
Signatures of Conical Intersection Dynamics in the Time-Resolved Photoelectron Spectrum of Furan: Theoretical Modeling with an Ensemble Density Functional Theory Method


Abstract
1. Introduction
2. Computational Methods
2.1. SSR Method
2.2. Computational Details
3. Results and Discussion
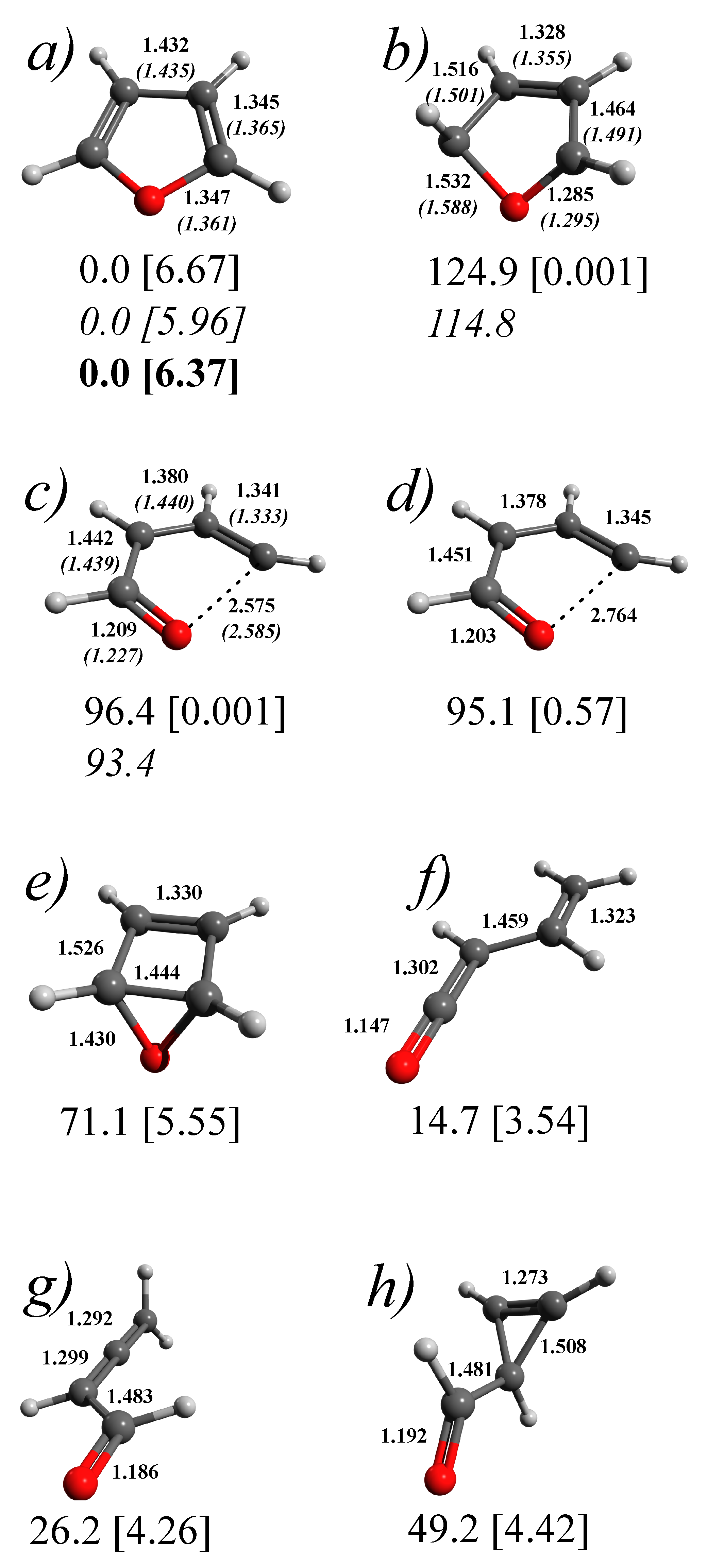
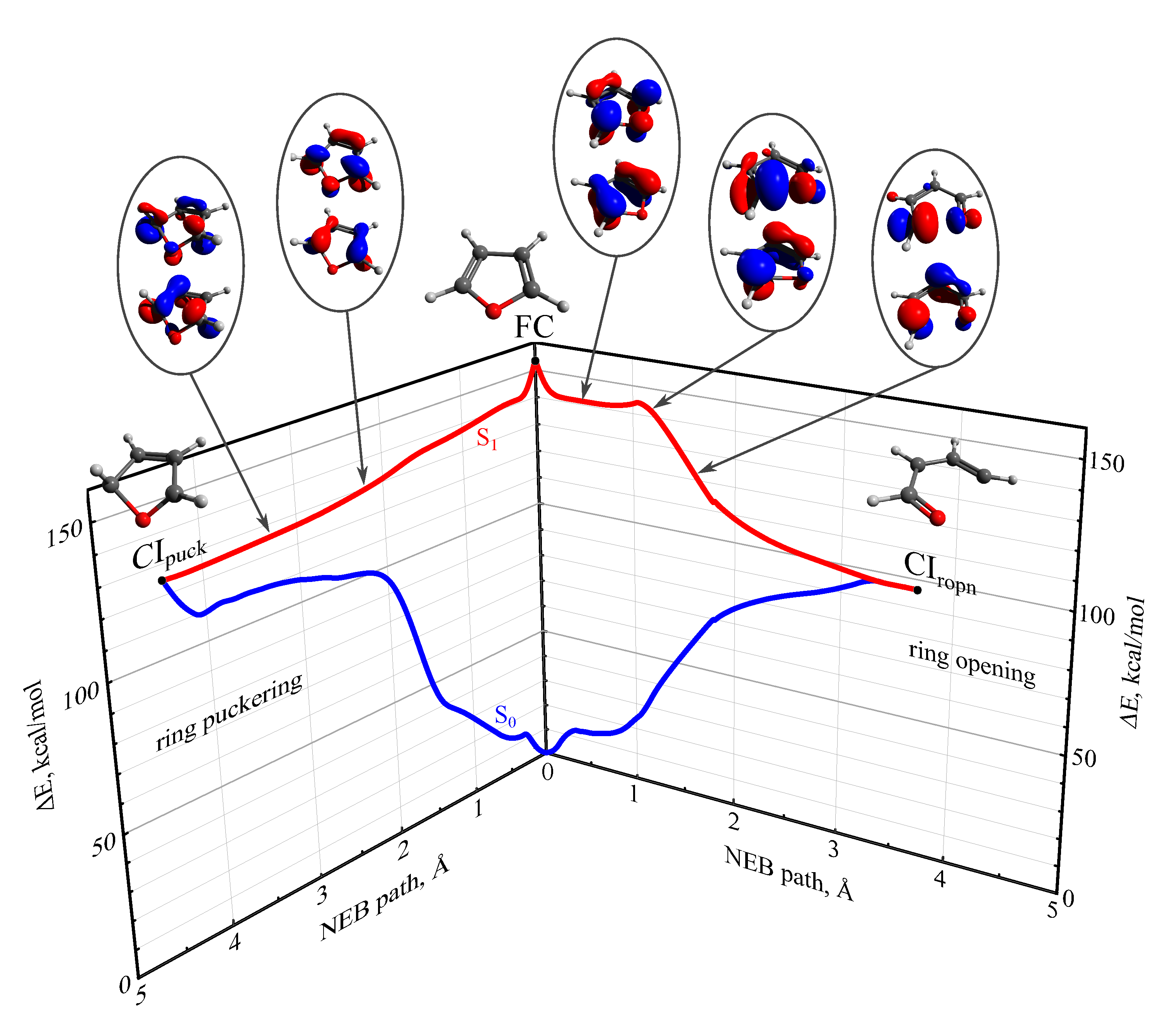
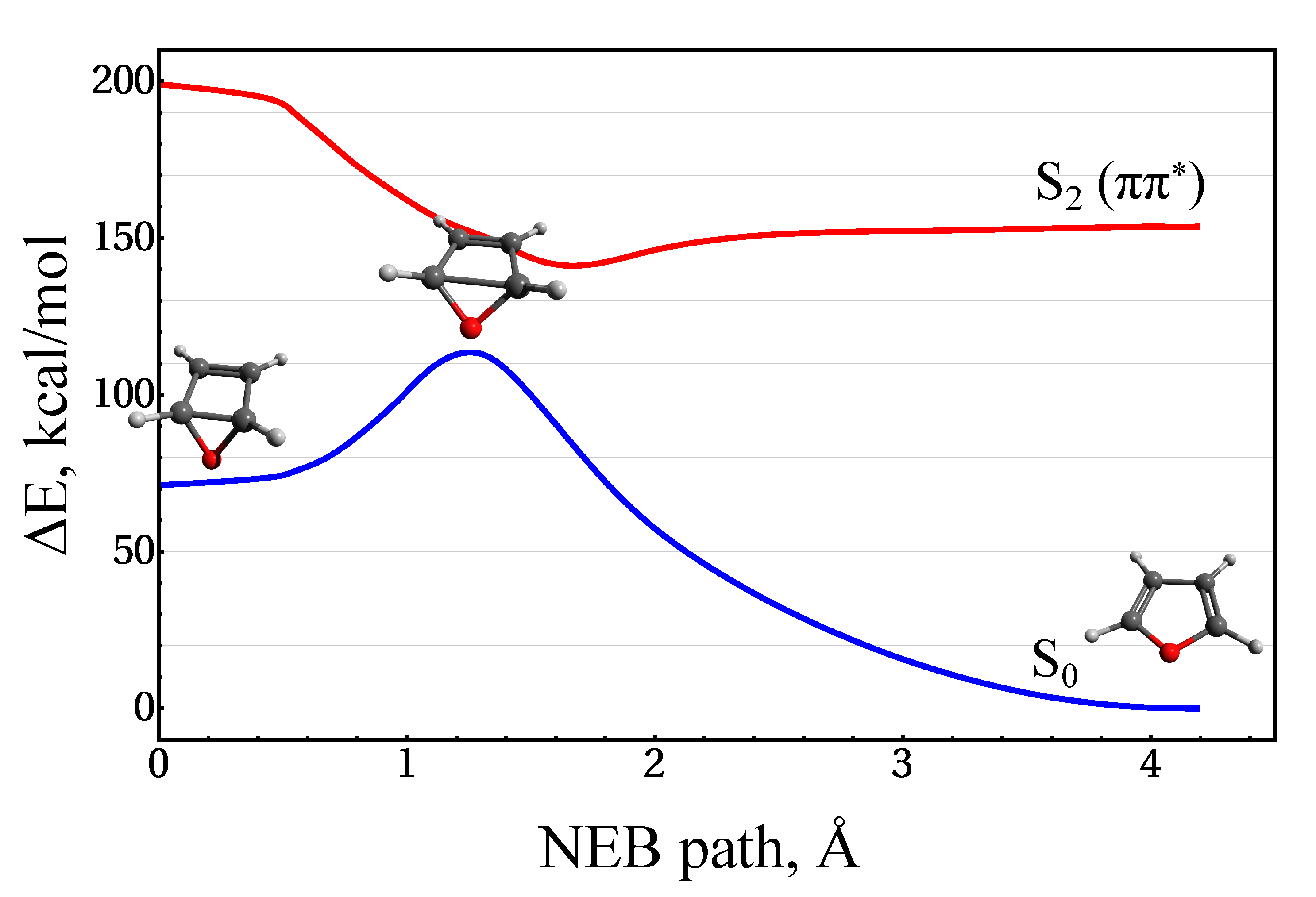
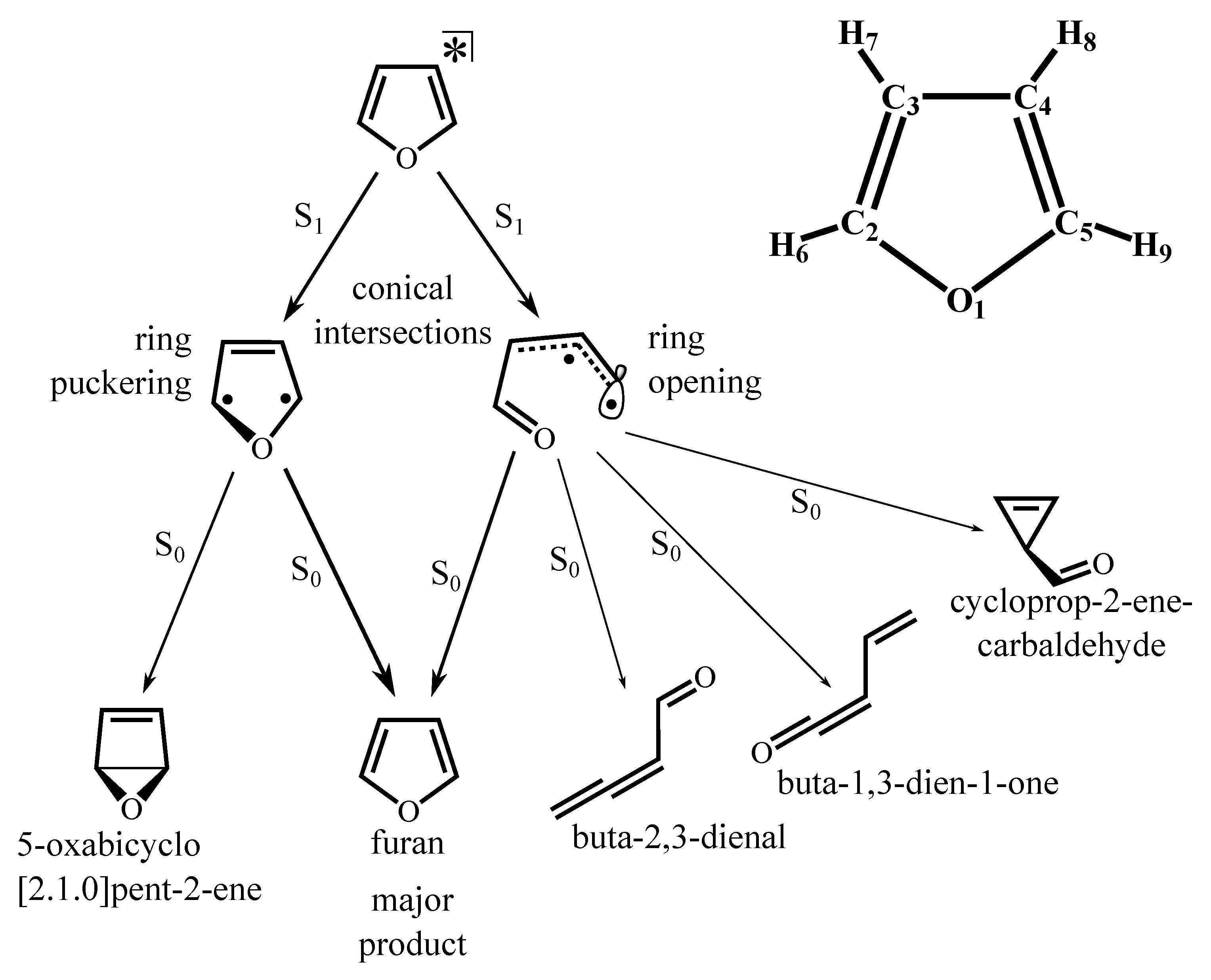
3.1. Stationary Points and Adiabatic Minimum Energy Paths (MEPs) on the Ground- and Excited-State PESs of Furan
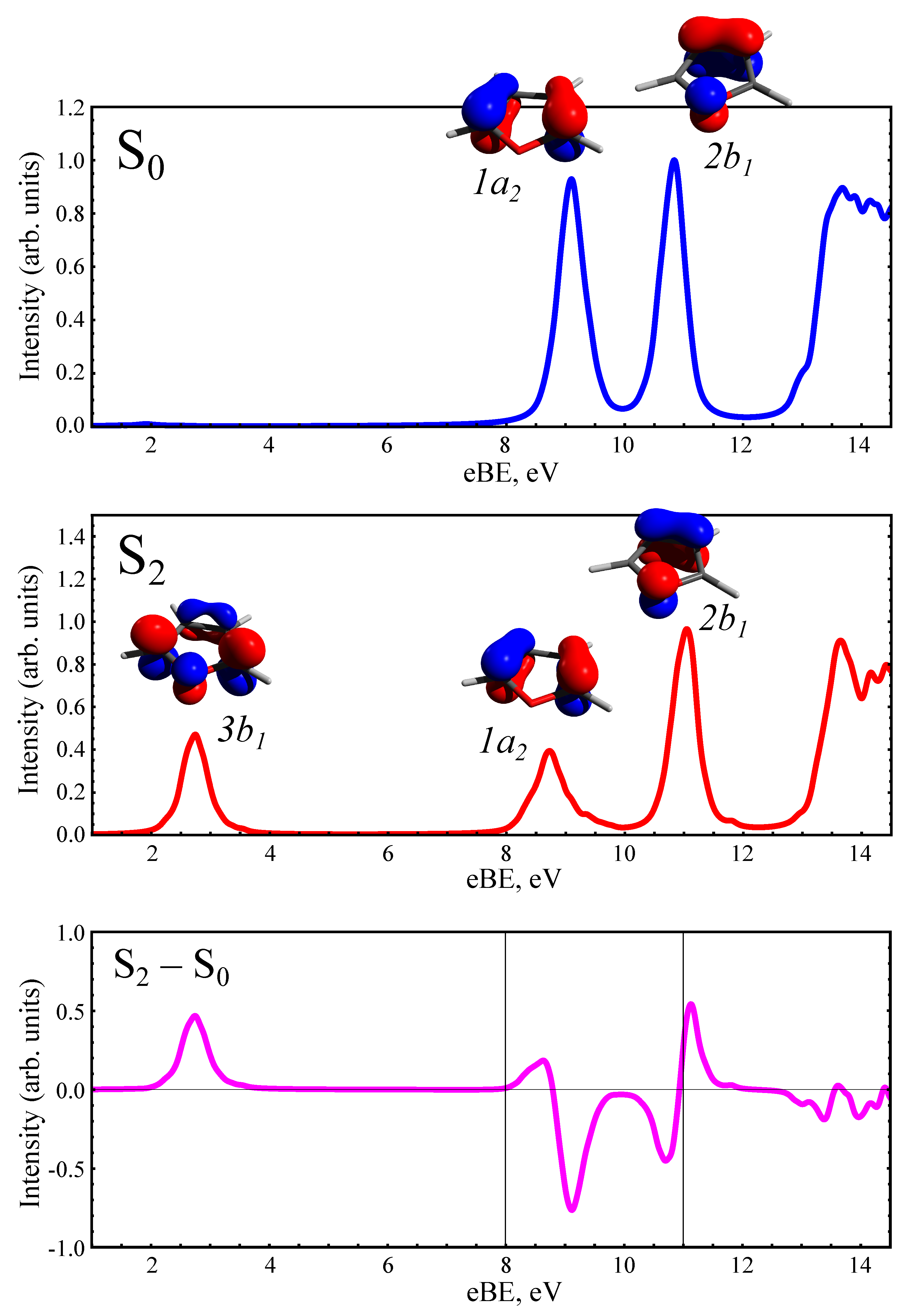
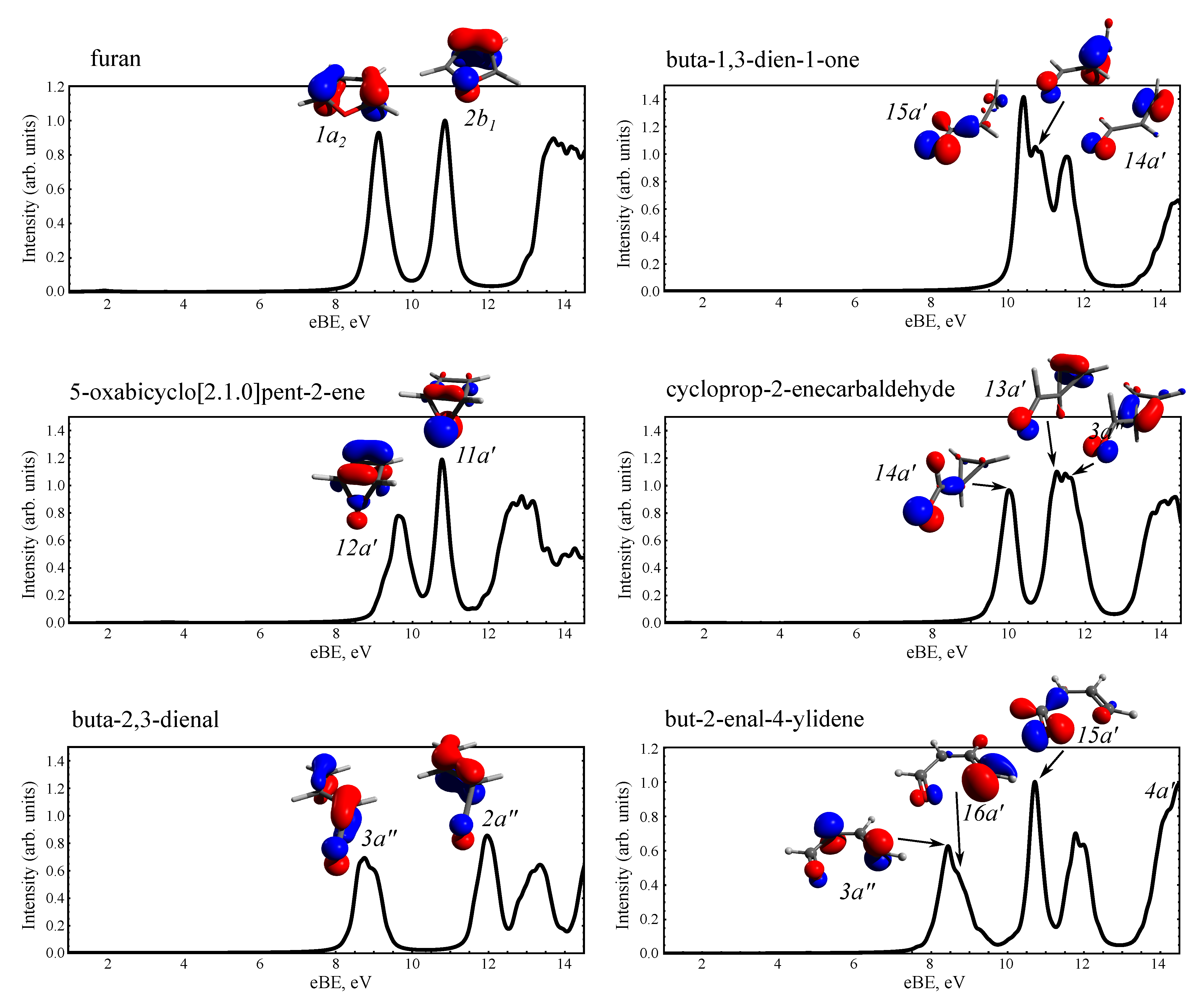
3.2. Static PE Spectra of Furan and the Photoproducts
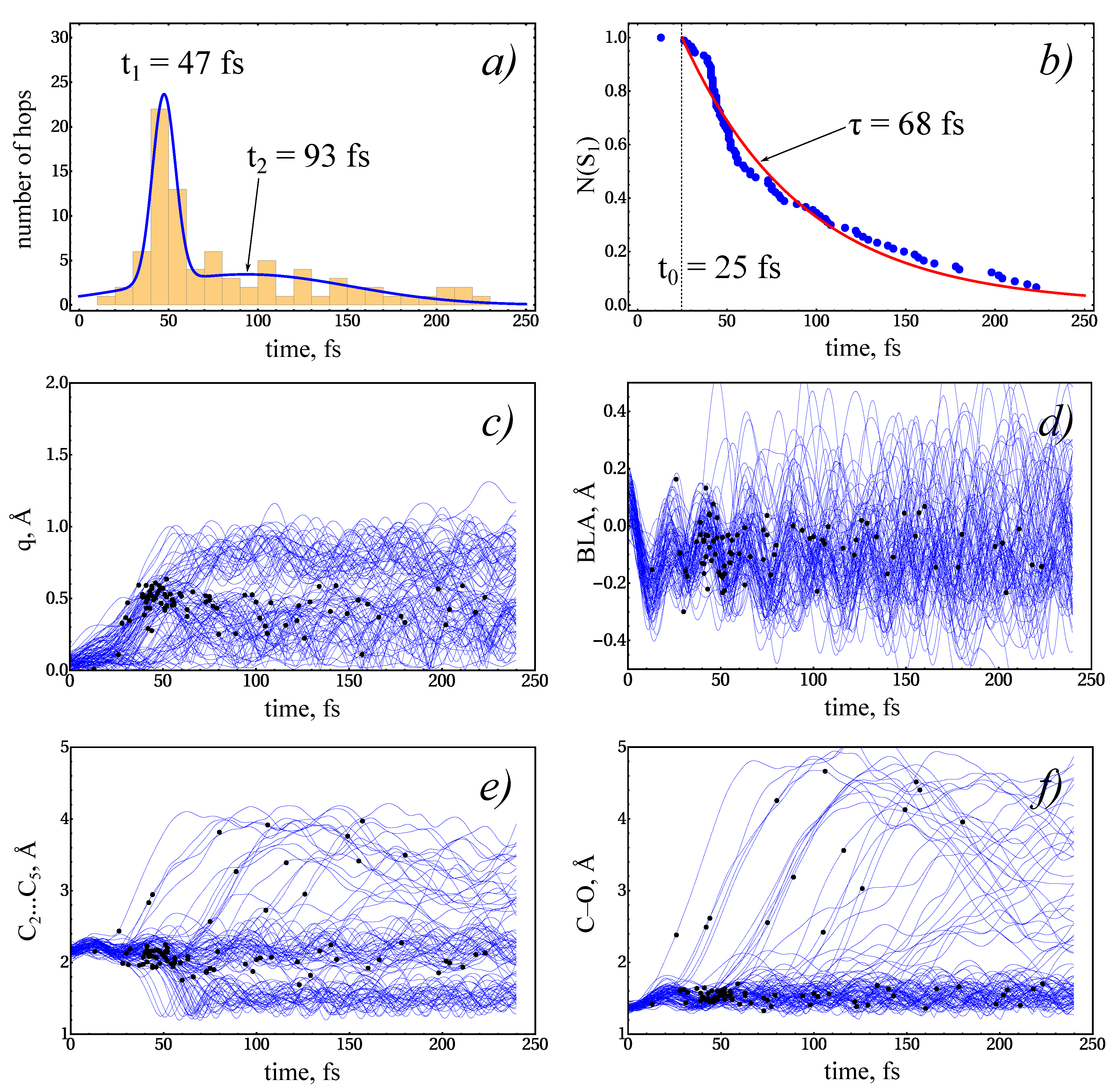
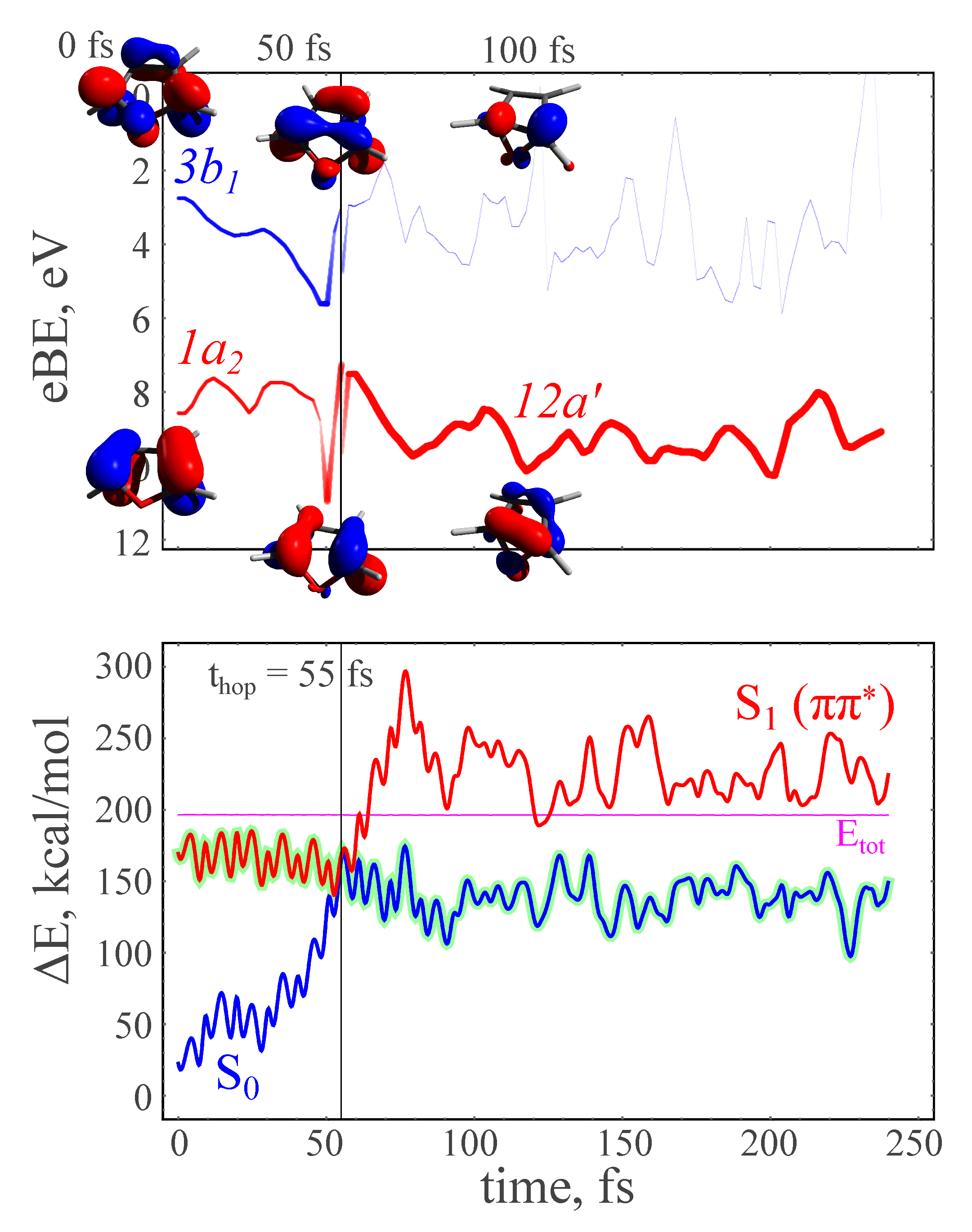
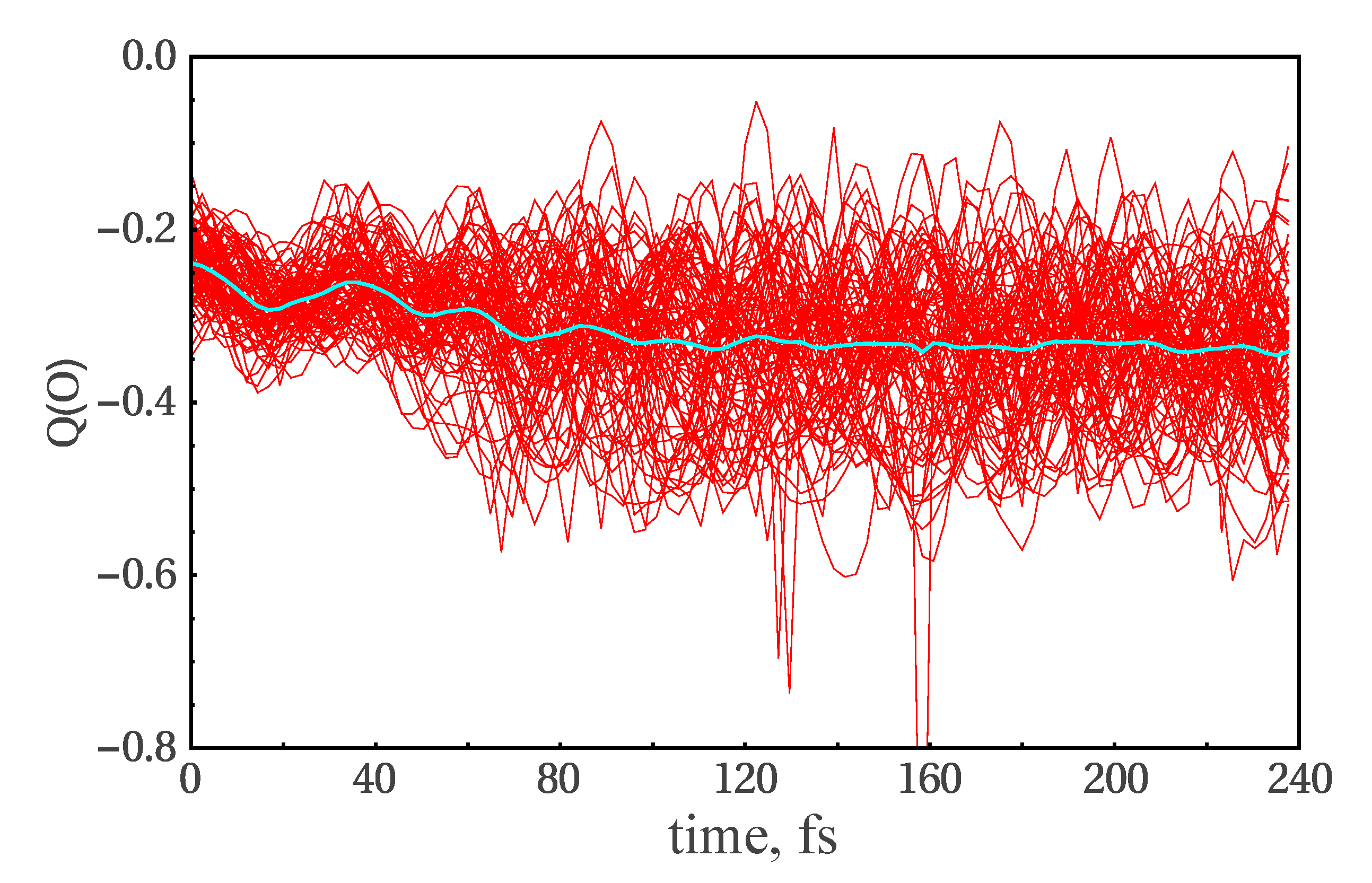
3.3. NAMD Simulations of the Relaxation
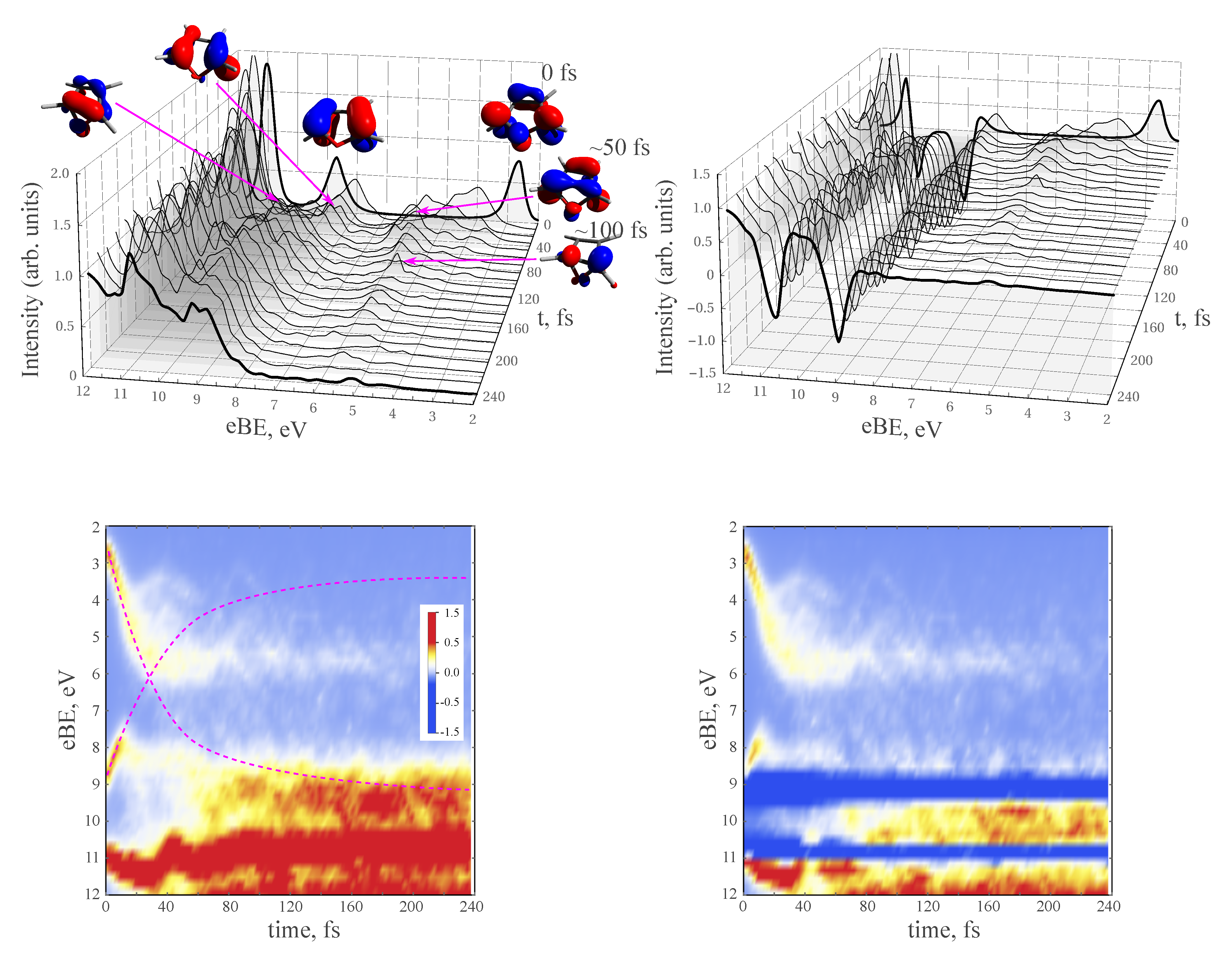
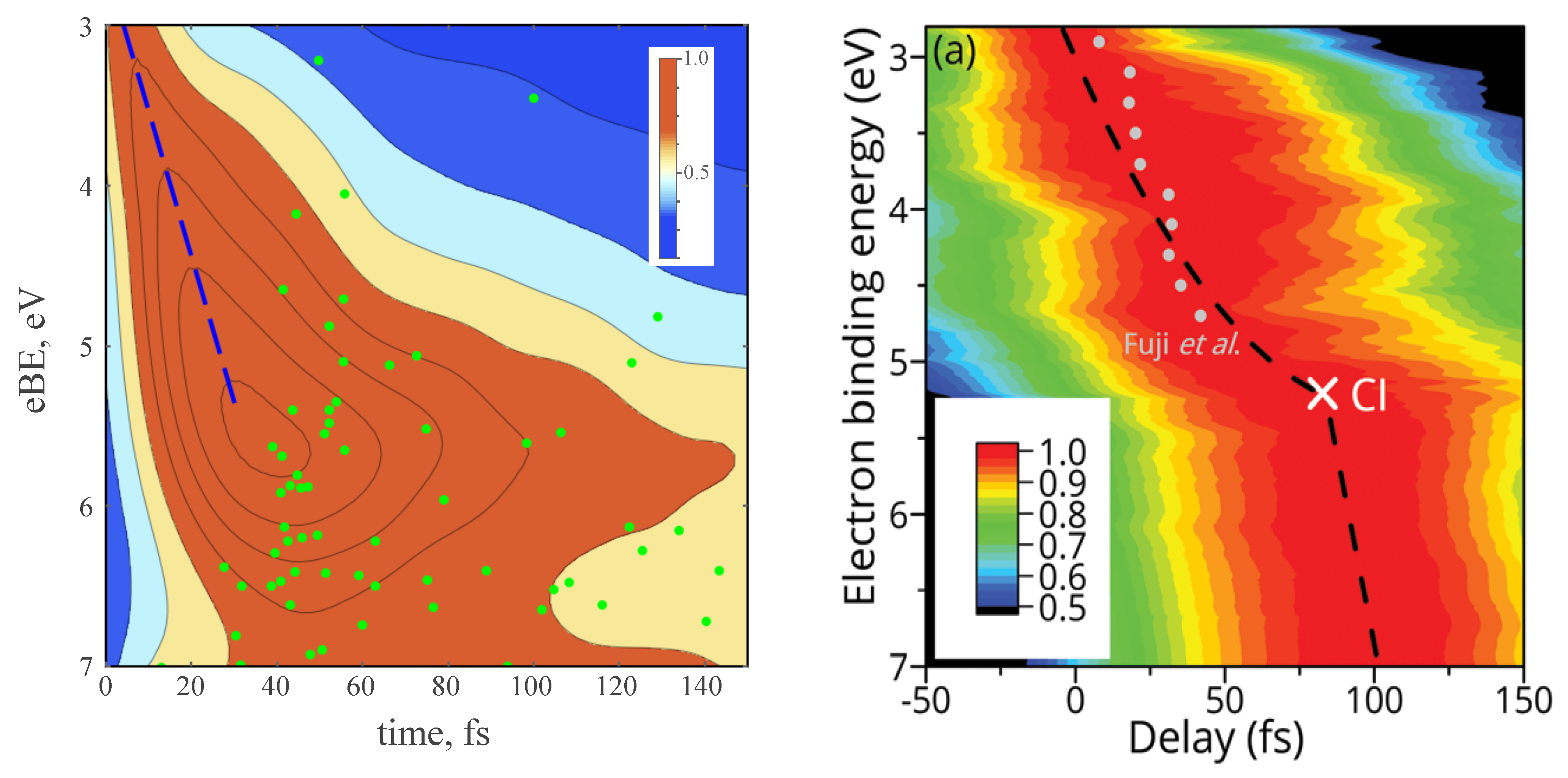
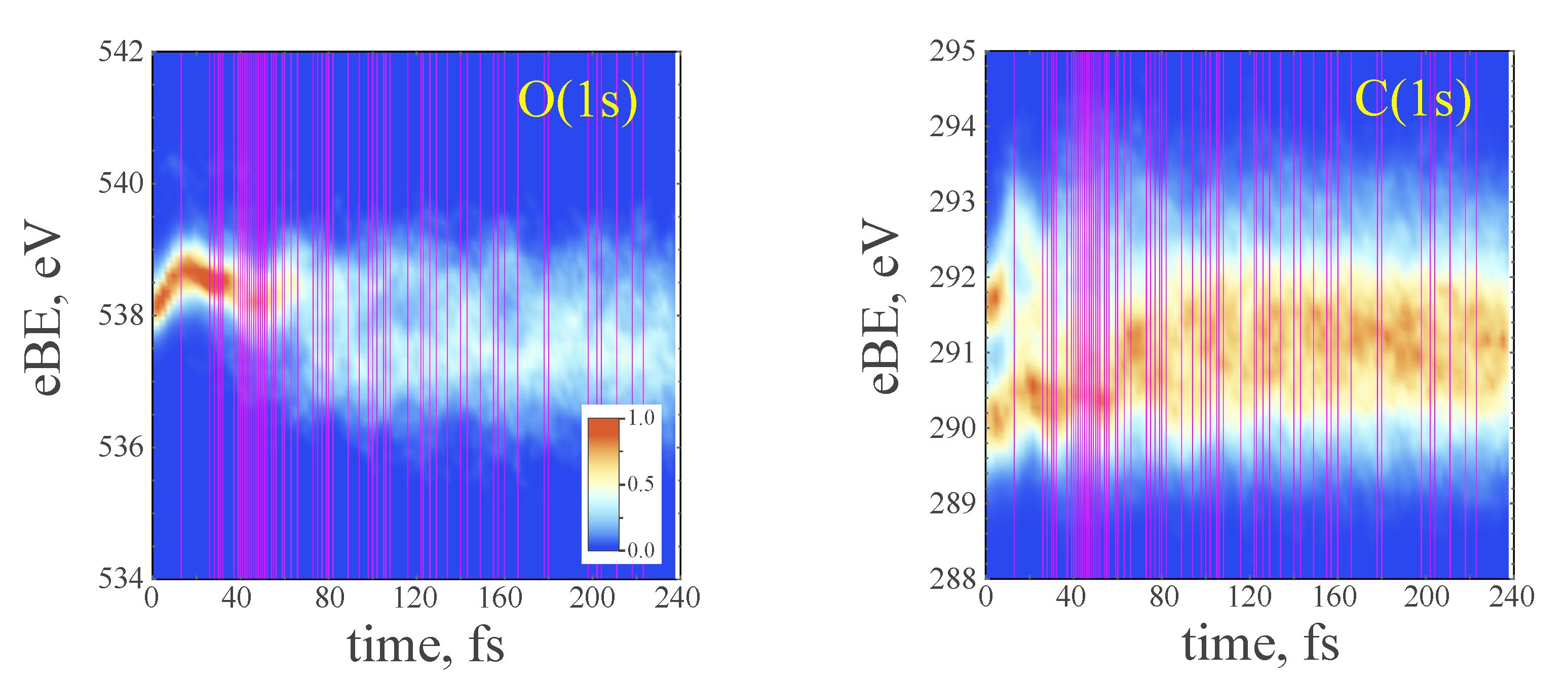
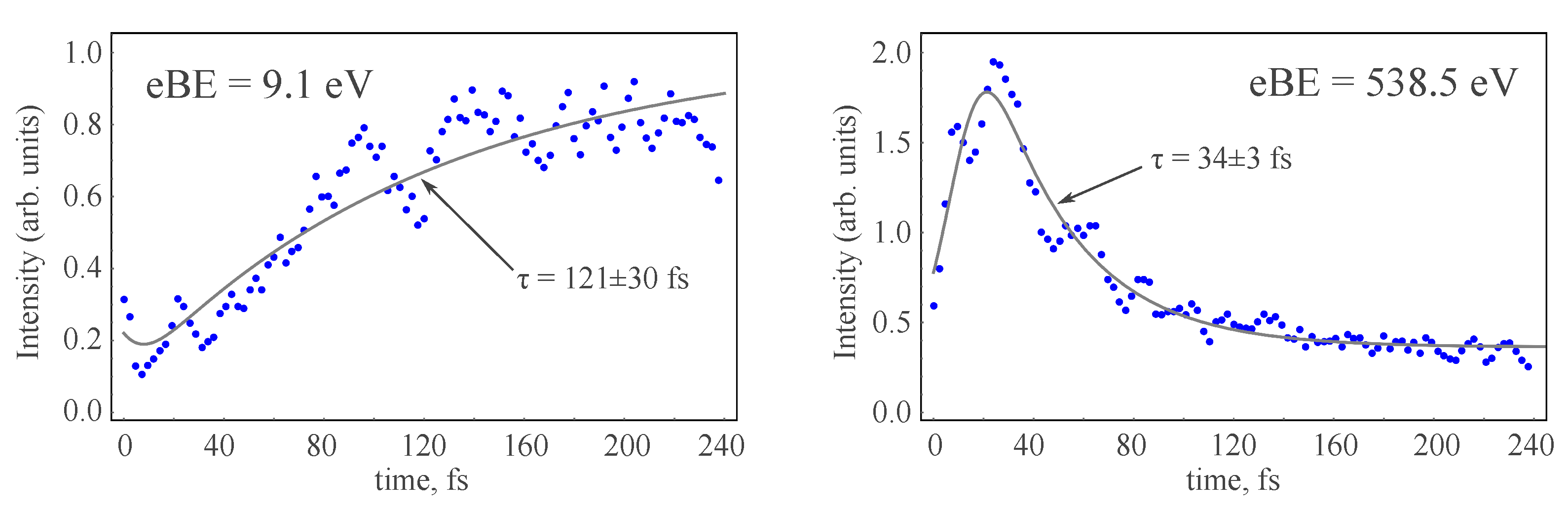
3.4. TRPE Spectra of Excited Furan
4. Conclusions
- The excited-state lifetime can be estimated from the dynamics of the recovery of the ground-state PE intensity.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neumark, D.M. Time-Resolved Photoelectron Spectroscopy of Molecules And Clusters. Annu. Rev. Phys. Chem. 2001, 52, 255–277. [Google Scholar] [CrossRef] [PubMed]
- Stolow, A. Femtosecond Time-Resolved Photoelectron Spectroscopy of Polyatomic Molecules. Annu. Rev. Phys. Chem. 2003, 54, 89–119. [Google Scholar] [CrossRef]
- Stolow, A.; Bragg, A.E.; Neumark, D.M. Femtosecond Time-Resolved Photoelectron Spectroscopy. Chem. Rev. 2004, 104, 1719–1758. [Google Scholar] [CrossRef]
- Fuji, T.; Suzuki, Y.I.; Horio, T.; Suzuki, T.; Mitrić, R.; Werner, U.; Bonačić-Koutecký, V. Ultrafast photodynamics of furan. J. Chem. Phys. 2010, 133, 234303. [Google Scholar] [CrossRef]
- Spesyvtsev, R.; Horio, T.; Suzuki, Y.I.; Suzuki, T. Excited-state dynamics of furan studied by sub-20-fs time-resolved photoelectron imaging using 159-nm pulses. J. Chem. Phys. 2015, 143, 014302. [Google Scholar] [CrossRef]
- Schalk, O.; Geng, T.; Hansson, T.; Thomas, R.D. The ring-opening channel and the influence of Rydberg states on the excited state dynamics of furan and its derivatives. J. Chem. Phys. 2018, 149, 084303. [Google Scholar] [CrossRef]
- Iikubo, R.; Sekikawa, T.; Harabuchi, Y.; Taketsugu, T. Structural dynamics of photochemical reactions probed by time-resolved photoelectron spectroscopy using high harmonic pulses. Faraday Discuss. 2016, 194, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Sato, M.; Suzuki, T. Direct Observation of Ground-State Product Formation in a 1,3-Cyclohexadiene Ring-Opening Reaction. J. Phys. Chem. Lett. 2015, 6, 343–346. [Google Scholar] [CrossRef]
