
Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke



{kind=link}
{kind=link}
Abstract
1. Introduction
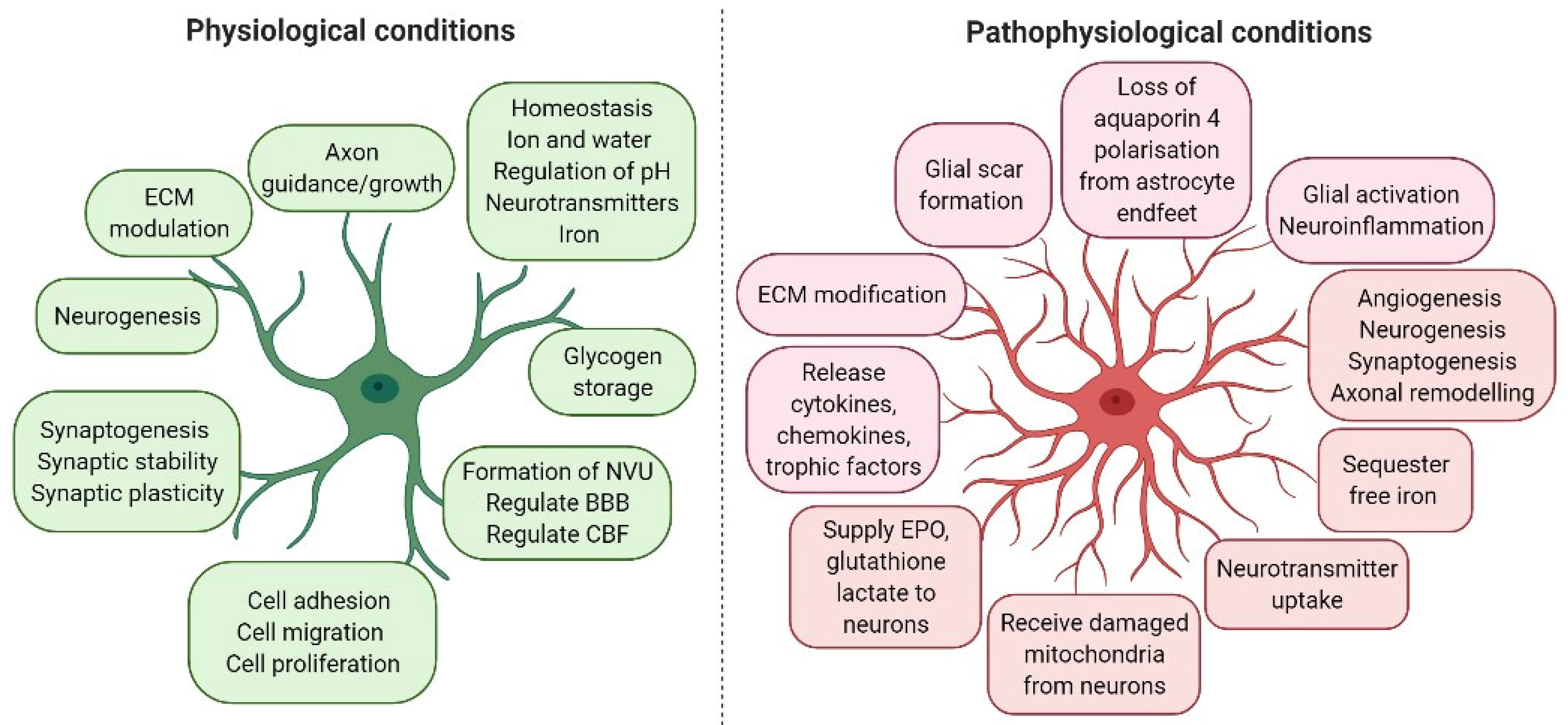
2. Astrocyte Activation in Neuroprotection, Neurotoxicity and Neurorestoration
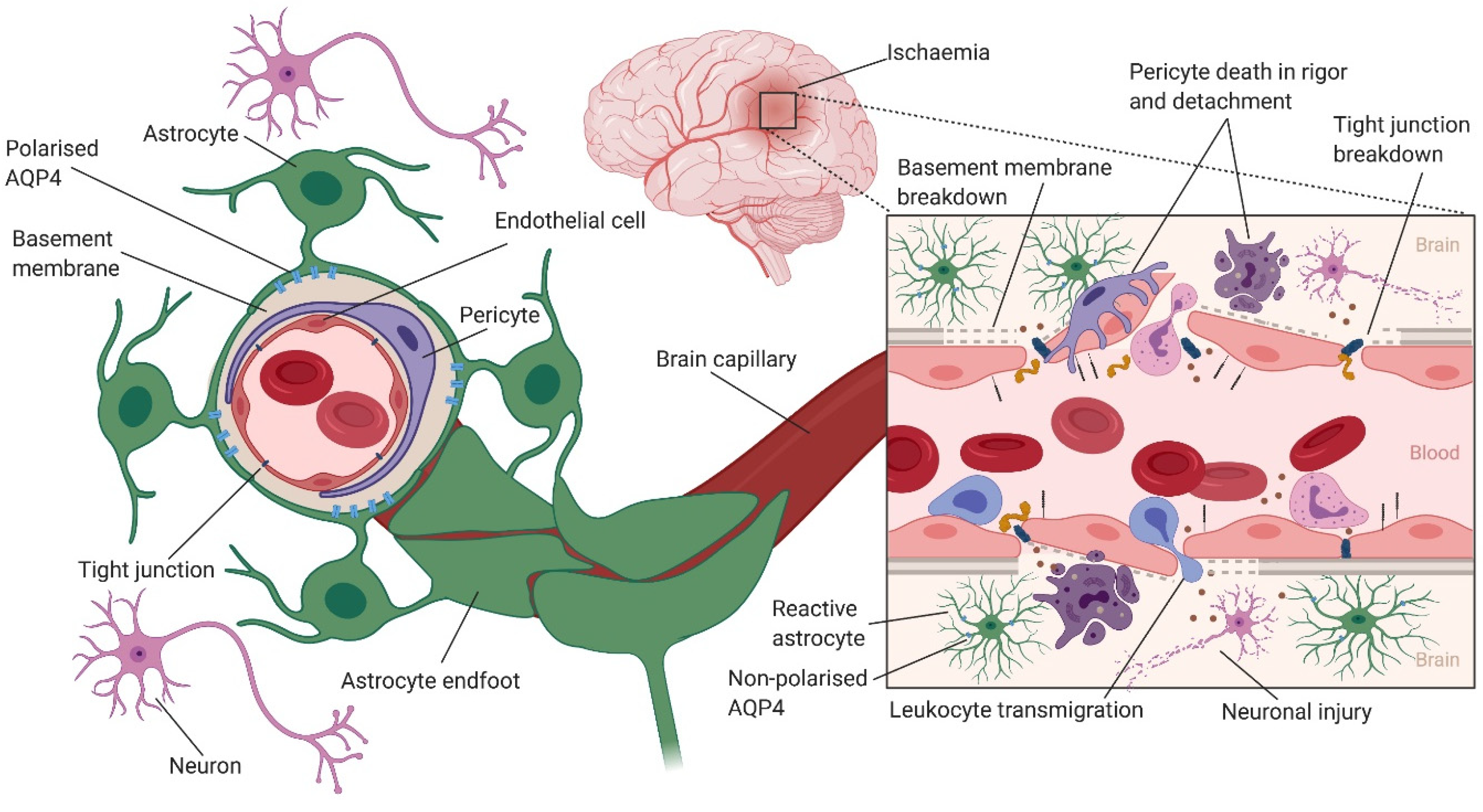
3. Astrocytes Modulate Cerebral Blood Flow, Angiogenesis and the Blood–Brain Barrier
4. The Role of Astrocytic Aquaporin 4 in Cerebral Oedema Formation
5. The Role of Astrocyte Activation in Neuroinflammation after Ischaemic Stroke
6. Astrocyte Activation and Glial Scar Formation
7. The Role of Astrocyte Secretions in Ischaemic Stroke
8. Targeting Astrocytes as a New Therapy for Stroke
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- GBD 2016 Stroke Collaborators. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef]
- Gribkoff, V.K.; Starrett, J.E., Jr.; Dworetzky, S.I.; Hewawasam, P.; Boissard, C.G.; Cook, D.A.; Frantz, S.W.; Heman, K.; Hibbard, J.R.; Huston, K.; et al. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat. Med. 2001, 7, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cabrer, P.; Campos, F.; Sobrino, T.; Castillo, J. Targeting the ischemic penumbra. Stroke 2011, 42, S7–S11. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.V.; Ma, H.; Ringleb, P.A.; Parsons, M.W.; Churilov, L.; Bendszus, M.; Levi, C.R.; Hsu, C.; Kleinig, T.J.; Fatar, M.; et al. Extending thrombolysis to 4·5-9 h and wake-up stroke using perfusion imaging: A systematic review and meta-analysis of individual patient data. Lancet 2019, 394, 139–147. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; Donnan, G.A.; Lees, K.R.; Hacke, W.; Khatri, P.; Hill, M.D.; Goyal, M.; Mitchell, P.J.; Saver, J.L.; Diener, H.C.; et al. Endovascular stent thrombectomy: The new standard of care for large vessel ischaemic Stroke. Lancet Neurol. 2015, 14, 846–854. [Google Scholar] [CrossRef]
- Campbell, B.C.; Mitchell, P.J.; Kleinig, T.J.; Dewey, H.M.; Churilov, L.; Yassi, N.; Yan, B.; Dowling, R.J.; Parsons, M.W.; Oxley, T.J.; et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N. Engl. J. Med. 2015, 372, 1009–1018. [Google Scholar] [CrossRef]
- Sardar, P.; Chatterjee, S.; Giri, J.; Kundu, A.; Tandar, A.; Sen, P.; Nairooz, R.; Huston, J.; Ryan, J.J.; Bashir, R.; et al. Endovascular therapy for acute ischaemic stroke: A systematic review and meta-analysis of randomized trials. Eur. Heart J. 2015, 36, 2373–2380. [Google Scholar] [CrossRef]
- Zerna, C.; Thomalla, G.; Campbell, B.C.V.; Rha, J.H.; Hill, M.D. Current practice and future directions in the diagnosis and acute treatment of ischaemic Stroke. Lancet 2018, 392, 1247–1256. [Google Scholar] [CrossRef]
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Neuhaus, A.A.; Couch, Y.; Hadley, G.; Buchan, A.M. Neuroprotection in stroke: The importance of collaboration and reproducibility. Brain 2017, 140, 2079–2092. [Google Scholar] [CrossRef]
- Hill, M.; Goyal, M.; Menon, B.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- García-Cabezas, M.A.; John, Y.J.; Barbas, H.; Zikopoulos, B. Distinction of Neurons, Glia and Endothelial Cells in the Cerebral Cortex: An Algorithm Based on Cytological Features. Front. Neuroanat. 2016, 10, 107. [Google Scholar] [CrossRef]
- Lent, R.; Azevedo, F.; Andrade-Moraes, C.H.; Pinto, A.V.O. How many neurons do you have? Some dogmas of quantitative neuroscience under revision. Eur. J. Neurosci. 2012, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Sherwood, C.C.; Stimpson, C.D.; Raghanti, M.A.; Wildman, D.E.; Uddin, M.; Grossman, L.I.; Goodman, M.; Redmond, J.C.; Bonar, C.J.; Erwin, J.M.; et al. Evolution of increased glia-neuron ratios in the human frontal cortex. Proc. Natl. Acad. Sci. USA 2006, 103, 13606–13611. [Google Scholar] [CrossRef]
- Pakkenberg, B.; Gundersen, H. Total number of neurons and glial cells in human brain nuclei estimated by the disector and the fractionator. J. Microsc. 1988, 150, 1–20. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Anderson, M.F.; Blomstrand, F.; Blomstrand, C.; Eriksson, P.S.; Nilsson, M. Astrocytes and stroke: Networking for survival? Neurochem. Res. 2003, 28, 293–305. [Google Scholar] [CrossRef]
- Liu, Z.; Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic Stroke. Prog. Neurobiol. 2016, 144, 103–120. [Google Scholar] [CrossRef]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxid. Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.H.; Cho, J.; Kwon, D.H.; Park, J.Y.; Woo, J.; Lee, J.M.; Lee, S.; Ko, H.Y.; Won, W.; Kim, R.G.; et al. Excessive Astrocytic GABA Causes Cortical Hypometabolism and Impedes Functional Recovery after Subcortical Stroke. Cell Rep. 2020, 32, 107861. [Google Scholar] [CrossRef]
- Cai, W.; Liu, H.; Zhao, J.; Chen, L.Y.; Chen, J.; Lu, Z.; Hu, X. Pericytes in Brain Injury and Repair After Ischemic Stroke. Trans. Stroke Res. 2017, 8, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Özen, I.; Deierborg, T.; Miharada, K.; Padel, T.; Englund, E.; Genové, G.; Paul, G. Brain pericytes acquire a microglial phenotype after Stroke. Acta Neuropathol. 2014, 128, 381–396. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.Q.; Ma, X.T.; Hu, Z.W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.J.; Tian, D.S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef]
