
Transcriptome Profiling of Cucumber (Cucumis sativus L.) Early Response to Pseudomonas syringae pv. lachrymans


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
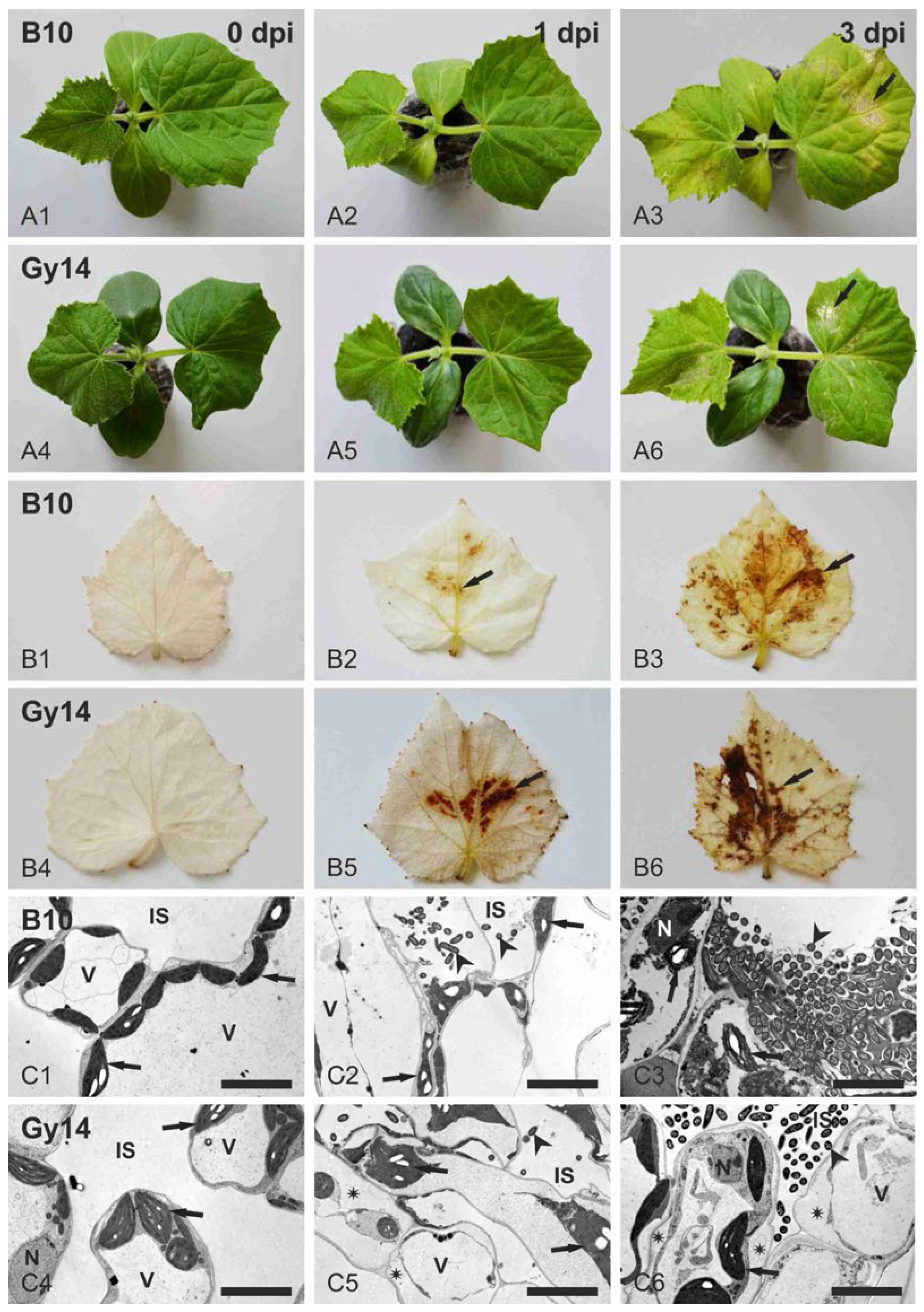
2.1. Cucumber Response to P. syringae pv. lachrymans
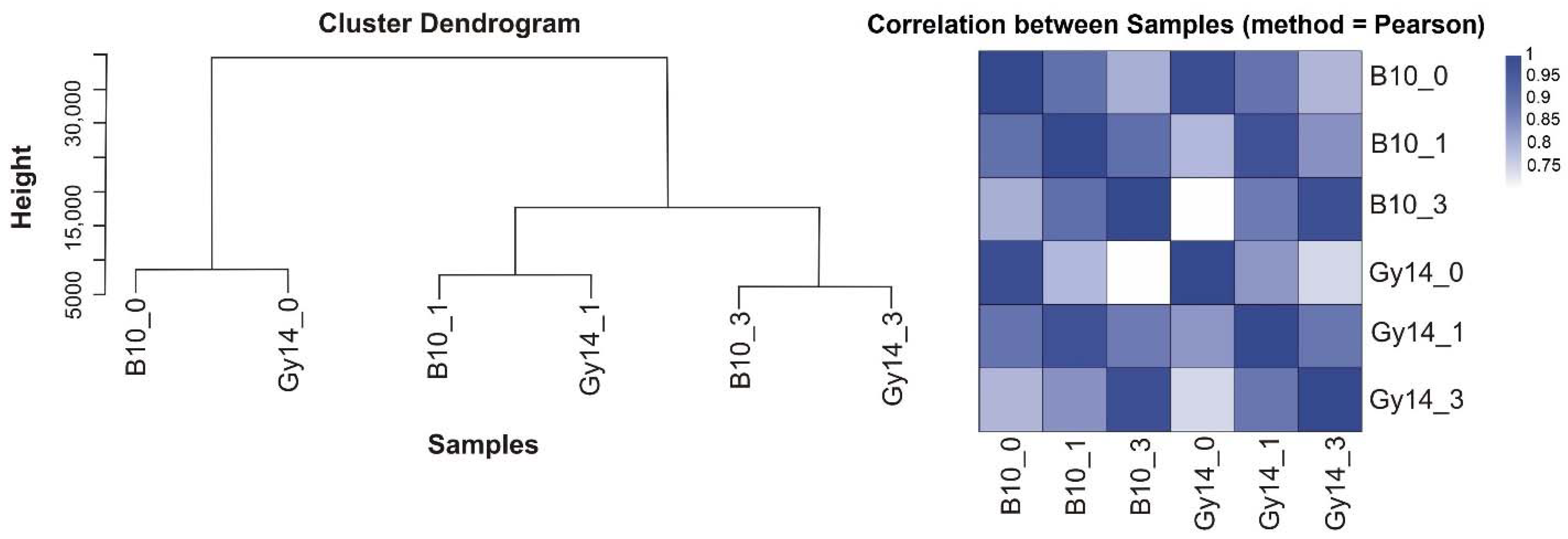
2.2. Transcriptome Profiling
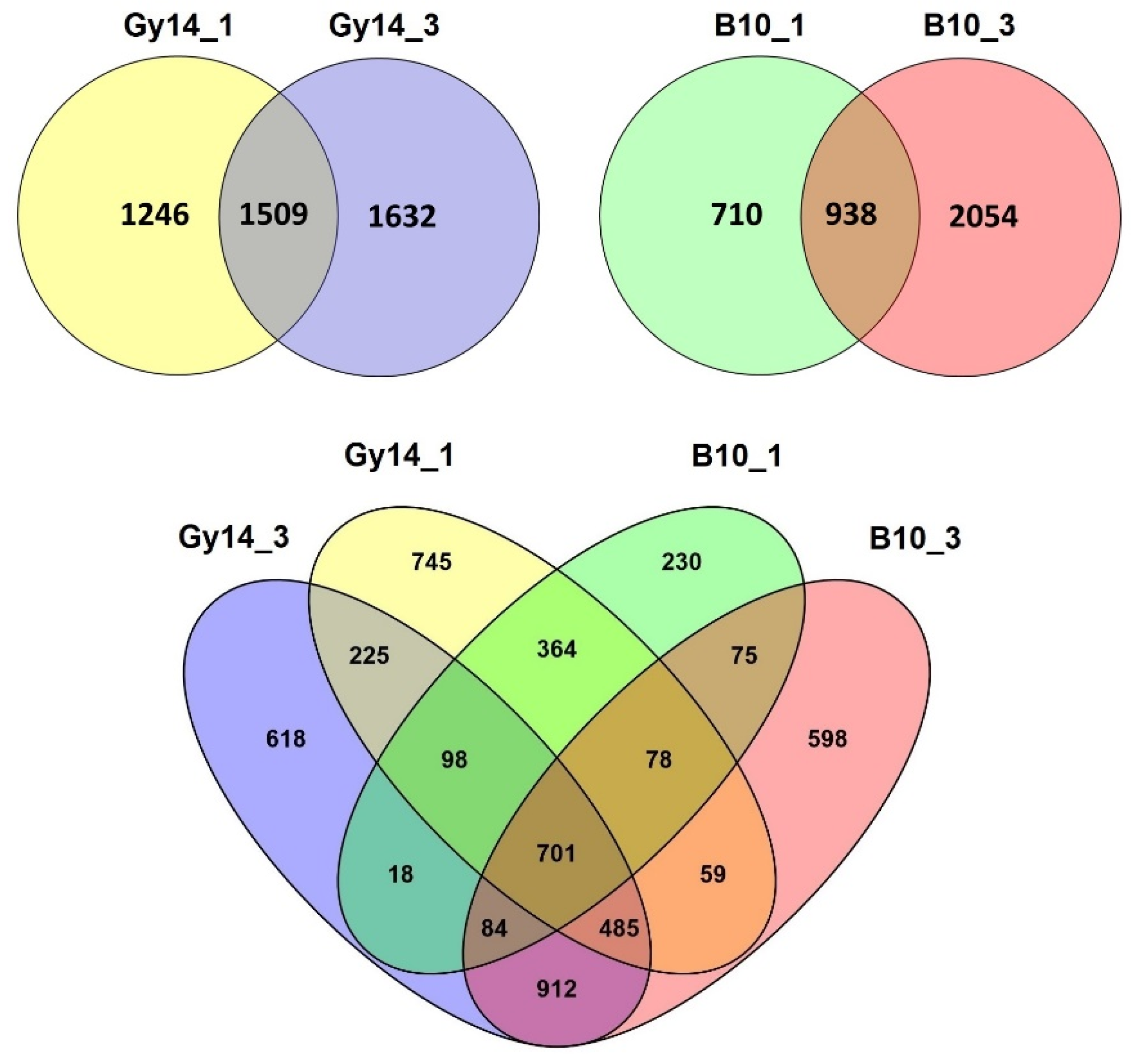
2.3. Differentially Expressed Genes
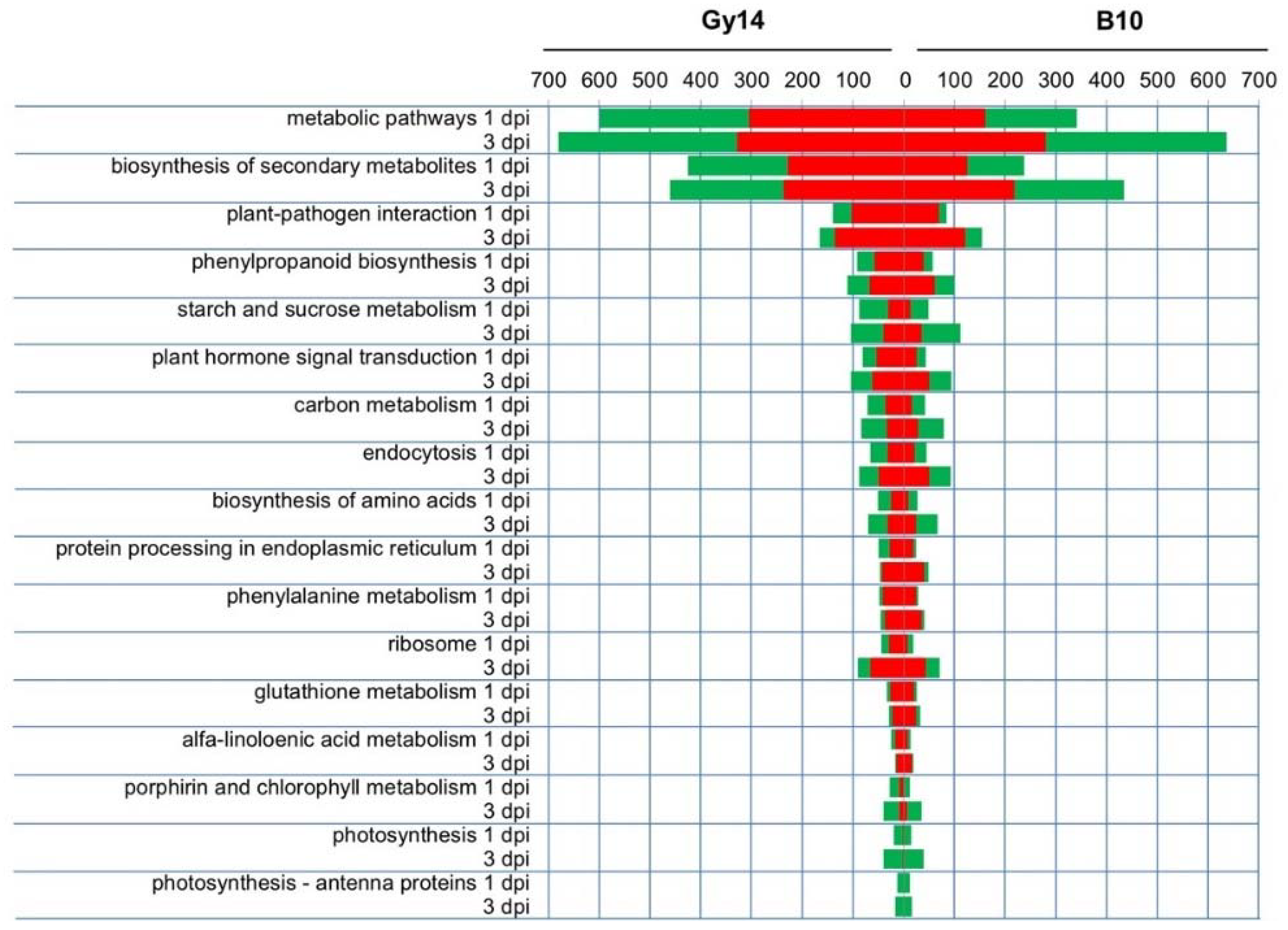
2.4. Functional Categories of DEGs
2.5. Genes Related to Defense Response
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Inoculation
4.2. Histochemical Staining
4.3. Microscopic Examination of Leaf Tissue
4.4. RNA Isolation, cDNA Library Construction and RNA-seq