- Adachi, S.; Schatteburg, T.; Humeniuk, A.; Mitrić, R.; Suzuki, T. Probing ultrafast dynamics during and after passing through conical intersections. Phys. Chem. Chem. Phys. 2019, 21, 13902–13905. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.F.; Reduzzi, M.; Wang, H.; Poullain, S.M.; Kobayashi, Y.; Barreau, L.; Prendergast, D.; Neumark, D.M.; Leone, S.R. Revealing electronic state-switching at conical intersections in alkyl iodides by ultrafast XUV transient absorption spectroscopy. Nat. Commun. 2020, 11, 4042. [Google Scholar] [CrossRef]
- Pathak, S.; Ibele, L.M.; Boll, R.; Callegari, C.; Demidovich, A.; Erk, B.; Feifel, R.; Forbes, R.; Di Fraia, M.; Giannessi, L.; et al. Tracking the ultraviolet-induced photochemistry of thiophenone during and after ultrafast ring opening. Nat. Chem. 2020, 12, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Neppl, S.; Gessner, O. Time-resolved X-ray photoelectron spectroscopy techniques for the study of interfacial charge dynamics. J. Electron Spectrosc. Relat. Phenom. 2015, 200, 64–77. [Google Scholar] [CrossRef]
- Attar, A.R.; Bhattacherjee, A.; Pemmaraju, C.D.; Schnorr, K.; Closser, K.D.; Prendergast, D.; Leone, S.R. Femtosecond X-ray spectroscopy of an electrocyclic ring-opening reaction. Science 2017, 356, 54–59. [Google Scholar] [CrossRef]
- Bressler, C.; Chergui, M. Ultrafast X-ray Absorption Spectroscopy. Chem. Rev. 2004, 104, 1781–1812. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.X.; Zhang, X.; Shelby, M.L. Recent advances on ultrafast X-ray spectroscopy in the chemical sciences. Chem. Sci. 2014, 5, 4136–4152. [Google Scholar] [CrossRef]
- Roebber, J.; Gerrity, D.; Hemley, R.; Vaida, V. Electronic spectrum of furan from 2200 to 1950 Å. Chem. Phys. Lett. 1980, 75, 104–106. [Google Scholar] [CrossRef]
- Gromov, E.V.; Trofimov, A.B.; Gatti, F.; Köppel, H. Theoretical study of photoinduced ring-opening in furan. J. Chem. Phys. 2010, 133, 164309. [Google Scholar] [CrossRef]
- Stenrup, M.; Larson, Å. A computational study of radiationless deactivation mechanisms of furan. Chem. Phys. 2011, 379, 6–12. [Google Scholar] [CrossRef]
- Hua, W.; Oesterling, S.; Biggs, J.D.; Zhang, Y.; Ando, H.; de Vivie-Riedle, R.; Fingerhut, B.P.; Mukamel, S. Monitoring conical intersections in the ring opening of furan by attosecond stimulated X-ray Raman spectroscopy. Struct. Dyn. 2016, 3, 023601. [Google Scholar] [CrossRef]