- Poskanzer, K.E.; Molofsky, A.V. Dynamism of an Astrocyte In Vivo: Perspectives on Identity and Function. Annu. Rev. Physiol. 2018, 80, 143–157. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.A.; Schnell, O.; Pöschl, J.; Eigenbrod, S.; Kretzschmar, H.A.; Tonn, J.C.; Schüller, U. ALDH1A1 is a marker of astrocytic differentiation during brain development and correlates with better survival in glioblastoma patients. Brain Pathol. 2012, 22, 788–797. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Dallérac, G.; Rouach, N. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef]
- Rosenberg, P.A.; Amin, S.; Leitner, M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J. Neurosci. 1992, 12, 56–61. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef]
- Aubert, A.; Costalat, R.; Magistretti, P.J.; Pellerin, L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc. Natl. Acad. Sci. USA 2005, 102, 16448–16453. [Google Scholar] [CrossRef]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Ring, A.; Sonnewald, U.; Waagepetersen, H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009, 109, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Cruz, N.F. Contributions of Glycogen to Astrocytic Energetics during Brain Activation. Metab. Brain Dis. 2015, 30, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.A. Physiologic coupling of glial glycogen metabolism to neuronal activity in brain. Can. J. Physiol. Pharmacol. 1992, 70, S138–S144. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Bergher, J.P.; Anderson, C.M.; Treadway, J.L.; Fosgerau, K.; Swanson, R.A. Astrocyte glycogen sustains neuronal activity during hypoglycemia: Studies with the glycogen phosphorylase inhibitor CP-316, 819 ([R-R*, S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide). J. Pharmacol. Exp. Ther. 2007, 321, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.B.; Heimbürger, C.M.; Bouman, S.D.; Schousboe, A.; Waagepetersen, H.S. Robust glycogen shunt activity in astrocytes: Effects of glutamatergic and adrenergic agents. Neuroscience 2009, 158, 284–292. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after Stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Heales, S.J.; Peuchen, S.; Barker, J.E.; Land, J.M.; Clark, J.B. Nitric oxide-mediated mitochondrial damage: A potential neuroprotective role for glutathione. Free Radic Biol Med. 1996, 21, 995–1001. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Martinez, A.D.; Retamal, M.A. Gap junction channels and hemichannels in the CNS: Regulation by signaling molecules. Neuropharmacology 2013, 75, 567–582. [Google Scholar] [CrossRef]
- Li, X.; Zhao, H.; Tan, X.; Kostrzewa, R.M.; Du, G.; Chen, Y.; Zhu, J.; Miao, Z.; Yu, H.; Kong, J.; et al. Inhibition of connexin43 improves functional recovery after ischemic brain injury in neonatal rats. Glia 2015, 63, 1553–1567. [Google Scholar] [CrossRef]
- Davidson, J.O.; Green, C.R.; Bennet, L.; Nicholson, L.F.; Danesh-Meyer, H.; O’Carroll, S.J.; Gunn, A.J. A key role for connexin hemichannels in spreading ischemic brain injury. Curr. Drug Targets 2013, 14, 36–46. [Google Scholar] [CrossRef]
- Kim, Y.; Davidson, J.O.; Green, C.R.; Nicholson, L.F.B.; O’Carroll, S.J.; Zhang, J. Connexins and Pannexins in cerebral ischemia. Biochim. Biophys. Acta Biomembr. 2018, 1860, 224–236. [Google Scholar] [CrossRef]
- Zündorf, G.; Kahlert, S.; Reiser, G. Gap-junction blocker carbenoxolone differentially enhances NMDA-induced cell death in hippocampal neurons and astrocytes in co-culture. J. Neurochem. 2007, 102, 508–521. [Google Scholar] [CrossRef]
- Takano, K.; Ogawa, M.; Kawabe, K.; Moriyama, M.; Nakamura, Y. Inhibition of gap junction elevates glutamate uptake in cultured astrocytes. Neurochem. Res. 2018, 43, 59–65. [Google Scholar] [CrossRef]
- Xu, L.J.; Emery, J.F.; Ouyang, Y.B.; Voloboueva, L.A.; Giffard, R.G. Astrocyte Targeted Overexpression of Hsp72 or SOD2 Reduces Neuronal Vulnerability to Forebrain Ischemia. Glia 2010, 58, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated micro. Glia Nat. 2017, 541, 481–487. [Google Scholar]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Pettigrew, L.C.; Kasner, S.E.; Gorman, M.; Atkinson, R.P.; Funakoshi, Y.; Ishibashi, H. Arundic Acid (ONO-2506) Stroke Study Group. Effect of arundic acid on serum S-100beta in ischemic Stroke. J. Neurol. Sci. 2006, 251, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Buffo, A.; Rolando, C.; Ceruti, S. Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 2010, 79, 77–89. [Google Scholar] [CrossRef]
- Barreto, G.E.; Gonzalez, J.; Torres, Y.; Morales, L. Astrocytic-neuronal crosstalk: Implications for neuroprotection from brain injury. Neurosci. Res. 2011, 71, 107–113. [Google Scholar] [CrossRef]
- Jean-Claude, P.; Bordey, A. The multifaceted subventricular zone astrocyte: From a metabolic and pro-neurogenic role to acting as a neural stem cell. Neuroscience 2016, 323, 20–28. [Google Scholar]
