
Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins

 , ,
, ,  ,
, 
Abstract
1. Intermediate Filament Proteins
2. Striated Muscle Intermediate Filament Proteins
2.1. Expression of Muscle Intermediate Filament Proteins
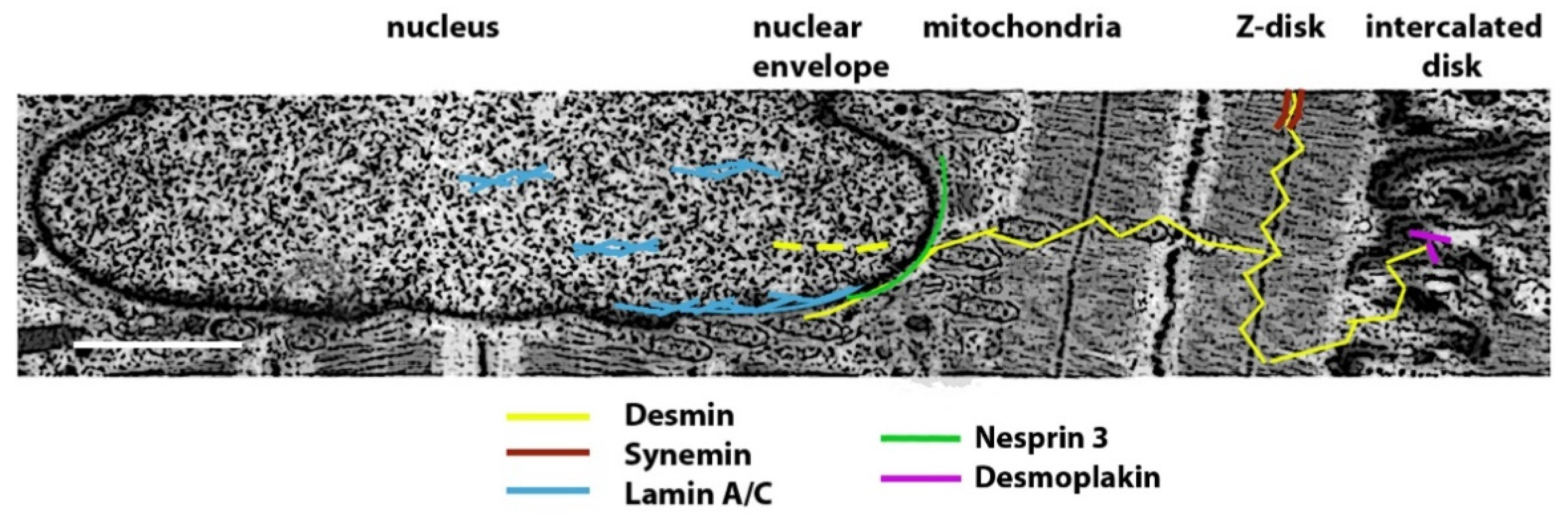
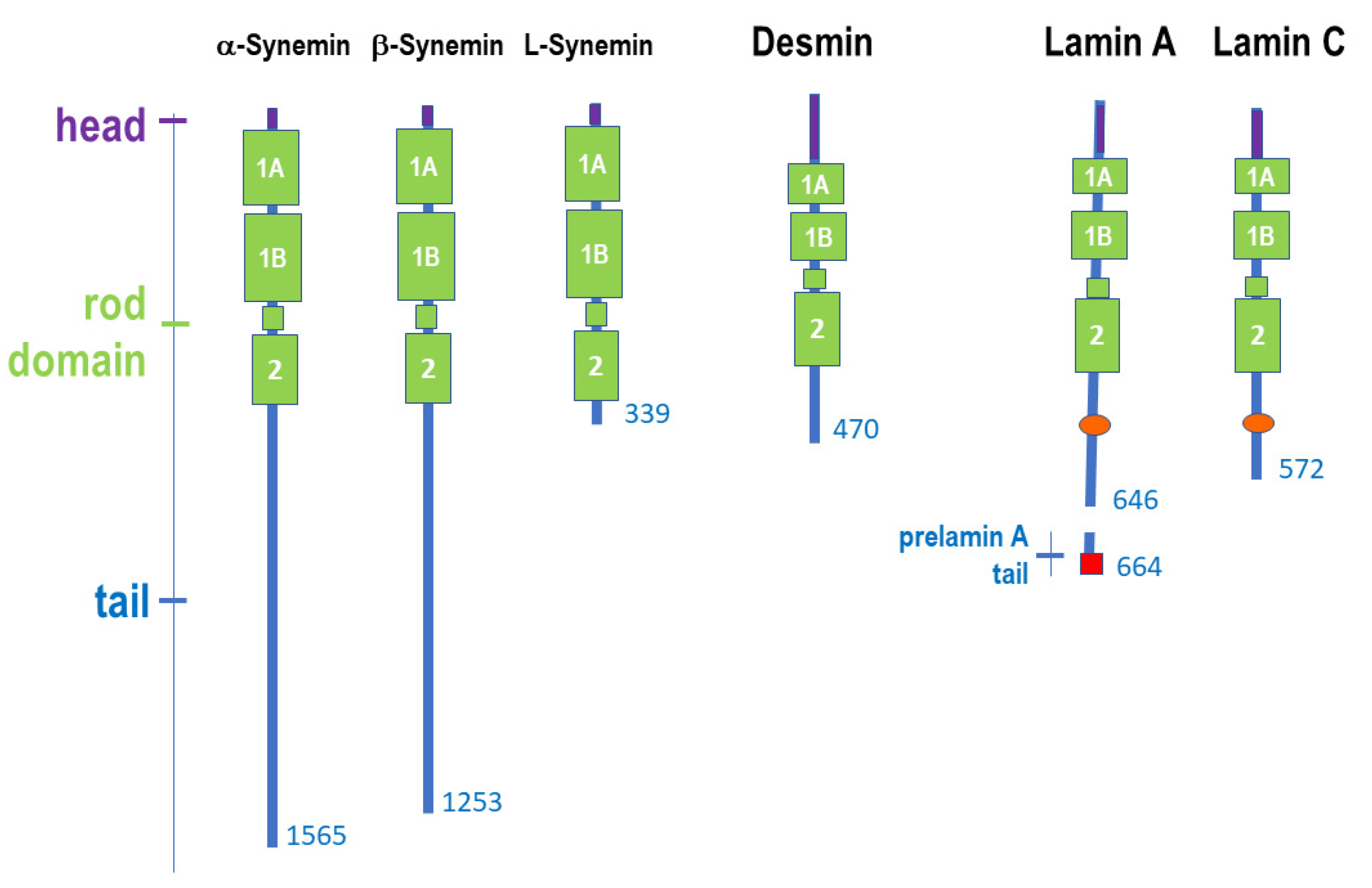
2.2. Structure of Muscle Intermediate Filaments
3. Striated Muscle Disorders Caused by Mutations in Intermediate Filament Proteins
4. Synemin-Related Cardiac Disorders
5. Desminopathies
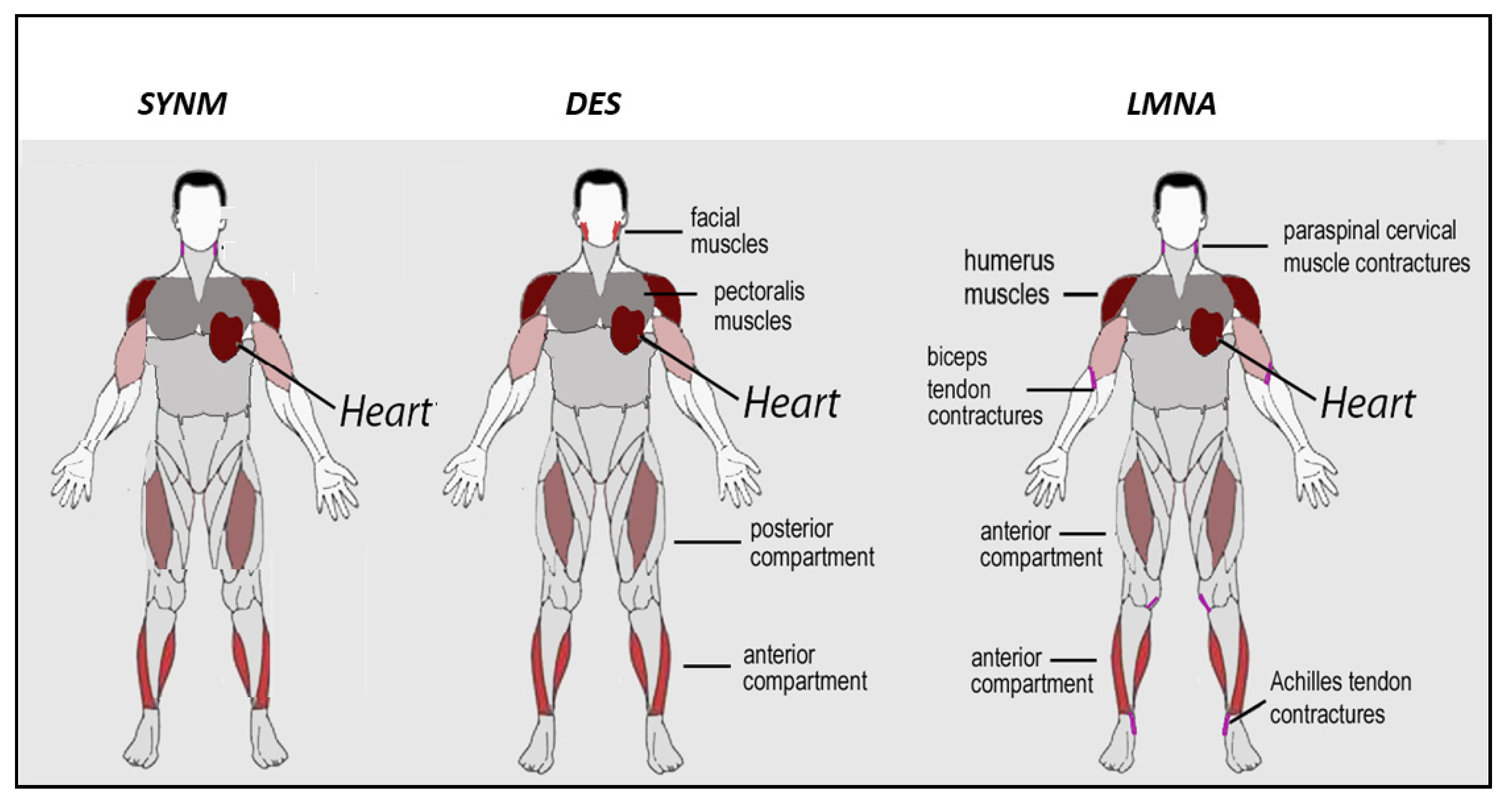
5.1. Clinical Aspects of Skeletal Muscle Involvement in Desminopathies
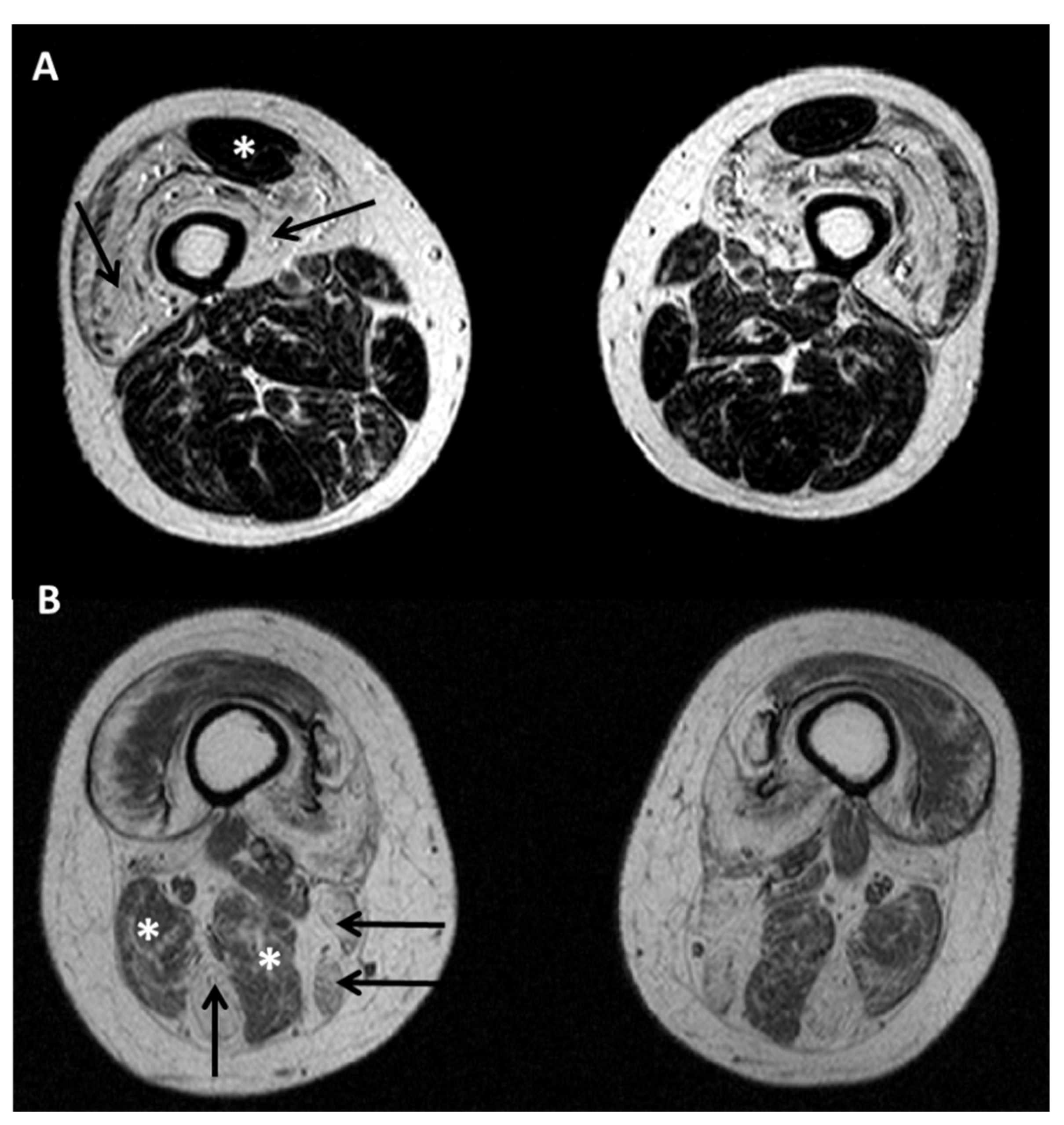
5.2. Diagnostic Protocols for Skeletal Muscle in Desminopathies
5.3. Clinical Aspects of Cardiac Involvement in Desminopathies
5.4. Genotype–Phenotype Correlation in Desminopathies
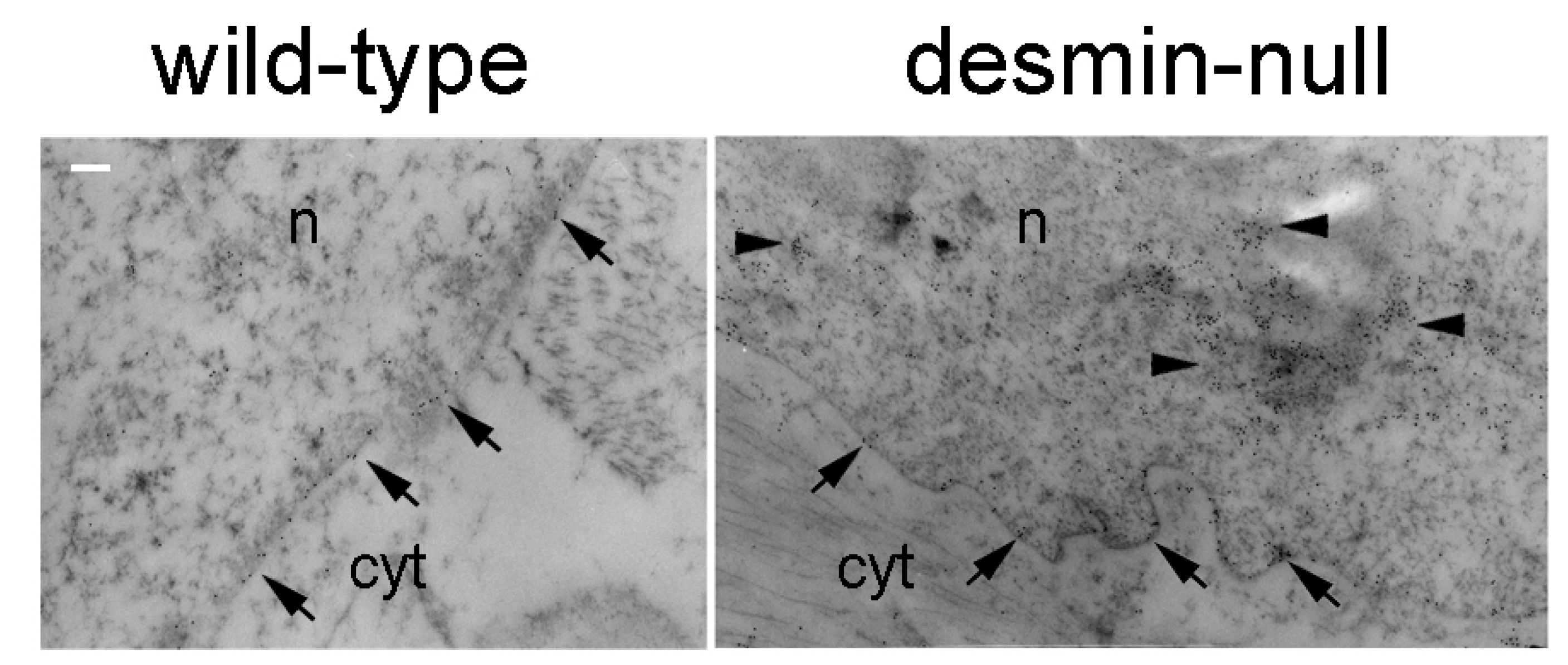
5.5. Pathogenesis of Desminopathies
6. Muscular Laminopathies
6.1. Clinical Aspects of Skeletal Muscle Involvement in Laminopathies
6.2. Clinical Aspects of Cardiac Muscle Involvement in Laminopathies
6.3. Genotype–Phenotype Correlation in Muscular Laminopathies
6.4. Pathogenesis of Muscular Laminopathies
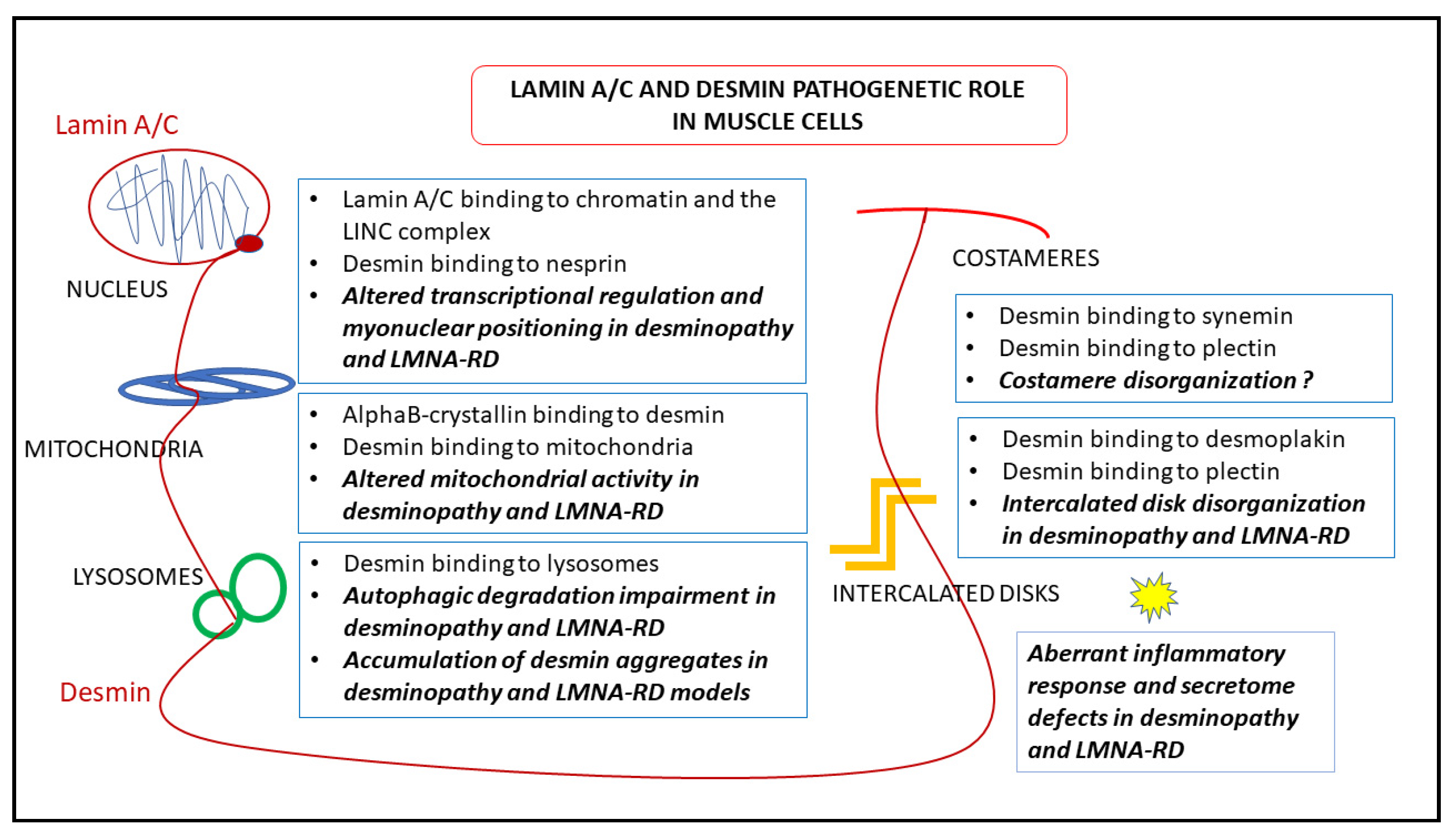
7. Pathogenetic Hypotheses Based on Desmin-Lamin A/C Interplay
8. Therapeutic Perspectives
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gruenbaum, Y.; Aebi, U. Intermediate filaments: A dynamic network that controls cell mechanics. F1000Prime Rep. 2014, 6, 54. [Google Scholar] [CrossRef]
- Singh, S.R.; Kadioglu, H.; Patel, K.; Carrier, L.; Agnetti, G. Is Desmin Propensity to Aggregate Part of its Protective Function? Cells 2020, 9, 491. [Google Scholar] [CrossRef] [PubMed]
- Tsikitis, M.; Galata, Z.; Mavroidis, M.; Psarras, S.; Capetanaki, Y. Intermediate filaments in cardiomyopathy. Biophys. Rev. 2018, 10, 1007–1031. [Google Scholar] [CrossRef] [PubMed]
- Eldirany, S.A.; Lomakin, I.B.; Ho, M.; Bunick, C.G. Recent insight into intermediate filament structure. Curr. Opin. Cell Biol. 2021, 68, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Cleland, M.M.; Murthy, S.N.; Mahammad, S.; Kuczmarski, E.R. Inroads into the structure and function of intermediate filament networks. J. Struct. Biol. 2012, 177, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Capetanaki, Y. Type III Intermediate Filaments Desmin, Glial Fibrillary Acidic Protein (GFAP), Vimentin, and Peripherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a021642. [Google Scholar] [CrossRef] [PubMed]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef]
- Heffler, J.; Shah, P.P.; Robison, P.; Phyo, S.; Veliz, K.; Uchida, K.; Bogush, A.; Rhoades, J.; Jain, R.; Prosser, B.L. A Balance Between Intermediate Filaments and Microtubules Maintains Nuclear Architecture in the Cardiomyocyte. Circ. Res. 2020, 126, e10–e26. [Google Scholar] [CrossRef]
- Redmond, C.J.; Coulombe, P.A. Intermediate filaments as effectors of differentiation. Curr. Opin. Cell Biol. 2020, 68, 155–162. [Google Scholar] [CrossRef]
- Van Bodegraven, E.J.; Etienne-Manneville, S. Intermediate filaments against actomyosin: The david and goliath of cell migration. Curr. Opin. Cell Biol. 2020, 66, 79–88. [Google Scholar] [CrossRef]
- Russell, M.A. Synemin Redefined: Multiple Binding Partners Results in Multifunctionality. Front. Cell Dev. Biol. 2020, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Sward, K.; Krawczyk, K.K.; Moren, B.; Zhu, B.; Matic, L.; Holmberg, J.; Hedin, U.; Uvelius, B.; Stenkula, K.; Rippe, C. Identification of the intermediate filament protein synemin/SYNM as a target of myocardin family coactivators. Am. J. Physiol. Cell Physiol. 2019, 317, C1128–C1142. [Google Scholar] [CrossRef] [PubMed]
- Deville, S.S.; Vehlow, A.; Forster, S.; Dickreuter, E.; Borgmann, K.; Cordes, N. The Intermediate Filament Synemin Regulates Non-Homologous End Joining in an ATM-Dependent Manner. Cancers 2020, 12, 1717. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Lammerding, J. Connecting the plasma membrane to the nucleus by intermediate filaments. Mol. Biol. Cell 2017, 28, 695–696. [Google Scholar] [CrossRef]
- Fuchs, C.; Gawlas, S.; Heher, P.; Nikouli, S.; Paar, H.; Ivankovic, M.; Schultheis, M.; Klammer, J.; Gottschamel, T.; Capetanaki, Y.; et al. Desmin enters the nucleus of cardiac stem cells and modulates Nkx2.5 expression by participating in transcription factor complexes that interact with the nkx2.5 gene. Biol. Open 2016, 5, 140–153. [Google Scholar] [CrossRef]