- Oesterling, S.; Schalk, O.; Geng, T.; Thomas, R.D.; Hansson, T.; de Vivie-Riedle, R. Substituent effects on the relaxation dynamics of furan, furfural and β-furfural: A combined theoretical and experimental approach. Phys. Chem. Chem. Phys. 2017, 19, 2025–2035. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.G.; Ko, C.; Quenneville, J.; Martínez, T.J. Conical intersections and double excitations in time-dependent density functional theory. Mol. Phys. 2006, 104, 1039–1051. [Google Scholar] [CrossRef]
- Nikiforov, A.; Gamez, J.A.; Thiel, W.; Huix-Rotllant, M.; Filatov, M. Assessment of approximate computational methods for conical intersections and branching plane vectors in organic molecules. J. Chem. Phys. 2014, 141, 124122. [Google Scholar] [CrossRef]
- Huix-Rotllant, M.; Nikiforov, A.; Thiel, W.; Filatov, M. Description of Conical Intersections with Density Functional Methods. In Density-Functional Methods for Excited States; Ferré, N., Filatov, M., Huix-Rotllant, M., Eds.; Springer: Heidelberg, Germany, 2016; Volume 368, pp. 445–476. [Google Scholar]
- Filatov, M.; Shaik, S. A spin-restricted ensemble-referenced Kohn-Sham method and its application to diradicaloid situations. Chem. Phys. Lett. 1999, 304, 429–437. [Google Scholar] [CrossRef]
- Moreira, I.d.P.R.; Costa, R.; Filatov, M.; Illas, F. Restricted ensemble-referenced Kohn-Sham versus broken symmetry approaches in density functional theory: Magnetic coupling in Cu binuclear complexes. J. Chem. Theory Comput. 2007, 3, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Kazaryan, A.; Heuver, J.; Filatov, M. Excitation Energies from Spin-Restricted Ensemble-Referenced Kohn-Sham Method: A State-Average Approach. J. Phys. Chem. A 2008, 112, 12980–12988. [Google Scholar] [CrossRef]
- Filatov, M. Assessment of density functional methods for obtaining geometries at conical intersections in organic molecules. J. Chem. Theory Comput. 2013, 9, 4526–4541. [Google Scholar] [CrossRef]
- Filatov, M. Spin-restricted ensemble-referenced Kohn-Sham method: Basic principles and application to strongly correlated ground and excited states of molecules. WIREs Comput. Mol. Sci. 2015, 5, 146–167. [Google Scholar] [CrossRef]
- Filatov, M. Ensemble DFT Approach to Excited States of Strongly Correlated Molecular Systems. In Density-Functional Methods for Excited States; Ferré, N., Filatov, M., Huix-Rotllant, M., Eds.; Springer: Heidelberg, Germany, 2016; Volume 368, pp. 97–124. [Google Scholar]
- Huix-Rotllant, M.; Filatov, M.; Gozem, S.; Schapiro, I.; Olivucci, M.; Ferré, N. Assessment of density functional theory for describing the correlation effects on the ground and excited state potential energy surfaces of a retinal chromophore model. J. Chem. Theory Comput. 2013, 9, 3917–3932. [Google Scholar] [CrossRef]