- Acaz-Fonseca, E.; Ortiz-Rodriguez, A.; Azcoitia, I.; Garcia-Segura, L.M.; Arevalo, M.A. Notch signaling in astrocytes mediates their morphological response to an inflammatory challenge. Cell Death Discov. 2019, 5, 85. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Bill, R.M.; Conner, A.C.; Heath, P.R.; Conner, M.T. Transcriptome analysis of gene expression provides new insights into the effect of mild therapeutic hypothermia on primary human cortical astrocytes cultured under hypoxia. Front. Cell Neurol. 2017, 11, 386. [Google Scholar] [CrossRef]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D.; et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Fernandez, E.; Staursky, D.; Lucas, K.; Nguyen, B.V.; Li, M.; Liu, Y.; Washington, C.; Coolen, L.M.; Fan, F.; Roman, R.J. 20-HETE Enzymes and Receptors in the Neurovascular Unit: Implications in Cerebrovascular Disease. Front. Neurol. 2020, 11, 983. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Mechanisms of Astrocyte-Mediated Cerebral Edema. Neurochem. Res. 2015, 40, 317–328. [Google Scholar] [CrossRef]
- Sykova, E. Glial diffusion barriers during aging and pathological states. Prog. Brain Res. 2001, 132, 339–363. [Google Scholar]
- Chen, R.L.; Lai, U.H.; Zhu, L.L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species (ROS) formation in the brain at different oxygen Levels: Role of hypoxia inducible factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef]
- Sutherland, B.A.; Papadakis, M.; Chen, R.L.; Buchan, A.M. Cerebral blood flow alteration in neuroprotection following cerebral ischaemia. J. Physiol. 2011, 589, 4105–4114. [Google Scholar] [CrossRef]
- Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. [Google Scholar] [CrossRef]
- Cárdenas-Rivera, A.; Campero-Romero, A.N.; Heras-Romero, Y.; Penagos-Puig, A.; Rincón-Heredia, R.; Tovar-Y.-Romo, L.B. Early Post-stroke Activation of Vascular Endothelial Growth Factor Receptor 2 Hinders the Receptor 1-Dependent Neuroprotection Afforded by the Endogenous Ligand. Front. Cell Neurosci. 2019, 13, 270. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Takahashi, T.; Ishikawa, M.; Onodera, O.; Shimohata, T.; Del Zoppo, G.J. Angiogenesis in the ischemic core: A potential treatment target? J. Cereb. Blood Flow Metab. 2019, 39, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood–brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Jickling, G.C.; Liu, D.; Stamova, B.; Ander, B.P.; Zhan, X.; Lu, A.; Sharp, F.R. Hemorrhagic transformation after ischemic stroke in animals and humans. J. Cereb. Blood Flow Metab. 2014, 34, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Davson, H.; Oldendorf, W.H. Symposium on membrane transport. Transport in the central nervous system. Proc. R. Soc. Med. 1967, 60, 326–329. [Google Scholar]
- Stewart, P.A.; Wiley, M.J. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: A study using quail-chick transplantation chimeras. Dev. Biol. 1981, 84, 183–192. [Google Scholar] [CrossRef]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood–brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Wilkinson, M.; Hume, R.; Strange, R.; Bell, J.E. Glial and neuronal differentiation in the human fetal brain 9–23 weeks of gestation. Neuropathol. Appl. Neurobiol. 1990, 16, 193–204. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Kacem, K.; Lacombe, P.; Seylaz, J.; Bonvento, G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: A confocal microscopy study. Glia 1998, 23, 1–10. [Google Scholar] [CrossRef]
- Lee, S.-W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef]
- Tao-Cheng, J.H.; Nagy, Z.; Brightman, M.W. Tight junctions of brain endothelium in vitro are enhanced by astro. Glia J. Neurosci. Off. J. Soc. Neurosci. 1987, 7, 3293–3299. [Google Scholar] [CrossRef]
- Rubin, L.L.; Staddon, J.M. The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 1999, 22, 11–28. [Google Scholar] [CrossRef]
- Patabendige, A.; Skinner, R.A.; Morgan, L.; Abbott, N.J. A detailed method for preparation of a functional and flexible blood-brain barrier model using porcine brain endothelial cells. Brain Res. 2013, 1521, 16–30. [Google Scholar] [CrossRef] [PubMed]
- McAllister, M.S.; Krizanac-Bengez, L.; Macchia, F.; Naftalin, R.J.; Pedley, K.C.; Mayberg, M.R.; Marroni, M.; Leaman, S.; Stanness, K.A.; Janigro, D.; et al. Mechanisms of glucose transport at the blood-brain barrier: An in vitro study. Brain Res. 2001, 904, 20–30. [Google Scholar] [CrossRef]
- Helms, H.C.; Madelung, R.; Waagepetersen, H.S.; Nielsen, C.U.; Brodin, B. In Vitro evidence for the brain glutamate efflux hypothesis: Brain endothelial cells cocultured with astrocytes display a polarized brain-to-blood transport of glutamate. Glia 2012, 60, 882–893. [Google Scholar] [CrossRef]
- Baello, S.; Iqbal, M.; Gibb, W.; Matthews, S.G. Astrocyte-mediated regulation of multidrug resistance p-glycoprotein in fetal and neonatal brain endothelial cells: Age-dependent effects. Physiol. Rep. 2016, 4, e12853. [Google Scholar] [CrossRef]