- Staszewska, I.; Fischer, I.; Wiche, G. Plectin isoform 1-dependent nuclear docking of desmin networks affects myonuclear architecture and expression of mechanotransducers. Hum. Mol. Genet. 2015, 24, 7373–7389. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Schneider, M.; Neumann, S.; Jaeger, V.M.; Taranum, S.; Munck, M.; Cartwright, S.; Richardson, C.; Carthew, J.; Noh, K.; et al. Nesprin interchain associations control nuclear size. Cell Mol. Life Sci. 2012, 69, 3493–3509. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Cabet, E.; Chevalier, N.R.; Moosmann, J.; Schultheis, D.; Haas, J.; Schowalter, M.; Berwanger, C.; Weyerer, V.; Agaimy, A.; et al. Dual Functional States of R406W-Desmin Assembly Complexes Cause Cardiomyopathy With Severe Intercalated Disc Derangement in Humans and in Knock-In Mice. Circulation 2020, 142, 2155–2171. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, O.; Brodehl, A.; Kiseleva, A.; Myasnikov, R.; Meshkov, A.; Stanasiuk, C.; Gartner, A.; Divashuk, M.; Sotnikova, E.; Koretskiy, S.; et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes 2021, 12, 121. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Green, K.J. Desmosomes in the heart: A review of clinical and mechanistic analyses. Cell Commun. Adhes. 2014, 21, 109–128. [Google Scholar] [CrossRef]
- Macquart, C.; Juttner, R.; Morales Rodriguez, B.; Le Dour, C.; Lefebvre, F.; Chatzifrangkeskou, M.; Schmitt, A.; Gotthardt, M.; Bonne, G.; Muchir, A. Microtubule cytoskeleton regulates Connexin 43 localization and cardiac conduction in cardiomyopathy caused by mutation in A-type lamins gene. Hum. Mol. Genet. 2019, 28, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; Capanni, C.; Mattioli, E.; Schena, E.; Squarzoni, S.; Bacalini, M.G.; Garagnani, P.; Salvioli, S.; Franceschi, C.; Lattanzi, G. Lamin A involvement in ageing processes. Ageing Res. Rev. 2020, 62, 101073. [Google Scholar] [CrossRef] [PubMed]
- Camozzi, D.; Capanni, C.; Cenni, V.; Mattioli, E.; Columbaro, M.; Squarzoni, S.; Lattanzi, G. Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus 2014, 5, 427–440. [Google Scholar] [CrossRef]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Maraldi, N.M.; Cenni, V.; Scotlandi, K.; Marino, M.T.; Merlini, L.; Squarzoni, S.; Lattanzi, G. Prelamin A-mediated recruitment of SUN1 to the nuclear envelope directs nuclear positioning in human muscle. Cell Death Differ. 2011, 18, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Meinke, P.; Mattioli, E.; Haque, F.; Antoku, S.; Columbaro, M.; Straatman, K.R.; Worman, H.J.; Gundersen, G.G.; Lattanzi, G.; Wehnert, M.; et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet. 2014, 10, e1004605. [Google Scholar] [CrossRef]
- Zhou, X.; Lin, Y.; Kato, M.; Mori, E.; Liszczak, G.; Sutherland, L.; Sysoev, V.O.; Murray, D.T.; Tycko, R.; McKnight, S.L. Transiently structured head domains control intermediate filament assembly. Proc. Natl. Acad. Sci. USA 2021, 118, 8. [Google Scholar] [CrossRef]
- Cenni, V.; D’Apice, M.R.; Garagnani, P.; Columbaro, M.; Novelli, G.; Franceschi, C.; Lattanzi, G. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res. Rev. 2018, 42, 1–13. [Google Scholar] [CrossRef]
- Makarov, A.A.; Zou, J.; Houston, D.R.; Spanos, C.; Solovyova, A.S.; Cardenal-Peralta, C.; Rappsilber, J.; Schirmer, E.C. Lamin A molecular compression and sliding as mechanisms behind nucleoskeleton elasticity. Nat. Commun. 2019, 10, 3056. [Google Scholar] [CrossRef]
- Ahn, J.; Lee, J.; Jeong, S.; Kang, S.M.; Park, B.J.; Ha, N.C. Beta-strand-mediated dimeric formation of the Ig-like domains of human lamin A/C and B1. Biochem. Biophys. Res. Commun. 2021, 550, 191–196. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Ralston, E.; Lu, Z.; Biscocho, N.; Soumaka, E.; Mavroidis, M.; Prats, C.; Lomo, T.; Capetanaki, Y.; Ploug, T. Blood vessels and desmin control the positioning of nuclei in skeletal muscle fibers. J. Cell Physiol. 2006, 209, 874–882. [Google Scholar] [CrossRef]
- Owens, D.J.; Messeant, J.; Moog, S.; Viggars, M.; Ferry, A.; Mamchaoui, K.; Lacene, E.; Romero, N.; Brull, A.; Bonne, G.; et al. Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth. Int. J. Mol. Sci. 2020, 22, 306. [Google Scholar] [CrossRef]
- Cenni, V.; Kojic, S.; Capanni, C.; Faulkner, G.; Lattanzi, G. Ankrd2 in Mechanotransduction and Oxidative Stress Response in Skeletal Muscle: New Cues for the Pathogenesis of Muscular Laminopathies. Oxid. Med. Cell Longev. 2019, 2019, 7318796. [Google Scholar] [CrossRef]
- Chiarini, F.; Evangelisti, C.; Cenni, V.; Fazio, A.; Paganelli, F.; Martelli, A.M.; Lattanzi, G. The Cutting Edge: The Role of mTOR Signaling in Laminopathies. Int. J. Mol. Sci. 2019, 20, 847. [Google Scholar] [CrossRef]
- Janin, A.; Bauer, D.; Ratti, F.; Valla, C.; Bertrand, A.; Christin, E.; Chopin, E.; Streichenberger, N.; Bonne, G.; Gache, V.; et al. SMAD6 overexpression leads to accelerated myogenic differentiation of LMNA mutated cells. Sci. Rep. 2018, 8, 5618. [Google Scholar] [CrossRef]
- Muchir, A.; Worman, H.J. Targeting Mitogen-Activated Protein Kinase Signaling in Mouse Models of Cardiomyopathy Caused by Lamin A/C Gene Mutations. Methods Enzymol. 2016, 568, 557–580. [Google Scholar] [PubMed]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef]
- Mestroni, L.; Sbaizero, O. Arrhythmogenic Cardiomyopathy: Mechanotransduction Going Wrong. Circulation 2018, 137, 1611–1613. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Park, K.Y.; Semino-Mora, C.; Lee, H.S.; Sivakumar, K.; Goldfarb, L.G. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med. 2000, 342, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Di Barletta, M.R.; Varnous, S.; Becane, H.M.; Hammouda, E.H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.; Benedetti, S.; Petrini, S.; Sambuughin, N.; Boldrini, R.; Menditto, I.; Ferrari, M.; Verardo, M.; Goldfarb, L.; Bertini, E. Major myofibrillar changes in early onset myopathy due to de novo heterozygous missense mutation in lamin A/C gene. Neuromuscul. Disord. 2005, 15, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Quijano-Roy, S. Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb. Clin. Neurol. 2013, 113, 1367–1376. [Google Scholar] [PubMed]