- Gozem, S.; Melaccio, F.; Valentini, A.; Filatov, M.; Huix-Rotllant, M.; Ferré, N.; Frutos, L.M.; Angeli, C.; Krylov, A.I.; Granovsky, A.A.; et al. Shape of Multireference, Equation-of-Motion Coupled-Cluster, and Density Functional Theory Potential Energy Surfaces at a Conical Intersection. J. Chem. Theory Comput. 2014, 10, 3074–3084. [Google Scholar] [CrossRef] [PubMed]
- Filatov, M.; Lee, S.; Choi, C.H. Computation of Molecular Ionization Energies Using an Ensemble Density Functional Theory Method. J. Chem. Theory Comput. 2020, 16, 4489–4504. [Google Scholar] [CrossRef]
- Filatov, M.; Lee, S.; Nakata, H.; Choi, C.H. Computation of Molecular Electron Affinities Using an Ensemble Density Functional Theory Method. J. Phys. Chem. A 2020, 124, 7795–7804. [Google Scholar] [CrossRef]
- Tao, H.; Allison, T.K.; Wright, T.W.; Stooke, A.M.; Khurmi, C.; van Tilborg, J.; Liu, Y.; Falcone, R.W.; Belkacem, A.; Martínez, T.J. Ultrafast internal conversion in ethylene. I. The excited state lifetime. J. Chem. Phys. 2011, 134, 244306. [Google Scholar] [CrossRef] [PubMed]
- Filatov, M.; Lee, S.; Nakata, H.; Choi, C.H. Structural or population dynamics: What is revealed by the time-resolved photoelectron spectroscopy of 1,3-cyclohexadiene? A study with an ensemble density functional theory method. Phys. Chem. Chem. Phys. 2020, 22, 17567–17573. [Google Scholar] [CrossRef]
- Filatov, M.; Min, S.K.; Kim, K.S. Non-adiabatic dynamics of ring opening in cyclohexa-1,3-diene described by an ensemble density-functional theory method. Mol. Phys. 2019, 117, 1128–1141. [Google Scholar] [CrossRef]
- Valone, S.M. A one-to-one mapping between one-particle densities and some n-particle ensembles. J. Chem. Phys. 1980, 73, 4653–4655. [Google Scholar] [CrossRef]
- Lieb, E.H. Density functionals for Coulomb systems. Int. J. Quantum Chem. 1983, 24, 243–277. [Google Scholar] [CrossRef]
- Perdew, J.P.; Parr, R.G.; Levy, M.; Balduz, J.L., Jr. Density-Functional Theory for Fractional Particle Number: Derivative Discontinuities of the Energy. Phys. Rev. Lett. 1982, 49, 1691–1694. [Google Scholar] [CrossRef]
- Englisch, H.; Englisch, R. Hohenberg-Kohn Theorem and Non-V-Representable Densities. Physica 1983, A121, 253–268. [Google Scholar] [CrossRef]
- Englisch, H.; Englisch, R. Exact Density Functionals for Ground-State Energies. I. General Results. Phys. Stat. Sol. 1984, 123, 711–721. [Google Scholar] [CrossRef]
- Englisch, H.; Englisch, R. Exact Density Functionals for Ground-State Energies II. Details and Remarks. Phys. Stat. Sol. 1984, 124, 373–379. [Google Scholar] [CrossRef]