- Demeuse, P.; Kerkhofs, A.; Struys-Ponsar, C.; Knoops, B.; Remacle, C.; van den Bosch de Aguilar, P. Compartmentalized coculture of rat brain endothelial cells and astrocytes: A syngenic model to study the blood-brain barrier. J. Neurosci. Methods 2002, 121, 21–31. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, T.; Ting, P.; Martinez, H.; Klatzo, I. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol. 1985, 68, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.A.; Mesquita, M.; Ashioti, M.; Beech, J.S.; Williams, S.C.R.; Irving, E.; Cash, D. Late changes in blood-brain barrier permeability in a rat tMCAO model of stroke detected by gadolinium-enhanced MRI. Neurol. Res. 2020, 42, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.G.; Xue, D.; Preston, E.; Karbalai, H.; Buchan, A.M. Biphasic opening of the blood–brain barrier following transient focal ischemia: Effects of hypothermia. Can. J. Neurol. Sci. 1999, 26, 298–304. [Google Scholar] [CrossRef]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D.; et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in Stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef]
- Sorby-Adams, A.J.; Marcoionni, A.M.; Dempsey, E.R.; Woenig, J.A.; Turner, R.J. The Role of Neurogenic Inflammation in Blood-Brain Barrier Disruption and Development of Cerebral Oedema Following Acute Central Nervous System (CNS) Injury. Int. J. Mol. Sci. 2017, 18, 1788. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Flores, S.; Berge, E.; Whittle, I.R. Surgical decompression for cerebral oedema in acute ischaemic Stroke. Cochr. Datab. Syst. Rev. 2012, 1, Cd003435. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, L.; Kardos, L.; Szász, J.; Szatmári, S.; Bereczki, D.; Hegedüs, K.; Csiba, L. Factors influencing hemorrhagic transformation in ischemic stroke: A clinicopathological comparison. Eur. J. Neurol. 2006, 13, 1251–1255. [Google Scholar] [CrossRef]
- Szepesi, R.; Csokonay, Á.; Murnyák, B.; Kouhsari, M.C.; Hofgárt, G.; Csiba, L.; Hortobágyi, T. Haemorrhagic transformation in ischaemic stroke is more frequent than clinically suspected—A neuropathological study. J. Neurol. Sci. 2016, 368, 4–10. [Google Scholar] [CrossRef]
- Balami, J.S.; Chen, R.L.; Grunwald, I.Q.; Buchan, A.M. Neurological complications of acute ischaemic Stroke. Lancet Neurol. 2011, 10, 357–371. [Google Scholar] [CrossRef]
- Simard, J.M.; Kent, T.A.; Chen, M.; Tarasov, K.V.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef]
- Verkman, A.S. Aquaporins. Curr. Biol. 2013, 23, R52–R55. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Verkman, A.S. Physiological roles of glycerol-transporting aquaporins: The aquaglyceroporins. Cell Mol. Life Sci. 2006, 63, 1386–1392. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Salman, M.M.; Conner, M.T.; Bill, R.M.; Conner, A.C. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta 2015, 1850, 2410–2421. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Pickel, S.U.; Jennings, J.; Törnroth-Horsefield, S.; Conner, M.T.; Bill, R.M.; Conner, A.C. Water channel pore size determines exclusion properties but not solute selectivity. Sci. Rep. 2019, 9, 20369. [Google Scholar] [CrossRef]
- Verkman, A.S.; Smith, A.J.; Phuan, P.W.; Tradtrantip, L.; Anderson, M.O. The aquaporin-4 water channel as a potential drug target in neurological disorders. Exp. Opin. Ther. Targets 2017, 21, 1161–1170. [Google Scholar] [CrossRef]
- Liang, D.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Cytotoxic edema: Mechanisms of pathological cell swelling. Neurosurg. Focus 2007, 22, E2. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef]
- Frydenlund, D.S.; Bhardwaj, A.; Otsuka, T.; Mylonakou, M.N.; Yasumura, T.; Davidson, K.G.; Zeynalov, E.; Skare, O.; Laake, P.; Haug, F.M.; et al. Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 13532–13536. [Google Scholar] [CrossRef] [PubMed]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic Stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Hirt, L.; Fukuda, A.M.; Ambadipudi, K.; Rashid, F.; Binder, D.; Verkman, A.; Ashwal, S.; Obenaus, A.; Badaut, J. Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J. Cereb. Blood Flow Metab. 2017, 37, 277–290. [Google Scholar] [CrossRef]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef]
- Abir-Awan, M.; Kitchen, P.; Salman, M.M.; Conner, M.T.; Conner, A.C.; Bill, R.M. Inhibitors of Mammalian Aquaporin Water Channels. Int. J. Mol. Sci. 2019, 20, 1589. [Google Scholar] [CrossRef]
- Kourghi, M.; Pei, J.V.; De Ieso, M.L.; Flynn, G.; Yool, A.J. Bumetanide Derivatives AqB007 and AqB011 Selectively Block the Aquaporin-1 Ion Channel Conductance and Slow Cancer Cell Migration. Mol. Pharmacol. 2016, 89, 133–140. [Google Scholar] [CrossRef]
- Migliati, E.; Meurice, N.; DuBois, P.; Fang, J.S.; Somasekharan, S.; Beckett, E.; Flynn, G.; Yool, A.J. Inhibition of aquaporin-1 and aquaporin-4 water permeability by a derivative of the loop diuretic bumetanide acting at an internal pore-occluding binding site. Mol. Pharmacol. 2009, 76, 105–112. [Google Scholar] [CrossRef]
