances osmotic tolerance in transgenic Arabidopsis. Plant Cell Rep. 2011, 30, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Yin, Z.; Jiang, J.; Zhang, M.; Guo, X.; Ye, Z.; Zhao, Y.; Xiong, H.; Zhang, Z. OsASR5 enhances drought tolerance through a stomatal closure pathway associated with ABA and H2O2 signalling in rice. Plant Biotechnol. J. 2017, 15, 183–196. [Google Scholar] [CrossRef]
- Jha, B.; Lal, S.; Tiwari, V.; Yadav, S.K.; Agarwal, P.K. The SbASR-1 gene cloned from an extreme halophyte Salicornia brachiata enhances salt tolerance in transgenic tobacco. Mar. Biotechnol. 2012, 14, 782–792. [Google Scholar] [CrossRef]
- Tiwari, V.; Chaturvedi, A.K.; Mishra, A.; Jha, B. Introgression of the SbASR-1 gene cloned from a halophyte Salicornia brachiata enhances salinity and drought endurance in transgenic groundnut (Arachis hypogaea) and acts as a transcription factor. PLoS ONE 2015, 10, e0131567. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, K.; Salladini, E.; O’Brien, D.P.; Brier, S.; Chenal, A.; Yacoubi, I.; Longhi, S. Structural disorder and induced folding within two cereal, ABA stress and ripening (ASR) proteins. Sci. Rep. 2017, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Kim, J.Y.; Lee, M.B.; Seo, Y.W. Over-expression of the Brachypodium ASR gene, BdASR4, enhances drought tolerance in Brachypodium distachyon. Plant Cell Rep. 2019, 38, 1109–1125. [Google Scholar] [CrossRef] [PubMed]
- Wetzler, D.E.; Wightman, F.F.; Bucci, H.A.; Rinaldi, J.; Caramelo, J.J.; Iusem, N.D.; Ricardi, M.M. Conformational plasticity of the intrinsically disordered protein ASR1 modulates its function as a drought stress-responsive gene. PLoS ONE 2018, 13, e0202808. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed]
- Olvera-Carrillo, Y.; Luis Reyes, J.; Covarrubias, A.A. Late embryogenesis abundant proteins: Versatile players in the plant adaptation to water limiting environments. Plant Signal Behav. 2011, 6, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Rikkerink, E.H.; Jones, W.T.; Uversky, V.N. Multifarious roles of intrinsic disorder in proteins illustrate its broad impact on plant biology. Plant Cell 2013, 25, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Olvera-Carrillo, Y.; Garciarrubio, A.; Campos, F.; Covarrubias, A.A. The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 2008, 148, 6–24. [Google Scholar] [CrossRef]
- Caramelo, J.J.; Iusem, N.D. When cells lose water: Lessons from biophysics and molecular biology. Prog. Biophys. Mol. Biol. 2009, 99, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hunault, G.; Jaspard, E. LEAPdb: A database for the late embryogenesis abundant proteins. BMC Genom. 2010, 11, 221. [Google Scholar] [CrossRef] [PubMed]
- Jaspard, E.; Macherel, D.; Hunault, G. Computational and statistical analyses of amino acid usage and physico-chemical properties of the twelve late embryogenesis abundant protein classes. PLoS ONE 2012, 7, e36968. [Google Scholar] [CrossRef]
- Philippe, R.; Courtois, B.; McNally, K.L.; Mournet, P.; El-Malki, R.; Le Paslier, M.C.; Fabre, D.; Billot, C.; Brunel, D.; Glaszmann, J.-C. Structure, allelic diversity and selection of Asr genes, candidate for drought tolerance, in Oryza sativa L. and wild relatives. Theor. Appl. Genet. 2010, 121, 769–787. [Google Scholar] [CrossRef]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. TOP-IDP-scale: A new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef]
- Müller-Späth, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Rüegger, S.; Reymond, L.; Nettels, D.; Schuler, B. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614. [Google Scholar] [CrossRef]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef]
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef]