- Gross, E.K.U.; Oliveira, L.N.; Kohn, W. Rayleigh-Ritz variational principle for ensembles of fractionally occupied states. Phys. Rev. A 1988, 37, 2805–2808. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.K.U.; Oliveira, L.N.; Kohn, W. Density-functional theory for ensembles of fractionally occupied states. I. Basic formalism. Phys. Rev. A 1988, 37, 2809–2820. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.N.; Gross, E.K.U.; Kohn, W. Density-functional theory for ensembles of fractionally occupied states. II. Application to the He atom. Phys. Rev. A 1988, 37, 2821–2833. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.N.; Gross, E.K.U.; Kohn, W. Ensemble-Density Functional Theory. Int. J. Quantum Chem. Quantum Chem. Symp. 1990, 24, 707–716. [Google Scholar] [CrossRef]
- Schipper, P.R.T.; Gritsenko, O.V.; Baerends, E.J. One-determinantal pure state versus ensemble Kohn-Sham solutions in the case of strong electron correlation: CH2 and C2. Theor. Chem. Acc. 1998, 99, 329–343. [Google Scholar] [CrossRef]
- Schipper, P.R.T.; Gritsenko, O.V.; Baerends, E.J. Benchmark calculations of chemical reactions in density functional theory: Comparison of the accurate Kohn-Sham solution with generalized gradient approximations for the H2+H and H2+H2 reactions. J. Chem. Phys. 1999, 111, 4056–4067. [Google Scholar] [CrossRef]
- Morrison, R.C. Electron correlation and noninteracting v-representability in density functional theory: The Be isoelectronic series. J. Chem. Phys. 2002, 117, 10506–10511. [Google Scholar] [CrossRef]
- Filatov, M.; Martínez, T.J.; Kim, K.S. Using the GVB Ansatz to develop ensemble DFT method for describing multiple strongly correlated electron pairs. Phys. Chem. Chem. Phys. 2016, 18, 21040–21050. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, A.; Gamez, J.A.; Thiel, W.; Filatov, M. Computational Design of a Family of Light-Driven Rotary Molecular Motors with Improved Quantum Efficiency. J. Phys. Chem. Lett. 2016, 7, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Filatov, M.; Martínez, T.J.; Kim, K.S. Description of ground and excited electronic states by ensemble density functional method with extended active space. J. Chem. Phys. 2017, 147, 064104. [Google Scholar] [CrossRef] [PubMed]
- Hirao, K.; Nakatsuji, H. General SCF operator satisfying correct variational condition. J. Chem. Phys. 1973, 59, 1457–1462. [Google Scholar] [CrossRef]
- Senjean, B.; Fromager, E. Unified formulation of fundamental and optical gap problems in density-functional theory for ensembles. Phys. Rev. A 2018, 98, 022513. [Google Scholar] [CrossRef]