- Migliati, E.R.; Amiry-Moghaddam, M.; Froehner, S.C.; Adams, M.E.; Ottersen, O.P.; Bhardwaj, A. Na(+)-K (+)-2Cl (-) cotransport inhibitor attenuates cerebral edema following experimental stroke via the perivascular pool of aquaporin. Neurocrit. Care 2010, 13, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Detmers, F.J.; de Groot, B.L.; Müller, E.M.; Hinton, A.; Konings, I.B.; Sze, M.; Flitsch, S.L.; Grubmüller, H.; Deen, P.M. Quaternary ammonium compounds as water channel blockers. Specificity, potency, and site of action. J. Biol. Chem. 2006, 281, 14207–14214. [Google Scholar] [CrossRef]
- Popescu, E.S.; Pirici, I.; Ciurea, R.N.; Bălşeanu, T.A.; Cătălin, B.; Mărgăritescu, C.; Mogoantă, L.; Hostiuc, S.; Pirici, D. Three-dimensional organ scanning reveals brain edema reduction in a rat model of strok e treated with an aquaporin 4 inhibitor. Rom. J. Morphol. Embryol. 2017, 58, 59–66. [Google Scholar] [PubMed]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Brown, J.E.; Bill, R.M.; Conner, A.C.; Conner, M.T. Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)- and calmodulin-mediated mechanism. Eur. J. Neurosci. 2017, 46, 2542–2547. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Taylor, L.H.; Salman, M.M.; Bill, R.M.; Conner, M.T.; Conner, A.C. Identification and Molecular Mechanisms of the Rapid Tonicity-induced Relocalization of the Aquaporin 4 Channel. J. Biol. Chem. 2015, 290, 16873–16881. [Google Scholar] [CrossRef] [PubMed]
- Steiner, E.; Enzmann, G.U.; Lin, S.; Ghavampour, S.; Hannocks, M.J.; Zuber, B.; Rüegg, M.A.; Sorokin, L.; Engelhardt, B. Loss of astrocyte polarization upon transient focal brain ischemia as a possible mechanism to counteract early edema formation. Glia 2012, 60, 1646–1659. [Google Scholar] [CrossRef]
- Ciappelloni, S.; Bouchet, D.; Dubourdieu, N.; Boué-Grabot, E.; Kellermayer, B.; Manso, C.; Marignier, R.; Oliet, S.H.R.; Tourdias, T.; Groc, L. Aquaporin-4 Surface Trafficking Regulates Astrocytic Process Motility and Synaptic Activity in Health and Autoimmune Disease. Cell Rep. 2019, 27, 3860–3872.e4. [Google Scholar] [CrossRef]
- Neely, J.D.; Amiry-Moghaddam, M.; Ottersen, O.P.; Froehner, S.C.; Agre, P.; Adams, M.E. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14108–14113. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef]
- Shinjyo, N.; de Pablo, Y.; Pekny, M.; Pekna, M. Complement peptide C3a promotes astrocyte survival in response to ischemic stress. Mol. Neurobiol. 2016, 53, 3076–3087. [Google Scholar] [CrossRef]
- Priego, N.; Valiente, M. The Potential of Astrocytes as Immune Modulators in Brain Tumors. Front. Immunol. 2019. [Google Scholar] [CrossRef]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 2, 28. [Google Scholar] [CrossRef]
- Damisah, E.C.; Hill, R.A.; Rai, A.; Chen, F.; Rothlin, C.V.; Ghosh, S.; Grutzendler, J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 2020, 6, eaba3239. [Google Scholar] [CrossRef]
- Rawlinson, C.; Jenkins, S.; Thei, L.; Dallas, M.; Chen, R.L. Post-ischaemic immunological response in the brain: Targeting microglial activation in ischaemic stroke therapy. Brain Sci. Neurogl. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Xu, J.; Zheng, Y.; Wei, Y.; Zhu, X.; Lo, E.H.; Moskowitz, M.A.; Sims, J.R. HMGB1 promotes MMP-9 upregulation through TLR4 after cerebral ischemia. Stroke 2010, 41, 2077–2082. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Hong, J.; Cho, I.; Jang, Y.H.; Lee, H.; Kim, D.; Yu, S.; Lee, S. TLR2-induced astrocyte MMP9 activation compromises the blood brain barrier and exacerbates intracerebral hemorrhage in animal models. Mol. Brain 2015, 8, 23. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Barakat, D.; Brambilla, R.; Agudelo, C.; Hernandez, E.; Bethea, J.R.; Shestopalov, V.I.; Ivanov, D. Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. Eur. J. Neurosci. 2009, 30, 175–185. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.; Weinstein, J.R. Neuroimmune Response in Ischemic Preconditioning. Neurotherapeutics 2016, 13, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Deng, J.; Wang, B.; Cheng, D.; Yang, Q.; Dong, H.; Xiong, L. Reactive Oxygen Species Scavenger Inhibits STAT3 Activation After Transient Focal Cerebral Ischemia–Reperfusion Injury in Rats. Anest. Analg. 2011, 113, 153–159. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, B.; Deng, Y.; Shangguan, S.; Zhou, F.; Zhou, W.; Li, X.; Li, Y.; Chen, G. Notch signaling in cerebrovascular diseases. Mol. Med. Rep. 2016, 14, 2883–2898. [Google Scholar] [CrossRef]
- Zhang, S.; An, Q.; Wang, T.; Gao, S.; Zhou, G. Autophagy- and MMP-2/9-mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-increased Blood-Brain Barrier Permeability During Early Reperfusion in Stroke. Neuroscience 2018, 377, 126–137. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.; Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers. J. Cerebr. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Villringer, K.; Sanz Cuesta, B.E.; Ostwaldt, A.-C.; Grittner, U.; Brunecker, P.; Khalil, A.A.; Schindler, K.; Eisenblätter, O.; Audebert, H.; Fiebach, J.B.; et al. DCE-MRI blood–brain barrier assessment in acute ischemic Stroke. Neurology 2017, 88, 433–440. [Google Scholar] [CrossRef]