- Azibani, F.; Muchir, A.; Vignier, N.; Bonne, G.; Bertrand, A.T. Striated muscle laminopathies. Semin. Cell Dev. Biol. 2014, 29, 107–115. [Google Scholar] [CrossRef]
- Maggi, L.; D’Amico, A.; Pini, A.; Sivo, S.; Pane, M.; Ricci, G.; Vercelli, L.; D’Ambrosio, P.; Travaglini, L.; Sala, S.; et al. LMNA-associated myopathies: The Italian experience in a large cohort of patients. Neurology 2014, 83, 1634–1644. [Google Scholar] [CrossRef]
- Ditaranto, R.; Boriani, G.; Biffi, M.; Lorenzini, M.; Graziosi, M.; Ziacchi, M.; Pasquale, F.; Vitale, G.; Berardini, A.; Rinaldi, R.; et al. Differences in cardiac phenotype and natural history of laminopathies with and without neuromuscular onset. Orphanet. J. Rare Dis. 2019, 14, 263. [Google Scholar] [CrossRef]
- Paulin, D.; Hovhannisyan, Y.; Kasakyan, S.; Agbulut, O.; Li, Z.; Xue, Z. Synemin-related skeletal and cardiac myopathies: An overview of pathogenic variants. Am. J. Physiol. Cell Physiol. 2020, 318, C709–C718. [Google Scholar] [CrossRef]
- Zhang, S.B.; Liu, Y.X.; Fan, L.L.; Huang, H.; Li, J.J.; Jin, J.Y.; Xiang, R. A novel heterozygous variant p.(Trp538Arg) of SYNM is identified by whole-exome sequencing in a Chinese family with dilated cardiomyopathy. Ann. Hum. Genet. 2019, 83, 95–99. [Google Scholar] [CrossRef]
- Zlotina, A.; Kiselev, A.; Sergushichev, A.; Parmon, E.; Kostareva, A. Rare Case of Ulnar-Mammary-Like Syndrome With Left Ventricular Tachycardia and Lack of TBX3 Mutation. Front. Genet. 2018, 9, 209. [Google Scholar] [CrossRef]
- Clemen, C.S.; Herrmann, H.; Strelkov, S.V.; Schroder, R. Desminopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 47–75. [Google Scholar] [CrossRef]
- Olive, M.; Odgerel, Z.; Martinez, A.; Poza, J.J.; Bragado, F.G.; Zabalza, R.J.; Jerico, I.; Gonzalez-Mera, L.; Shatunov, A.; Lee, H.S.; et al. Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul. Disord. 2011, 21, 533–542. [Google Scholar] [CrossRef]
- Selcen, D.; Engel, A.G. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann. Neurol. 2003, 54, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.A.S.; Lacene, E.; Brochier, G.; Labasse, C.; Madelaine, A.; Silva, V.G.D.; Corazzini, R.; Papadopoulos, K.; Behin, A.; Laforet, P.; et al. Genetic Mutations and Demographic, Clinical, and Morphological Aspects of Myofibrillar Myopathy in a French Cohort. Genet. Test Mol. Biomarkers. 2018, 22, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.; de Walle, H.E.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–866. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.A.; Salajegheh, M.; Judge, D.P.; Feldman, M.W.; Kuncl, R.W.; Waldon, Z.; Steen, H.; Wagner, K.R. Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann. Neurol. 2012, 71, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Murphy, A.; Udd, B.; LGMD Workshop Study Group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Kley, R.A.; Strach, K.; Meyer, C.; Sommer, T.; Eger, K.; Rolfs, A.; Meyer, W.; Pou, A.; Pradas, J.; et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology 2008, 71, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Claeys, K.G.; van der Ven, P.F.; Behin, A.; Stojkovic, T.; Eymard, B.; Dubourg, O.; Laforet, P.; Faulkner, G.; Richard, P.; Vicart, P.; et al. Differential involvement of sarcomeric proteins in myofibrillar myopathies: A morphological and immunohistochemical study. Acta Neuropathol. 2009, 117, 293–307. [Google Scholar] [CrossRef]
- Wahbi, K.; Behin, A.; Charron, P.; Dunand, M.; Richard, P.; Meune, C.; Vicart, P.; Laforet, P.; Stojkovic, T.; Becane, H.M.; et al. High cardiovascular morbidity and mortality in myofibrillar myopathies due to DES gene mutations: A 10-year longitudinal study. Neuromuscul. Disord. 2012, 22, 211–218. [Google Scholar] [CrossRef]
- Delort, F.; Segard, B.D.; Hakibilen, C.; Bourgois-Rocha, F.; Cabet, E.; Vicart, P.; Huang, M.E.; Clary, G.; Lilienbaum, A.; Agbulut, O.; et al. Alterations of redox dynamics and desmin post-translational modifications in skeletal muscle models of desminopathies. Exp. Cell Res. 2019, 383, 111539. [Google Scholar] [CrossRef]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [PubMed]
- Paller, M.S.; Martin, C.M.; Pierpont, M.E. Restrictive cardiomyopathy: An unusual phenotype of a lamin A variant. ESC Heart Fail 2018, 5, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019, 40, 734–741. [Google Scholar] [CrossRef]
- Van Waning, J.I.; Moesker, J.; Heijsman, D.; Boersma, E.; Majoor-Krakauer, D. Systematic Review of Genotype-Phenotype Correlations in Noncompaction Cardiomyopathy. J. Am. Heart Assoc. 2019, 8, e012993. [Google Scholar] [CrossRef]
- Taylor, M.R.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251. [Google Scholar] [CrossRef]