- Senjean, B.; Fromager, E. N-centered ensemble density-functional theory for open systems. Int. J. Quantum Chem. 2020, e26190. [Google Scholar] [CrossRef]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Morrell, M.M.; Parr, R.G.; Levy, M. Calculation of ionization potentials from density matrices and natural functions, and the long-range behavior of natural orbitals and electron density. J. Chem. Phys. 1975, 62, 549–554. [Google Scholar] [CrossRef]
- Smith, D.W.; Day, O.W. Extension of Koopmans’ theorem. I. Derivation. J. Chem. Phys. 1975, 62, 113–114. [Google Scholar] [CrossRef]
- Cioslowski, J.; Piskorz, P.; Liu, G. Ionization potentials and electron affinities from the extended Koopmans’ theorem applied to energy-derivative density matrices: The EKTMPn and EKTQCISD methods. J. Chem. Phys. 1997, 107, 6804–6811. [Google Scholar] [CrossRef]
- Welden, A.R.; Phillips, J.J.; Zgid, D. Ionization potentials and electron affinities from the extended Koopmans’ theorem in self-consistent Green’s function theory. arXiv 2015, arXiv:1505.05575v1. [Google Scholar]
- Spanner, M.; Patchkovskii, S.; Zhou, C.; Matsika, S.; Kotur, M.; Weinacht, T.C. Dyson norms in XUV and strong-field ionization of polyatomics: Cytosine and uracil. Phys. Rev. A 2012, 86, 053406. [Google Scholar] [CrossRef]
- Oppenheim, I.; Ross, J. Temperature Dependence of Distribution Functions in Quantum Statistical Mechanics. Phys. Rev. 1957, 107, 28–32. [Google Scholar] [CrossRef]
- Davies, R.W.; Davies, K.T.R. On the Wigner Distribution Function for an Oscillator. Ann. Phys. 1975, 89, 261–273. [Google Scholar] [CrossRef]
- Thompson, A.L.; Martínez, T.J. Time-resolved photoelectron spectroscopy from first principles: Excited state dynamics of benzene. Faraday Discuss. 2011, 150, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.J.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.; Schmidt, M. Theory and Applications of Computational Chemistry, the First Forty Years; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Filatov, M.; Liu, F.; Martínez, T.J. Analytical derivatives of the individual state energies in ensemble density functional theory method. I. General formalism. J. Chem. Phys. 2017, 147, 034113. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Kästner, J.; Carr, J.M.; Keal, T.W.; Thiel, W.; Wander, A.; Sherwood, P. DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations. J. Phys. Chem. A 2009, 113, 11856–11865. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; Berne, B.J., Ciccotti, G., Coker, D.F., Eds.; World Scientific: Singapore, 1998; Chapter 16; pp. 385–404. [Google Scholar] [CrossRef]