- Arba, F.; Leigh, R.; Inzitari, D.; Warach, S.J.; Luby, M.; Lees, K.R. Blood–Brain barrier leakage increases with small vessel disease in acute ischemic Stroke. Neurology 2017, 89, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Gauberti, M.; Montagne, A.; Marcos-Contreras, O.A.; Béhot, A.L.; Maubert, E.; Vivien, D. Ultra-Sensitive Molecular MRI of Vascular Cell Adhesion Molecule-1 Reveals a Dynamic Inflammatory Penumbra After Strokes. Stroke 2013, 44, 1988–1996. [Google Scholar] [CrossRef]
- Quenault, A.; Martinez de Lizarrondo, S.; Etard, O.; Gauberti, M.; Orset, C.; Haelewyn, B.; Segal, H.C.; Rothwell, P.M.; Vivien, D.; Touzé, E.; et al. Molecular magnetic resonance imaging discloses endothelial activation after transient ischaemic attack. Brain 2016, 140, 146–157. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Raker, C.; Schleif, M.; Blank, N.; Matušková, H.; Ulas, T.; Händler, K.; Torres, S.V.; Schumacher, T.; Tai, K.; Schultze, J.L.; et al. Stroke target identification guided by astrocyte transcriptome analysis. Glia 2018, 67, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Capetanaki, Y. Type III Intermediate Filaments Desmin, Glial Fibrillary Acidic Protein (GFAP), Vimentin, and Peripherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a021642. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.P.; Cekanaviciute, E.; Mamer, L.E.; Buckwalter, M.S. TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after Stroke. J. Neuroinflamm. 2010, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Cua, R.C.; Lau, L.W.; Keough, M.B.; Midha, R.; Apte, S.S.; Yong, V.W. Overcoming Neurite-Inhibitory Chondroitin Sulfate Proteoglycans in the Astrocyte Matrix. Glia 2013, 61, 972–984. [Google Scholar] [CrossRef]
- Yang, T.; Dai, Y.J.; Chen, G.; Cui, S.S. Dissecting the Dual Role of the Glial Scar and Scar-Forming Astrocytes in Spinal Cord Injury. Front. Cell Neurosci. 2020, 14, 78. [Google Scholar] [CrossRef]
- Yiu, G.; He, Z. Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 2006, 7, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, M.; Tian, L.; Chen, J.; Liu, R.; Ning, B. Reactive Astrogliosis: Implications in Spinal Cord Injury Progression and Therapy. Oxid. Med. Cell Longev. 2020, 2020, 9494352. [Google Scholar] [CrossRef]
- Rocamonde, B.; Paradells, S.; Barcia, C.; Garcia Esparza, A.; Soria, J.M. Lipoic acid treatment after brain injury: Study of the glial reaction. Clin. Dev. Immunol. 2013, 2013, 521939. [Google Scholar] [CrossRef]
- Abeysinghe, H.C.S.; Bokhari, L.; Dusting, G.J.; Roulston, C.L. Cyclosporine A Reduces Glial Scarring and Facilitates Functional Recovery Following Transient Focal Ischemia. J. Neurol. Neurophysiol. 2015, 6, 2. [Google Scholar]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids CNS axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef]
- Raposo, C.; Schwartz, M. Glial scar and immune cell involvement in tissue remodeling and repair following acute CNS injuries. Glia 2014, 62, 1895–1904. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.P.; Zorec, R. Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 135, a020370. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Dowell, J.A.; Johnson, J.A.; Li, L. Identification of Astrocyte Secreted Proteins with a Combination of Shotgun Proteomics and Bioinformatics. J. Proteome Res. 2009, 8, 4135–4143. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Elshourbagy, N.A.; Liao, W.S.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc. Natl. Acad. Sci. USA 1985, 82, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Bortolotto, V.; Canonico, P.L.; Sortino, M.A.; Grilli, M. Astrocyte-Derived Paracrine Signals: Relevance for Neurogenic Niche Regulation and Blood–Brain Barrier Integrity. Front. Pharmacol. 2019, 10, 1346. [Google Scholar] [CrossRef] [PubMed]
- Deli, M.A.; Abrahám, C.S.; Kataoka, Y.; Niwa, M. Permeability studies on in vitro blood-brain barrier models: Physiology, pathology, and pharmacology. Cell. Mol. Neurobiol. 2005, 25, 59–127. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A. The value of in vitro models of the blood-brain barrier and their uses. Altern. Lab. Anim. 2012, 40, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Mun-Bryce, S.; Rosenberg, G.A. Matrix metalloproteinases in cerebrovascular disease. J. Cereb. Blood Flow Metab. 1998, 18, 1163–1172. [Google Scholar] [CrossRef]
- Li, Y.N.; Pan, R.; Qin, X.J.; Yang, W.L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 2014, 129, 120–129. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonniere, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1717–1731. [Google Scholar] [CrossRef]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef]