- Bermudez-Jimenez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.A.; Alvarez, M.; Lopez-Fernandez, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The novel desmin variant p.Leu115Ile is associated with a unique form of biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2020. [Google Scholar] [CrossRef]
- Muntoni, F.; Bonne, G.; Goldfarb, L.G.; Mercuri, E.; Piercy, R.J.; Burke, M.; Yaou, R.B.; Richard, P.; Recan, D.; Shatunov, A.; et al. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain 2006, 129 Pt 5, 1260–1268. [Google Scholar] [CrossRef]
- Eiber, N.; Frob, F.; Schowalter, M.; Thiel, C.; Clemen, C.S.; Schroder, R.; Hashemolhosseini, S. Lack of Desmin in Mice Causes Structural and Functional Disorders of Neuromuscular Junctions. Front. Mol. Neurosci. 2020, 13, 567084. [Google Scholar] [CrossRef]
- Henderson, M.; De Waele, L.; Hudson, J.; Eagle, M.; Sewry, C.; Marsh, J.; Charlton, R.; He, L.; Blakely, E.L.; Horrocks, I.; et al. Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol. 2013, 125, 917–919. [Google Scholar] [CrossRef]
- Cetin, N.; Balci-Hayta, B.; Gundesli, H.; Korkusuz, P.; Purali, N.; Talim, B.; Tan, E.; Selcen, D.; Erdem-Ozdamar, S.; Dincer, P. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: Distinct histopathological outcomes compared with desminopathies. J. Med. Genet. 2013, 50, 437–443. [Google Scholar] [CrossRef]
- Durmus, H.; Ayhan, O.; Cirak, S.; Deymeer, F.; Parman, Y.; Franke, A.; Eiber, N.; Chevessier, F.; Schlotzer-Schrehardt, U.; Clemen, C.S.; et al. Neuromuscular endplate pathology in recessive desminopathies: Lessons from man and mice. Neurology 2016, 87, 799–805. [Google Scholar] [CrossRef]
- Mejat, A.; Decostre, V.; Li, J.; Renou, L.; Kesari, A.; Hantai, D.; Stewart, C.L.; Xiao, X.; Hoffman, E.; Bonne, G.; et al. Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J. Cell Biol. 2009, 184, 31–44. [Google Scholar] [CrossRef]
- Bar, H.; Mucke, N.; Katus, H.A.; Aebi, U.; Herrmann, H. Assembly defects of desmin disease mutants carrying deletions in the alpha-helical rod domain are rescued by wild type protein. J. Struct. Biol. 2007, 158, 107–115. [Google Scholar] [CrossRef]
- Ulbricht, A.; Eppler, F.J.; Tapia, V.E.; van der Ven, P.F.; Hampe, N.; Hersch, N.; Vakeel, P.; Stadel, D.; Haas, A.; Saftig, P.; et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr. Biol. 2013, 23, 430–435. [Google Scholar] [CrossRef]
- Selcen, D.; Muntoni, F.; Burton, B.K.; Pegoraro, E.; Sewry, C.; Bite, A.V.; Engel, A.G. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 2009, 65, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Sarparanta, J.; Jonson, P.H.; Kawan, S.; Udd, B. Neuromuscular Diseases Due to Chaperone Mutations: A Review and Some New Results. Int. J. Mol. Sci. 2020, 21, 1409. [Google Scholar] [CrossRef] [PubMed]
- Bar, H.; Schopferer, M.; Sharma, S.; Hochstein, B.; Mucke, N.; Herrmann, H.; Willenbacher, N. Mutations in desmin’s carboxy-terminal “tail” domain severely modify filament and network mechanics. J. Mol. Biol. 2010, 397, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Jacko, D.; Bersiner, K.; Schulz, O.; Przyklenk, A.; Spahiu, F.; Hohfeld, J.; Bloch, W.; Gehlert, S. Coordinated alpha-crystallin B phosphorylation and desmin expression indicate adaptation and deadaptation to resistance exercise-induced loading in human skeletal muscle. Am. J. Physiol. Cell Physiol. 2020, 319, C300–C312. [Google Scholar] [CrossRef] [PubMed]
- Rapti, K.; Diokmetzidou, A.; Kloukina, I.; Milner, D.J.; Varela, A.; Davos, C.H.; Capetanaki, Y. Opposite effects of catalase and MnSOD ectopic expression on stress induced defects and mortality in the desmin deficient cardiomyopathy model. Free Radic. Biol. Med. 2017, 110, 206–218. [Google Scholar] [CrossRef]
- Weisleder, N.; Taffet, G.E.; Capetanaki, Y. Bcl-2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc. Natl. Acad. Sci. USA 2004, 101, 769–774. [Google Scholar] [CrossRef]
- Diokmetzidou, A.; Soumaka, E.; Kloukina, I.; Tsikitis, M.; Makridakis, M.; Varela, A.; Davos, C.H.; Georgopoulos, S.; Anesti, V.; Vlahou, A.; et al. Desmin and alphaB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J. Cell Sci. 2016, 129, 3705–3720. [Google Scholar] [CrossRef]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin related disease: A matter of cell survival failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef]
- Capetanaki, Y. Desmin cytoskeleton: A potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc. Med. 2002, 12, 339–348. [Google Scholar] [CrossRef]
- Capetanaki, Y.; Bloch, R.J.; Kouloumenta, A.; Mavroidis, M.; Psarras, S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell Res. 2007, 313, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Galata, Z.; Kloukina, I.; Kostavasili, I.; Varela, A.; Davos, C.H.; Makridakis, M.; Bonne, G.; Capetanaki, Y. Amelioration of desmin network defects by alphaB-crystallin overexpression confers cardioprotection in a mouse model of dilated cardiomyopathy caused by LMNA gene mutation. J. Mol. Cell. Cardiol. 2018, 125, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Tannous, P.; Zhu, H.; Johnstone, J.L.; Shelton, J.M.; Rajasekaran, N.S.; Benjamin, I.J.; Nguyen, L.; Gerard, R.D.; Levine, B.; Rothermel, B.A.; et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 9745–9750. [Google Scholar] [CrossRef]