- Levine, B.; Coe, J.D.; Martínez, T.J. Optimizing conical intersections without derivative coupling vectors: Application to multistate multireference second-order perturbation theory (MS-CASPT2). J. Phys. Chem. B 2008, 112, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.K.; Lee, I.S.; Min, S.K. Surface Hopping Dynamics beyond Nonadiabatic Couplings for Quantum Coherence. J. Phys. Chem. Lett. 2018, 9, 1097–1104. [Google Scholar] [CrossRef]
- Hunter, G. Conditional probability amplitudes in wave mechanics. Int. J. Quantum Chem. 1975, 9, 237–242. [Google Scholar] [CrossRef]
- Abedi, A.; Maitra, N.T.; Gross, E.K.U. Exact factorization of the time-dependent electron-nuclear wave function. Phys. Rev. Lett. 2010, 105, 123002. [Google Scholar] [CrossRef] [PubMed]
- Abedi, A.; Maitra, N.T.; Gross, E.K.U. Correlated electron-nuclear dynamics: Exact factorization of the molecular wave-function. J. Chem. Phys. 2012, 137, 22A530. [Google Scholar] [CrossRef]
- Abedi, A.; Agostini, F.; Suzuki, Y.; Gross, E.K.U. Dynamical steps that bridge piecewise adiabatic shapes in the exact time-dependent potential energy surface. Phys. Rev. Lett. 2013, 110, 263001. [Google Scholar] [CrossRef]
- Agostini, F.; Abedi, A.; Suzuki, Y.; Min, S.K.; Maitra, N.T.; Gross, E.K.U. The exact electronic back-reaction on classical nuclei in non-adiabatic charge transfer. J. Chem. Phys. 2015, 142, 084303. [Google Scholar] [CrossRef] [PubMed]
- Tully, J.C. Molecular dynamics with electronic transitions. J. Chem. Phys. 1990, 93, 1061. [Google Scholar] [CrossRef]
- Loos, P.F.; Lipparini, F.; Boggio-Pasqua, M.; Scemama, A.; Jacquemin, D. A Mountaineering Strategy to Excited States: Highly Accurate Energies and Benchmarks for Medium Sized Molecules. J. Chem. Theory Comput. 2020, 16, 1711–1741. [Google Scholar] [CrossRef]
- Politzer, P.; Abu-Awwad, F. A comparative analysis of Hartree-Fock and Kohn-Sham orbital energies. Theor. Chem. Acc. 1998, 99, 83–87. [Google Scholar] [CrossRef]
- Chong, D.P.; Gritsenko, O.V.; Baerends, E.J. Interpretation of the Kohn-Sham orbital energies as approximate vertical ionization potentials. J. Chem. Phys. 2002, 116, 1760–1772. [Google Scholar] [CrossRef]
- Ranasinghe, D.S.; Margraf, J.T.; Perera, A.; Bartlett, R.J. Vertical valence ionization potential benchmarks from equation-of-motion coupled cluster theory and QTP functionals. J. Chem. Phys. 2019, 150, 074108. [Google Scholar] [CrossRef]