- Dhandapani, K.M.; Hadman, M.; De Sevilla, L.; Wade, M.F.; Mahesh, V.B.; Brann, D.W. Astrocyte protection of neurons: Role of transforming growth factor-beta signaling via a c-Jun-AP-1 protective pathway. J. Biol. Chem. 2003, 278, 43329–43339. [Google Scholar] [CrossRef]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. The Role of Astrocytes in neuroprotection after Brain Stroke: Potential in Cell Therapy. Front. Mol. Neurosci. 2017, 10, 88. [Google Scholar] [CrossRef]
- Song, C.; Wu, Y.; Yang, Z.; Kalueff, A.; Tsao, Y.; Dong, Y.; Su, K. Astrocyte-Conditioned Medium Protects Prefrontal Cortical Neurons from Glutamate-Induced Cell Death by Inhibiting TNF-α Expression. Neuroimmunomodulation 2019, 26, 33–42. [Google Scholar] [CrossRef]
- Lu, X.; Al-Aref, R.; Zhao, D.; Shen, J.; Yan, Y.; Gao, Y. Astrocyte-conditioned medium attenuates glutamate-induced apoptotic cell death in primary cultured spinal cord neurons of rats. Neurol. Res. 2015, 37, 803–808. [Google Scholar] [CrossRef]
- Bicknell, B.A.; Pujic, Z.; Dayan, P.; Goodhill, G.J. Control of neurite growth and guidance by an inhibitory cell-body signal. PLoS Comput. Biol. 2018, 14, e1006218. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, G.; Dirnagl, U. Neuroprotective role of astrocytes in cerebral ischemia: Focus on ischemic preconditioning. Glia 2005, 50, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Helena, S.; Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016. [Google Scholar] [CrossRef]
- Chen, C.C.; Liu, L.; Ma, F.; Wong, C.W.; Guo, X.E.; Chacko, J.V.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Ségaliny, A.; et al. Elucidation of Exosome Migration across the Blood-Brain Barrier Model In Vitro. Cell. Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Yang, H.; Manaenko, A.; Lu, J.; Mei, Q.; Hu, Q. Potential of Exosomes for the Treatment of Stroke; SAGE Publications: Los Angeles, CA, USA, 2019. [Google Scholar]
- Venturini, A.; Passalacqua, M.; Pelassa, S.; Pastorino, F.; Tedesco, M.; Cortese, K.; Gagliani, M.C.; Leo, G.; Maura, G.; Guidolin, D.; et al. Exosomes from Astrocyte Processes: Signaling to Neurons. Front. Pharmacol. 2019, 10, 1452. [Google Scholar] [CrossRef]
- Pei, X.; Li, Y.; Zhu, L.; Zhou, Z. Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic Stroke. Exp. Cell Res. 2019, 382, 111474. [Google Scholar] [CrossRef] [PubMed]
- Hira, K.; Ueno, Y.; Tanaka, R.; Miyamoto, N.; Yamashiro, K.; Inaba, T.; Urabe, T.; Okano, H.; Hattori, N. Astrocyte-Derived Exosomes Treated with a Semaphorin 3A Inhibitor Enhance Stroke Recovery via Prostaglandin D2 Synthase. Stroke 2018, 49, 2483–2494. [Google Scholar] [CrossRef]
- Taylor, A.R.; Robinson, M.B.; Gifondorwa, D.J.; Tytell, M.; Milligan, C.E. Regulation of heat shock protein 70 release in astrocytes: Role of signaling kinases. Dev. Neurobiol. 2007, 67, 1815–1829. [Google Scholar] [CrossRef]
- Xu, L.; Cao, H.; Xie, Y.; Zhang, Y.; Du, M.; Xu, X.; Ye, R.; Liu, X. Exosome-shuttled miR-92b-3p from ischemic preconditioned astrocytes protects neurons against oxygen and glucose deprivation. Brain Res. 2019, 17, 66–73. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol. Ther. Nucl. Acids 2017, 7, 278–287. [Google Scholar] [CrossRef]
- Lafourcade, C.; Ramírez, J.P.; Luarte, A.; Fernández, A.; Wyneken, U. MiRNAs in Astrocyte-Derived Exosomes as Possible Mediators of Neuronal Plasticity. J. Exp. Neurosci. 2016, 10, 1–9. [Google Scholar] [CrossRef]
- Zagrean, A.M.; Hermann, D.M.; Opris, I.; Zagrean, L.; Popa-Wagner, A. Multicellular Crosstalk Between Exosomes and the Neurovascular Unit After Cerebral Ischemia. Therapeutic Implications. Front. Neurosci. 2018, 12, 811. [Google Scholar] [CrossRef]
- Paterno, R.; Chillon, J. Potentially Common therapeutic targets for multiple sclerosis and ischemic Stroke. Front. Physiol. 2018, 9, 855. [Google Scholar] [CrossRef]
- Verma, R.; Mishra, V.; Sasmal, D.; Raghubir, R. Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury. Eur. J. Pharmacol. 2010, 638, 65–71. [Google Scholar] [CrossRef]
- Ouyang, Y.; Voloboueva, L.A.; Xu, L.; Giffard, R.G. Selective Dysfunction of Hippocampal CA1 Astrocytes Contributes to Delayed Neuronal Damage after Transient Forebrain Ischemia. J. Neurosci. 2017, 27, 4253–4260. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Chen, D.; Gao, F.; Lv, H.; Zhang, G.; Sun, X.; Liu, L.; Mo, D.; Ma, N.; Song, L.; et al. Exosomes derived from microRNA-138-5p-overexpressing bone marrow derived mesenchymal stem cells confer neuroprotection to astrocytes following ischemic stroke via inhibition of LCN. J. Biol. Eng. 2019, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, R.; Kastrisianaki, E.; Giambanco, I.; Donato, R. S100B protein stimulates microglia migration via rage-dependent up-regulation of chemokine expression and release. J. Biol. Chem. 2011, 286, 7214–7226. [Google Scholar] [CrossRef]
- Matsui, T.; Mori, T.; Tateishi, N.; Kagamiishi, Y.; Satoh, S.; Katsube, N.; Morikawa, E.; Morimoto, T.; Ikuta, F.; Asano, T. Astrocytic Activation and Delayed Infarct Expansion after Permanent Focal Ischemia in Rats. Part I: Enhanced Astrocytic Synthesis of S-100β in the Periinfarct Area Precedes Delayed Infarct Expansion. J. Cerebr. Blood Flow Metab. 2002, 22, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Rempe, D. Targeting astrocytes for stroke therapy. Neurotherapeutics 2010, 7, 439–451. [Google Scholar] [CrossRef]