- Cabet, E.; Batonnet-Pichon, S.; Delort, F.; Gausseres, B.; Vicart, P.; Lilienbaum, A. Antioxidant Treatment and Induction of Autophagy Cooperate to Reduce Desmin Aggregation in a Cellular Model of Desminopathy. PLoS ONE 2015, 10, e0137009. [Google Scholar] [CrossRef]
- Mavroidis, M.; Davos, C.H.; Psarras, S.; Varela, A.; Athanasiadis, N.C.; Katsimpoulas, M.; Kostavasili, I.; Maasch, C.; Vater, A.; Van Tintelen, J.P.; et al. Complement system modulation as a target for treatment of arrhythmogenic cardiomyopathy. Basic Res. Cardiol. 2015, 110, 27. [Google Scholar] [CrossRef]
- Psarras, S.; Mavroidis, M.; Sanoudou, D.; Davos, C.H.; Xanthou, G.; Varela, A.E.; Panoutsakopoulou, V.; Capetanaki, Y. Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur. Heart J. 2012, 33, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Psarras, S.; Beis, D.; Nikouli, S.; Tsikitis, M.; Capetanaki, Y. Three in a Box: Understanding Cardiomyocyte, Fibroblast, and Innate Immune Cell Interactions to Orchestrate Cardiac Repair Processes. Front. Cardiovasc. Med. 2019, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, G.; Tonin, P.; Mora, M.; Filosto, M.; Morandi, L.; Savio, C.; Dal Pra, I.; Rizzuto, N.; Tomelleri, G. Expression of protein kinase C isoforms and interleukin-1beta in myofibrillar myopathy. Neurology 2004, 62, 1778–1782. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ebbinghaus, H.; Deutsch, M.A.; Gummert, J.; Gartner, A.; Ratnavadivel, S.; Milting, H. Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies. Int. J. Mol. Sci. 2019, 20, 4381. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.F.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Butler, A.W.; Ng, K.M.; Siu, C.W.; Simpson, M.A.; Lai, W.H.; Chan, Y.C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Mateddu, A.; Marrosu, G.; Cocco, E.; Marrosu, M.G. Genetic and clinical characteristics of skeletal and cardiac muscle in patients with lamin A/C gene mutations. Muscle. Nerve. 2013, 48, 161–170. [Google Scholar] [CrossRef]
- Maggi, L.; Carboni, N.; Bernasconi, P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells 2016, 5, 33. [Google Scholar] [CrossRef]
- Brodsky, G.L.; Muntoni, F.; Miocic, S.; Sinagra, G.; Sewry, C.; Mestroni, L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 2000, 101, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Poppe, M.; Quinlivan, R.; Messina, S.; Kinali, M.; Demay, L.; Bourke, J.; Richard, P.; Sewry, C.; Pike, M.; et al. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: From congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch. Neurol. 2004, 61, 690–694. [Google Scholar] [CrossRef]
- Peretto, G.; Di Resta, C.; Perversi, J.; Forleo, C.; Maggi, L.; Politano, L.; Barison, A.; Previtali, S.C.; Carboni, N.; Brun, F.; et al. Cardiac and Neuromuscular Features of Patients With LMNA-Related Cardiomyopathy. Ann. Intern. Med. 2019, 171, 458–463. [Google Scholar] [CrossRef]
- Fan, Y.; Tan, D.; Song, D.; Zhang, X.; Chang, X.; Wang, Z.; Zhang, C.; Chan, S.H.; Wu, Q.; Wu, L.; et al. Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L. Selectivity of muscle sparing in Emery-Dreifuss muscular dystrophy. Neuromuscul. Disord. 2009, 19, 500–501. [Google Scholar] [CrossRef]
- Emery, A.E. Emery-Dreifuss muscular dystrophy—A 40 year retrospective. Neuromuscul. Disord. 2000, 10, 228–232. [Google Scholar] [CrossRef]
- Boriani, G.; Biagini, E.; Ziacchi, M.; Malavasi, V.L.; Vitolo, M.; Talarico, M.; Mauro, E.; Gorlato, G.; Lattanzi, G. Cardiolaminopathies from bench to bedside: Challenges in clinical decision-making with focus on arrhythmia-related outcomes. Nucleus 2018, 9, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Boriani, G.; Gallina, M.; Merlini, L.; Bonne, G.; Toniolo, D.; Amati, S.; Biffi, M.; Martignani, C.; Frabetti, L.; Bonvicini, M.; et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: A long-term longitudinal study. Stroke 2003, 34, 901–908. [Google Scholar] [CrossRef]
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef]
- Bonne, G.; Yaou, R.B.; Beroud, C.; Boriani, G.; Brown, S.; de Visser, M.; Duboc, D.; Ellis, J.; Hausmanowa-Petrusewicz, I.; Lattanzi, G.; et al. 108th ENMC International Workshop, 3rd Workshop of the MYO-CLUSTER project: EUROMEN, 7th International Emery-Dreifuss Muscular Dystrophy (EDMD) Workshop, 13–15 September 2002, Naarden, The Netherlands. Neuromuscul. Disord. 2003, 13, 508–515. [Google Scholar] [CrossRef]
- Diaz-Manera, J.; Alejaldre, A.; Gonzalez, L.; Olive, M.; Gomez-Andres, D.; Muelas, N.; Vilchez, J.J.; Llauger, J.; Carbonell, P.; Marquez-Infante, C.; et al. Muscle imaging in muscle dystrophies produced by mutations in the EMD and LMNA genes. Neuromuscul. Disord. 2016, 26, 33–40. [Google Scholar] [CrossRef]
- Gueneau, L.; Bertrand, A.T.; Jais, J.P.; Salih, M.A.; Stojkovic, T.; Wehnert, M.; Hoeltzenbein, M.; Spuler, S.; Saitoh, S.; Verschueren, A.; et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2009, 85, 338–353. [Google Scholar] [CrossRef]