- Sell, J.A.; Kuppermann, A. Angular distributions in the photoelectron spectroscopy of furan, thiophene, and pyrrole. Chem. Phys. Lett. 1979, 61, 355–362. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Nangia, S.; Jasper, A.W.; Miller, T.F.; Truhlar, D.G. Army ants algorithm for rare event sampling of delocalized nonadiabatic transitions by trajectory surface hopping and the estimation of sampling errors by the bootstrap method. J. Chem. Phys. 2004, 120, 3586–3597. [Google Scholar] [CrossRef] [PubMed]
- Aryasetiawan, F.; Gunnarsson, O.; Rubio, A. Excitation energies from time-dependent density-functional formalism for small systems. Europhys. Lett. 2002, 57, 683–689. [Google Scholar] [CrossRef]
- Siegbahn, K. Electron Spectroscopy for Chemical Analysis (E.S.C.A.). Philos. Trans. R. Soc. Lond. Ser. A Math. Phys. Sci. 1970, 268, 33–57. [Google Scholar]
- Gelius, U.; Allan, C.J.; Johansson, G.; Siegbahn, H.; Allison, D.A.; Siegbahn, K. The ESCA Spectra of Benzene and the Iso-electronic Series, Thiophene, Pyrrole and Furan. Phys. Scr. 1971, 3, 237–242. [Google Scholar] [CrossRef]
- Chambers, S.A.; Thomas, T.D. Satellite structure in the x-ray photoelectron spectra of gaseous furan, pyrrole, and thiophen. J. Chem. Phys. 1977, 67, 2596–2603. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
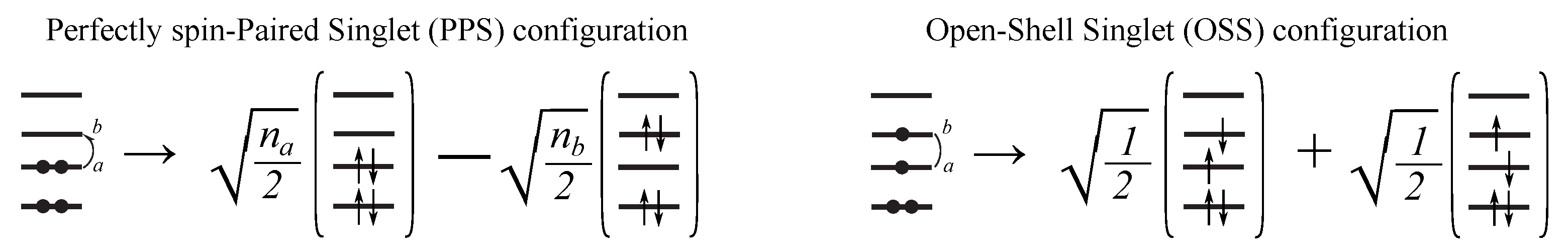{kind=link}
{kind=link}
| hop times 1 | c | , fs | , fs | , fs | , fs |
| 0.40 ± 0.08 2 | 47 ± 2 | 6 ± 2 | 93 ± 18 | 59 ± 11 | |
| S decay 3 | , fs | , fs | , fs | ||
| 68 ± 10 | 25 ± 3 | 93 ± 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filatov, M.; Lee, S.; Nakata, H.; Choi, C.-H. Signatures of Conical Intersection Dynamics in the Time-Resolved Photoelectron Spectrum of Furan: Theoretical Modeling with an Ensemble Density Functional Theory Method. Int. J. Mol. Sci. 2021, 22, 4276. https://doi.org/10.3390/ijms22084276
Filatov M, Lee S, Nakata H, Choi C-H. Signatures of Conical Intersection Dynamics in the Time-Resolved Photoelectron Spectrum of Furan: Theoretical Modeling with an Ensemble Density Functional Theory Method. International Journal of Molecular Sciences. 2021; 22(8):4276. https://doi.org/10.3390/ijms22084276
Chicago/Turabian StyleFilatov, Michael, Seunghoon Lee, Hiroya Nakata, and Cheol-Ho Choi. 2021. "Signatures of Conical Intersection Dynamics in the Time-Resolved Photoelectron Spectrum of Furan: Theoretical Modeling with an Ensemble Density Functional Theory Method" International Journal of Molecular Sciences 22, no. 8: 4276. https://doi.org/10.3390/ijms22084276
APA StyleFilatov, M., Lee, S., Nakata, H., & Choi, C.-H. (2021). Signatures of Conical Intersection Dynamics in the Time-Resolved Photoelectron Spectrum of Furan: Theoretical Modeling with an Ensemble Density Functional Theory Method. International Journal of Molecular Sciences, 22(8), 4276. https://doi.org/10.3390/ijms22084276