- Asano, T.; Mori, T.; Shimoda, T.; Shinagawa, R.; Satoh, S.; Yada, N.; Katsumata, S.; Matsuda, S.; Kagamiishi, Y.; Tateishi, N. Arundic acid (ONO-2506) ameliorates delayed ischemic brain damage by preventing astrocytic overproduction of S100B. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 127–142. [Google Scholar] [CrossRef]
- Tateishi, N.; Mori, T.; Kagamiishi, Y.; Satoh, S.; Katsube, N.; Morikawa, E.; Morimoto, T.; Matsui, T.; Asano, T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: Suppression of astrocytic activation by a novel agent (R)-()-2-propyloctanoic acid (ONO-2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J. Cereb. Blood Flow Metab. 2002, 22, 723–734. [Google Scholar] [PubMed]
- Zlokovic, B.V.; Gottesman, R.F.; Bernstein, K.E.; Seshadri, S.; McKee, A.; Snyder, H.; Greenberg, S.M.; Yaffe, K.; Schaffer, C.B.; Yuan, C.; et al. Vascular contributions to cognitive impairment and dementia (VCID): A report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke Workshop. Alzheimers Dement. 2020, 16, 1714–1733. [Google Scholar] [CrossRef]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. Neuroprotection Induced by Transplanted CDK5 Knockdown Astrocytes in Global Cerebral Ischemic Rats. Mol. Neurobiol. 2017, 54, 6681–6696. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vargas, J.A.; Múnera, A.; Cardona-Gómez, G.P. CDK5 knockdown prevents hippocampal degeneration and cognitive dysfunction produced by cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Posada-Duque, R.A.; Palacio-Castañeda, V.; Cardona-Gómez, G.P. CDK5 knockdown in astrocytes provide neuroprotection as a trophic source via RacMol. Cell. Neurosci. 2015, 68, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Calixto, A.; Posada-Duque, R.; Cardona-Gómez, G.P. Recovery of Neurovascular Unit Integrity by CDK5-KD Astrocyte Transplantation in a Global Cerebral Ischemia Model. Mol. Neurobiol. 2018, 55, 8563–8585. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Bacigaluppi, M.; Russo, G.L.; Peruzzotti-Jametti, L.; Rossi, S.; Sandrone, S.; Butti, E.; De Ceglia, R.; Bergamaschi, A.; Motta, C.; Gallizioli, M.; et al. Neural Stem Cell Transplantation Induces Stroke Recovery by Upregulating Glutamate Transporter GLT-1 in Astrocytes. J. Neurosci. 2016, 36, 10529–10544. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Guo, K.; Fan, W.; Lu, Y.; Chen, L.; Wang, Y.; Shao, Y.; Wu, G.; Xu, J.; Lü, L. Niche astrocytes promote the survival, proliferation and neuronal differentiation of co-transplanted neural stem cells following ischemic stroke in rats. Exp. Ther. Med. 2017, 13, 645–650. [Google Scholar] [CrossRef]
- Sivandzade, F.; Cucullo, L. In-Vitro blood-brain barrier modeling: A review of modern and fast-advancing technologies. J. Cereb. Blood Flow Metab. 2018, 38, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Park, T.E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Cucullo, L.; Hossain, M.; Tierney, W.; Janigro, D. A new dynamic in vitro modular capillaries-venules modular system: Cerebrovascular physiology in a box. BMC Neurosci. 2013, 14, 18. [Google Scholar] [CrossRef]
- Cucullo, L.; Couraud, P.O.; Weksler, B.; Romero, I.A.; Hossain, M.; Rapp, E.; Janigro, D. Immortalized human brain endothelial cells and flow-based vascular modeling: A marriage of convenience for rational neurovascular studies. J. Cereb. Blood Flow Metab. 2008, 28, 312–328. [Google Scholar] [CrossRef]
- Wang, S.N.; Wang, Z.; Xu, T.Y.; Cheng, M.H.; Li, W.L.; Miao, C.Y. Cerebral Organoids Repair Ischemic Stroke Brain Injury. Transl. Stroke Res. 2020, 11, 983–1000. [Google Scholar] [CrossRef]
- Nzou, G.; Wicks, R.T.; VanOstrand, N.R.; Mekky, G.A.; Seale, S.A.; El-Taibany, A.; Wicks, E.E.; Nechtman, C.M.; Marrotte, E.J.; Makani, V.S.; et al. Multicellular 3D Neurovascular Unit Model for Assessing Hypoxia and Neuroinflammation Induced Blood-Brain Barrier Dysfunction. Sci. Rep. 2020, 10, 9766. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patabendige, A.; Singh, A.; Jenkins, S.; Sen, J.; Chen, R. Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. Int. J. Mol. Sci. 2021, 22, 4280. https://doi.org/10.3390/ijms22084280
Patabendige A, Singh A, Jenkins S, Sen J, Chen R. Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. International Journal of Molecular Sciences. 2021; 22(8):4280. https://doi.org/10.3390/ijms22084280
Chicago/Turabian StylePatabendige, Adjanie, Ayesha Singh, Stuart Jenkins, Jon Sen, and Ruoli Chen. 2021. "Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke" International Journal of Molecular Sciences 22, no. 8: 4280. https://doi.org/10.3390/ijms22084280
APA StylePatabendige, A., Singh, A., Jenkins, S., Sen, J., & Chen, R. (2021). Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. International Journal of Molecular Sciences, 22(8), 4280. https://doi.org/10.3390/ijms22084280