- Muchir, A.; Bonne, G.; van der Kooi, A.J.; van Meegen, M.; Baas, F.; Bolhuis, P.A.; de Visser, M.; Schwartz, K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 2000, 9, 1453–1459. [Google Scholar] [CrossRef]
- Quijano-Roy, S.; Mbieleu, B.; Bonnemann, C.G.; Jeannet, P.Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef]
- Menezes, M.P.; Waddell, L.B.; Evesson, F.J.; Cooper, S.; Webster, R.; Jones, K.; Mowat, D.; Kiernan, M.C.; Johnston, H.M.; Corbett, A.; et al. Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy. Neurology 2012, 78, 1258–1263. [Google Scholar] [CrossRef]
- Choi, S.A.; Cho, A.; Kim, S.Y.; Kim, W.J.; Shim, Y.K.; Lee, J.S.; Jang, S.S.; Lim, B.C.; Kim, H.; Hwang, H.; et al. Importance of early diagnosis in LMNA-related muscular dystrophy for cardiac surveillance. Muscle. Nerve. 2019, 60, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Komaki, H.; Hayashi, Y.K.; Tsuburaya, R.; Sugie, K.; Kato, M.; Nagai, T.; Imataka, G.; Suzuki, S.; Saitoh, S.; Asahina, N.; et al. Inflammatory changes in infantile-onset LMNA-associated myopathy. Neuromuscul. Disord. 2011, 21, 563–568. [Google Scholar] [CrossRef]
- Hattori, A.; Komaki, H.; Kawatani, M.; Sakuma, H.; Saito, Y.; Nakagawa, E.; Sugai, K.; Sasaki, M.; Hayashi, Y.K.; Nonaka, I.; et al. A novel mutation in the LMNA gene causes congenital muscular dystrophy with dropped head and brain involvement. Neuromuscul. Disord. 2012, 22, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Carboni, N.; Mura, M.; Marrosu, G.; Cocco, E.; Marini, S.; Solla, E.; Mateddu, A.; Maioli, M.A.; Piras, R.; Mallarini, G.; et al. Muscle imaging analogies in a cohort of patients with different clinical phenotypes caused by LMNA gene mutations. Muscle. Nerve. 2010, 41, 458–463. [Google Scholar] [CrossRef]
- Mercuri, E.; Clements, E.; Offiah, A.; Pichiecchio, A.; Vasco, G.; Bianco, F.; Berardinelli, A.; Manzur, A.; Pane, M.; Messina, S.; et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann. Neurol. 2010, 67, 201–208. [Google Scholar] [CrossRef]
- Lin, H.T.; Liu, X.; Zhang, W.; Liu, J.; Zuo, Y.H.; Xiao, J.X.; Zhu, Y.; Yuan, Y.; Wang, Z.X. Muscle Magnetic Resonance Imaging in Patients with Various Clinical Subtypes of LMNA-Related Muscular Dystrophy. Chin. Med. J. 2018, 131, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, P.; Lattanzi, G.; Ognibene, A.; Columbaro, M.; Capanni, C.; Merlini, L.; Maraldi, N.M.; Squarzoni, S. Nuclear alterations in autosomal-dominant Emery-Dreifuss muscular dystrophy. Muscle. Nerve. 2001, 24, 826–829. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Chaouch, M.; Kozlov, S.; Vallat, J.M.; Tazir, M.; Kassouri, N.; Szepetowski, P.; Hammadouche, T.; Vandenberghe, A.; Stewart, C.L.; et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 2002, 70, 726–736. [Google Scholar] [CrossRef]
- Chaouch, M.; Allal, Y.; De Sandre-Giovannoli, A.; Vallat, J.M.; Amer-el-Khedoud, A.; Kassouri, N.; Chaouch, A.; Sindou, P.; Hammadouche, T.; Tazir, M.; et al. The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in Lamin A/C gene. Neuromuscul. Disord. 2003, 13, 60–67. [Google Scholar] [CrossRef]
- Tazir, M.; Azzedine, H.; Assami, S.; Sindou, P.; Nouioua, S.; Zemmouri, R.; Hamadouche, T.; Chaouch, M.; Feingold, J.; Vallat, J.M.; et al. Phenotypic variability in autosomal recessive axonal Charcot-Marie-Tooth disease due to the R298C mutation in lamin A/C. Brain 2004, 127 Pt 1, 154–163. [Google Scholar] [CrossRef]
- Goizet, C.; Yaou, R.B.; Demay, L.; Richard, P.; Bouillot, S.; Rouanet, M.; Hermosilla, E.; Le Masson, G.; Lagueny, A.; Bonne, G.; et al. A new mutation of the lamin A/C gene leading to autosomal dominant axonal neuropathy, muscular dystrophy, cardiac disease, and leuconychia. J. Med. Genet. 2004, 41, e29. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Vilar, D.; Lado-Abeal, J.; Palos-Paz, F.; Lattanzi, G.; Bandin, M.A.; Bellido, D.; Dominguez-Gerpe, L.; Calvo, C.; Perez, O.; Ramazanova, A.; et al. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin. Endocrinol. 2008, 69, 61–68. [Google Scholar] [CrossRef]
- Carboni, N.; Politano, L.; Floris, M.; Mateddu, A.; Solla, E.; Olla, S.; Maggi, L.; Antonietta Maioli, M.; Piras, R.; Cocco, E.; et al. Overlapping syndromes in laminopathies: A meta-analysis of the reported literature. Acta Myol. 2013, 32, 7–17. [Google Scholar] [PubMed]
- Lombardi, F.; Gullotta, F.; Columbaro, M.; Filareto, A.; D’Adamo, M.; Vielle, A.; Guglielmi, V.; Nardone, A.M.; Azzolini, V.; Grosso, E.; et al. Compound heterozygosity for mutations in LMNA in a patient with a myopathic and lipodystrophic mandibuloacral dysplasia type A phenotype. J. Clin. Endocrinol. Metab. 2007, 92, 4467–4471. [Google Scholar] [CrossRef]
- Van Berlo, J.H.; de Voogt, W.G.; van der Kooi, A.J.; van Tintelen, J.P.; Bonne, G.; Yaou, R.B.; Duboc, D.; Rossenbacker, T.; Heidbuchel, H.; de Visser, M.; et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 2005, 83, 79–83. [Google Scholar] [CrossRef]