- Lieutaud, P.; Canard, B.; Longhi, S. MeDor: A metaserver for predicting protein disorder. BMC Genom. 2008, 9 (Suppl. S2), S25. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Labesse, G.; Durand, P.; Poupon, A.; Canard, L.; Chomilier, J.; Henrissat, B.; Mornon, J.P. Deciphering protein sequence information through hydrophobic cluster analysis (HCA): Current status and perspectives. Cell Mol. Life Sci. 1997, 53, 621–645. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Schramm, A.; Bignon, C.; Brocca, S.; Grandori, R.; Santambrogio, C.; Longhi, S. An arsenal of methods for the experimental characterization of intrinsically disordered proteins–How to choose and combine them? Arch. Biochem. Biophys. 2019, 676, 108055. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef]
- Vincentelli, R.; Canaan, S.; Campanacci, V.; Valencia, C.; Maurin, D.; Frassinetti, F.; Scappucini-Calvo, L.; Bourne, Y.; Cambillau, C.; Bignon, C. High-throughput automated refolding screening of inclusion bodies. Protein Sci. 2004, 13, 2782–2792. [Google Scholar] [CrossRef]
- Kelly, S.M.; Price, N.C. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 2000, 1, 349–384. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.W. Circular dichroism of intrinsically disordered proteins. In Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 303–321. [Google Scholar]
- DeForte, S.; Uversky, V.N. Order, disorder, and everything in between. Molecules 2016, 21, 1090. [Google Scholar] [CrossRef]
- Hua, Q.-X.; Jia, W.-H.; Bullock, B.P.; Habener, J.F.; Weiss, M.A. Transcriptional activator-coactivator recognition: Nascent folding of a kinase-inducible transactivation domain predicts its structure on coactivator binding. Biochemistry 1998, 37, 5858–5866. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, M.; Mercurio, F.A.; Leone, M. About TFE: Old and New Findings. Curr. Protein Pept. Sci. 2019, 20, 425–451. [Google Scholar] [CrossRef] [PubMed]
- Receveur-Bréchot, V.; Bourhis, J.M.; Uversky, V.N.; Canard, B.; Longhi, S. Assessing protein disorder and induced folding. Proteins Struct. Funct. Bioinform. 2006, 62, 24–45. [Google Scholar] [CrossRef]
- Konrad, Z.; Bar-Zvi, D. Synergism between the chaperone-like activity of the stress regulated ASR1 protein and the osmolyte glycine-betaine. Planta 2008, 227, 1213–1219. [Google Scholar] [CrossRef]
- Hsu, Y.-F.; Yu, S.-C.; Yang, C.-Y.; Wang, C.-S. Lily ASR protein-conferred cold and freezing resistance in Arabidopsis. Plant Physiol. Biochem. 2011, 49, 937–945. [Google Scholar] [CrossRef]
- Williams, R.W.; Xue, B.; Uversky, V.N.; Dunker, A.K. Distribution and cluster analysis of predicted intrinsically disordered protein Pfam domains. Intrinsically Disord. Proteins 2013, 1, e25724. [Google Scholar] [CrossRef][Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Galea, C.A.; Nourse, A.; Wang, Y.; Sivakolundu, S.G.; Heller, W.T.; Kriwacki, R.W. Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27Kip1. J. Mol. Biol. 2008, 376, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Jeffrey, P.D.; Patten, A.K.; Massagué, J.; Pavletich, N.P. Crystal structure of the p27 Kip1 cyclin-dependent-kinase inibitor bound to the cyclin A–Cdk2 complex. Nature 1996, 382, 325–331. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9, S1. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.F.; Staby, L.; Bugge, K.; O’Shea, C.; Kragelund, B.B.; Skriver, K. Evolutionary conservation of the intrinsic disorder-based Radical-Induced Cell Death1 hub interactome. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.; Xu, Y.; Rouse, S.L.; Jaffray, E.G.; Plechanovová, A.; Matthews, S.J.; Penedo, J.C.; Hay, R.T. Functional 3D architecture in an intrinsically disordered E3 ligase domain facilitates ubiquitin transfer. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Shukla, S.; Kumar, S.; Suryawanshi, A.; Chaudhry, U.; Ramachandran, S.; Maiti, S. Intrinsically disordered protein from a pathogenic mesophile Mycobacterium tuberculosis adopts structured conformation at high temperature. Proteins Struct. Funct. Bioinform. 2008, 71, 1123–1133. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef]