- Nigro, G.; Russo, V.; Rago, A.; Papa, A.A.; Carbone, N.; Marchel, M.; Palladino, A.; Hausmanowa-Petrusewicz, I.; Russo, M.G.; Politano, L. Regional and transmural dispersion of repolarisation in patients with Emery-Dreifuss muscular dystrophy. Kardiol. Pol. 2012, 70, 1154–1159. [Google Scholar]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef]
- Liu, Z.; Shan, H.; Huang, J.; Li, N.; Hou, C.; Pu, J. A novel lamin A/C gene missense mutation (445 V > E) in immunoglobulin-like fold associated with left ventricular non-compaction. Europace 2016, 18, 617–622. [Google Scholar] [CrossRef]
- Baban, A.; Cicenia, M.; Magliozzi, M.; Gnazzo, M.; Cantarutti, N.; Silvetti, M.S.; Adorisio, R.; Dallapiccola, B.; Bertini, E.; Novelli, A.; et al. Cardiovascular Involvement in Pediatric Laminopathies. Report of Six Patients and Literature Revision. Front. Pediatr. 2020, 8, 374. [Google Scholar] [CrossRef]
- Arbustini, E.; Pilotto, A.; Repetto, A.; Grasso, M.; Negri, A.; Diegoli, M.; Campana, C.; Scelsi, L.; Baldini, E.; Gavazzi, A.; et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. J. Am. Coll. Cardiol. 2002, 39, 981–990. [Google Scholar] [CrossRef]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Van Rijsingen, I.A.; Nannenberg, E.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Grasso, M.; et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Heart Fail 2013, 15, 376–384. [Google Scholar] [CrossRef]
- Madej-Pilarczyk, A. Clinical aspects of Emery-Dreifuss muscular dystrophy. Nucleus 2018, 9, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.E.; Muchir, A.; Bonne, G. ‘State-of-the-heart’ of cardiac laminopathies. Curr. Opin. Cardiol. 2013, 28, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.T.; Chikhaoui, K.; Yaou, R.B.; Bonne, G. Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 2011, 39, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Granger, B.; Gueneau, L.; Drouin-Garraud, V.; Pedergnana, V.; Gagnon, F.; Ben Yaou, R.; Tezenas du Montcel, S.; Bonne, G. Modifier locus of the skeletal muscle involvement in Emery-Dreifuss muscular dystrophy. Hum. Genet. 2011, 129, 149–159. [Google Scholar] [CrossRef]
- Roncarati, R.; Viviani Anselmi, C.; Krawitz, P.; Lattanzi, G.; von Kodolitsch, Y.; Perrot, A.; di Pasquale, E.; Papa, L.; Portararo, P.; Columbaro, M.; et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1105–1111. [Google Scholar] [CrossRef]
- Meinke, P.; Kerr, A.R.W.; Czapiewski, R.; de Las Heras, J.I.; Dixon, C.R.; Harris, E.; Kolbel, H.; Muntoni, F.; Schara, U.; Straub, V.; et al. A multistage sequencing strategy pinpoints novel candidate alleles for Emery-Dreifuss muscular dystrophy and supports gene misregulation as its pathomechanism. EBioMedicine 2020, 51, 102587. [Google Scholar] [CrossRef]
- Frock, R.L.; Kudlow, B.A.; Evans, A.M.; Jameson, S.A.; Hauschka, S.D.; Kennedy, B.K. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006, 20, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, E.; Mozzetta, C.; Marullo, F.; Gregoretti, F.; Gargiulo, A.; Columbaro, M.; Cortesi, A.; Antonelli, L.; Di Pelino, S.; Squarzoni, S.; et al. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 2015, 211, 533–551. [Google Scholar] [CrossRef]
- Santi, S.; Cenni, V.; Capanni, C.; Lattanzi, G.; Mattioli, E. PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation. Cells 2020, 9, 1735. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Mozzetta, C.; Pegoli, G.; Lucini, F.; Valsoni, S.; Rosti, V.; Petrini, C.; Cortesi, A.; Gregoretti, F.; Antonelli, L.; et al. Dysfunctional polycomb transcriptional repression contributes to lamin A/C-dependent muscular dystrophy. J. Clin. Investig. 2020, 130, 2408–2421. [Google Scholar] [CrossRef]
- Bernasconi, P.; Carboni, N.; Ricci, G.; Siciliano, G.; Politano, L.; Maggi, L.; Mongini, T.; Vercelli, L.; Rodolico, C.; Biagini, E.; et al. Elevated TGF beta2 serum levels in Emery-Dreifuss Muscular Dystrophy: Implications for myocyte and tenocyte differentiation and fibrogenic processes. Nucleus 2018, 9, 292–304. [Google Scholar] [CrossRef]
- Cappelletti, C.; Tramacere, I.; Cavalcante, P.; Schena, E.; Politano, L.; Carboni, N.; Gambineri, A.; D’Amico, A.; Ruggiero, L.; Ricci, G.; et al. Cytokine Profile in Striated Muscle Laminopathies: New Promising Biomarkers for Disease Prediction. Cells 2020, 9, 1532. [Google Scholar] [CrossRef] [PubMed]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef]
- Cappelletti, C.; Salerno, F.; Canioni, E.; Mora, M.; Mantegazza, R.; Bernasconi, P.; Maggi, L. Up-regulation of Toll-like receptors 7 and 9 and its potential implications in the pathogenic mechanisms of LMNA-related myopathies. Nucleus 2018, 9, 398–409. [Google Scholar] [CrossRef]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 2267. [Google Scholar] [CrossRef]
- Shah, P.P.; Lv, W.; Rhoades, J.H.; Poleshko, A.; Abbey, D.; Caporizzo, M.A.; Linares-Saldana, R.; Heffler, J.G.; Sayed, N.; Thomas, D.; et al. Pathogenic LMNA variants disrupt cardiac lamina-chromatin interactions and de-repress alternative fate genes. Cell Stem Cell 2021. [Google Scholar] [CrossRef]
- Muchir, A.; Reilly, S.A.; Wu, W.; Iwata, S.; Homma, S.; Bonne, G.; Worman, H.J. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc. Res. 2012, 93, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Chatzifrangkeskou, M.; Yadin, D.; Marais, T.; Chardonnet, S.; Cohen-Tannoudji, M.; Mougenot, N.; Schmitt, A.; Crasto, S.; Di Pasquale, E.; Macquart, C.; et al. Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2018, 27, 3060–3078. [Google Scholar] [CrossRef]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Investig. 2004, 113, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.B.; Davis, J.; Weisleder, N.; Kostavassili, I.; McCulloch, A.D.; Ralston, E.; Capetanaki, Y.; Lieber, R.L. Structural and functional roles of desmin in mouse skeletal muscle during passive deformation. Biophys. J. 2004, 86, 2993–3008. [Google Scholar] [CrossRef]
- Li, H.; Choudhary, S.K.; Milner, D.J.; Munir, M.I.; Kuisk, I.R.; Capetanaki, Y. Inhibition of desmin expression blocks myoblast fusion and interferes with the myogenic regulators MyoD and myogenin. J. Cell Biol. 1994, 124, 827–841. [Google Scholar] [CrossRef]
- Weitzer, G.; Milner, D.J.; Kim, J.U.; Bradley, A.; Capetanaki, Y. Cytoskeletal control of myogenesis: A desmin null mutation blocks the myogenic pathway during embryonic stem cell differentiation. Dev. Biol. 1995, 172, 422–439. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hollrigl, A.; Puz, S.; Al-Dubai, H.; Kim, J.U.; Capetanaki, Y.; Weitzer, G. Amino-terminally truncated desmin rescues fusion of des(-/-) myoblasts but negatively affects cardiomyogenesis and smooth muscle development. FEBS Lett. 2002, 523, 229–233. [Google Scholar] [CrossRef]
- Mattioli, E.; Columbaro, M.; Jafferali, M.H.; Schena, E.; Hallberg, E.; Lattanzi, G. Samp1 Mislocalization in Emery-Dreifuss Muscular Dystrophy. Cells 2018, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Bernasconi, P.; Cavalcante, P.; Cappelletti, C.; D’Apice, M.R.; Sbraccia, P.; Novelli, G.; Prencipe, S.; Lemma, S.; Baldini, N.; et al. Modulation of TGFbeta 2 levels by lamin A in U2-OS osteoblast-like cells: Understanding the osteolytic process triggered by altered lamins. Oncotarget 2015, 6, 7424–7437. [Google Scholar] [CrossRef]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra102. [Google Scholar] [CrossRef]
- Liao, C.Y.; Anderson, S.S.; Chicoine, N.H.; Mayfield, J.R.; Garrett, B.J.; Kwok, C.S.; Academia, E.C.; Hsu, Y.M.; Miller, D.M.; Bair, A.M.; et al. Evidence that S6K1, but not 4E-BP1, mediates skeletal muscle pathology associated with loss of A-type lamins. Cell Discov. 2017, 3, 17039. [Google Scholar] [CrossRef] [PubMed]
- De La Banda, M.G.G.; Benito, D.N.-D.; Dabaj, I.; Ben Yaou, R.; Ortez, C.; Wahbi, K.; Maldonado, S.; Clarke, N.; Carlier, R.; Colomer, J.; et al. P.256Steroid treatment may change natural history in young children with LMNA mutations and dropped head syndrome. Neuromuscul. Disord. 2019, 29, S141. [Google Scholar] [CrossRef]
- Arimura, S.; Okada, T.; Tezuka, T.; Chiyo, T.; Kasahara, Y.; Yoshimura, T.; Motomura, M.; Yoshida, N.; Beeson, D.; Takeda, S.; et al. Neuromuscular disease. DOK7 gene therapy benefits mouse models of diseases characterized by defects in the neuromuscular junction. Science 2014, 345, 1505–1508. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | LMNA | DES |
|---|---|---|
| Inheritance | AD | AD |
| Age at onset | at birth to 4th decade | 3rd–4th decades |
| Distal muscle weakness | rare, frequent only in EDMD2 | frequent |
| Cranial nerve involvement | rare | relatively frequent |
| Contractures | frequent | very rare |
| Scoliosis/rigid spine | frequent | absent |
| Loss of independent ambulation | rare and late * | frequent and late |
| Respiratory insufficiency | rare, mainly in L-CMD | relatively frequent |
| Heart involvement | very frequent | very frequent |
| Cardiomyopathy | DCM, ACM, rarely LVNC | DCM, ACM, less frequently RCM, HCM, rarely LVNC |
| Need of ICD/PM | frequent, mainly ICD | frequent, mainly PM |
| Serum CK | normal or mildly elevated, high in L-CMD | normal or mildly elevated |
| Spontaneous activity at EMG | rare | frequent |
| Skeletal muscle histology | unspecific | MFM changes ** |
| CNS involvement | no | no |
| Type of predominant mutations | Missense | Missense |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maggi, L.; Mavroidis, M.; Psarras, S.; Capetanaki, Y.; Lattanzi, G. Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins. Int. J. Mol. Sci. 2021, 22, 4256. https://doi.org/10.3390/ijms22084256
Maggi L, Mavroidis M, Psarras S, Capetanaki Y, Lattanzi G. Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins. International Journal of Molecular Sciences. 2021; 22(8):4256. https://doi.org/10.3390/ijms22084256
Chicago/Turabian StyleMaggi, Lorenzo, Manolis Mavroidis, Stelios Psarras, Yassemi Capetanaki, and Giovanna Lattanzi. 2021. "Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins" International Journal of Molecular Sciences 22, no. 8: 4256. https://doi.org/10.3390/ijms22084256
APA StyleMaggi, L., Mavroidis, M., Psarras, S., Capetanaki, Y., & Lattanzi, G. (2021). Skeletal and Cardiac Muscle Disorders Caused by Mutations in Genes Encoding Intermediate Filament Proteins. International Journal of Molecular Sciences, 22(8), 4256. https://doi.org/10.3390/ijms22084256