- Zhou, J.; Oldfield, C.J.; Yan, W.; Shen, B.; Dunker, A.K. Intrinsically disordered domains: Sequence disorder function relationships. Protein Sci. 2019, 28, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Rom, S.; Gilad, A.; Kalifa, Y.; Konrad, Z.; Karpasas, M.M.; Goldgur, Y.; Bar-Zvi, D. Mapping the DNA-and zinc-binding domains of ASR1 (abscisic acid stress ripening), an abiotic-stress regulated plant specific protein. Biochimie 2006, 88, 621–628. [Google Scholar] [CrossRef]
- Tompa, P.; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J 2004, 18, 1169–1175. [Google Scholar] [CrossRef]
- Pazos, F.; Pietrosemoli, N.; Garcia-Martin, J.A.; Solano, R. Protein intrinsic disorder in plants. Front Plant Sci. 2013, 4, 363. [Google Scholar] [CrossRef]
- Uversky, V.N. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry 1993, 32, 13288–13298. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Forman-Kay, J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef]
- Brocca, S.; Testa, L.; Sobott, F.; Samalikova, M.; Natalello, A.; Papaleo, E.; Lotti, M.; De Gioia, L.; Doglia, S.M.; Alberghina, L.; et al. Compaction properties of an intrinsically disordered protein: Sic1 and its kinase-inhibitor domain. Biophys. J. 2011, 100, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Mery, J.; Heitz, A.; Heitz, F.; Divita, G. Design and synthesis of a peptide derived from positions 195–244 of human cdc25C phosphatase. J. Pept. Sci. 1999, 5, 263–271. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | N | MM | pI | f+ | f− | FCR | NCPR | κ | GRAVY | PDR |
|---|---|---|---|---|---|---|---|---|---|---|
| TtABA-WDS | 66 | 7192 | 6.12 | 0.136 | 0.197 | 0.333 | −0.061 | 0.096 | −0.904 | 2 |
| HvABA-WDS | 66 | 7226 | 6.24 | 0.151 | 0.197 | 0.348 | −0.045 | 0.087 | −0.851 | 2 |
| Protein | Mass (Da) | RSobs | RSNF | RSU | RSIDP | RSobs/RSU | RSobs/RSIDP | CI |
|---|---|---|---|---|---|---|---|---|
| TtABA-WDS | 9200 | 21.5 | 16.2 | 26.1 | 23.6 | 0.83 | 0.91 | 0.46 |
| HvABA-WDS | 9234 | 20.3 | 16.2 | 26.1 | 23.6 | 0.78 | 0.86 | 0.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yacoubi, I.; Hamdi, K.; Fourquet, P.; Bignon, C.; Longhi, S. Structural and Functional Characterization of the ABA-Water Deficit Stress Domain from Wheat and Barley: An Intrinsically Disordered Domain behind the Versatile Functions of the Plant Abscissic Acid, Stress and Ripening Protein Family. Int. J. Mol. Sci. 2021, 22, 2314. https://doi.org/10.3390/ijms22052314
Yacoubi I, Hamdi K, Fourquet P, Bignon C, Longhi S. Structural and Functional Characterization of the ABA-Water Deficit Stress Domain from Wheat and Barley: An Intrinsically Disordered Domain behind the Versatile Functions of the Plant Abscissic Acid, Stress and Ripening Protein Family. International Journal of Molecular Sciences. 2021; 22(5):2314. https://doi.org/10.3390/ijms22052314
Chicago/Turabian StyleYacoubi, Ines, Karama Hamdi, Patrick Fourquet, Christophe Bignon, and Sonia Longhi. 2021. "Structural and Functional Characterization of the ABA-Water Deficit Stress Domain from Wheat and Barley: An Intrinsically Disordered Domain behind the Versatile Functions of the Plant Abscissic Acid, Stress and Ripening Protein Family" International Journal of Molecular Sciences 22, no. 5: 2314. https://doi.org/10.3390/ijms22052314
APA StyleYacoubi, I., Hamdi, K., Fourquet, P., Bignon, C., & Longhi, S. (2021). Structural and Functional Characterization of the ABA-Water Deficit Stress Domain from Wheat and Barley: An Intrinsically Disordered Domain behind the Versatile Functions of the Plant Abscissic Acid, Stress and Ripening Protein Family. International Journal of Molecular Sciences, 22(5), 2314. https://doi.org/10.3390/ijms22052314