4.5. Identification of Differentially Expressed Genes (DEGs)
4.6. Functional Classification of DEGs
4.7. RT-qPCR Analysis
4.8. Validation of RNA-seq Results
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAOSTAT Database. Available online: http://www.fao.org/faostat/en/#home (accessed on 17 January 2021).
- Ainsworth, G.C. Introduction to the History of Plant Pathology; Cambridge University Press: Cambridge, UK, 1981; p. 68. [Google Scholar]
- Janse, J.D. Phytobacteriology: Principles and Practice; CABI Publishing: Wallingford, UK, 2005; pp. 252–289. [Google Scholar]
- Bradbury, J.F. Guide to Plant Pathogenic Bacteria; CAB International Mycological Institute: Kew, Surrey, UK, 1986. [Google Scholar]
- Bhat, N.A.; Bhat, K.A.; Zargar, M.Y.; Teli, M.A.; Nazir, M.; Zargar, S.M. Current status of angular leaf spot (Pseudomonas syringae pv. lachrymans) of cucumber: A review. Int. J. Curr. Res. 2010, 8, 1–11. [Google Scholar]
- Dessert, J.M.; Baker, L.R.; Fobes, J.F. Inheritance of reaction to Pseudomonas lachrymans in pickling cucumber. Euphytica 1982, 31, 847–855. [Google Scholar] [CrossRef]
- Słomnicka, R.; Olczak-Woltman, H.; Bartoszewski, G.; Niemirowicz-Szczytt, K. Genetic and pathogenic diversity of Pseudomonas syringae strains isolated from cucurbits. Eur. J. Plant Pathol. 2015, 141, 1–14. [Google Scholar] [CrossRef]
- Keinath, A.P.; Wintermantel, W.M.; Zitter, T.A. Compendium of Cucurbit Diseases and Pests; American Phytopathological Society: St. Paul, MN, USA, 2017. [Google Scholar]
- Leben, C. Survival of Pseudomonas syringae pv. lachrymans with cucumber seed. Can. J. Plant Pathol. 1981, 3, 247–249. [Google Scholar] [CrossRef]
- Shila, S.J.; Islam, M.R.; Ahmed, N.N.; Dastogeer, K.M.G.; Meah, M.B. Detection of Pseudomonas syringae pv. lachymans associated with the seeds of cucurbits. J. Agric. Res. 2013, 1, 1–8. [Google Scholar]
- Chand, J.N.; Walker, J.C. Inheritance of resistance to angular leaf spot of cucumber. Phytopathology 1964, 54, 51–53. [Google Scholar]
- Olczak-Woltman, H.; Bartoszewski, G.; Madry, W.; Niemirowicz-Szczytt, K. Inheritance of resistance to angular leaf spot (Pseudomonas syringae pv. lachrymans) in cucumber and identification of molecular markers linked to resistance. Plant Pathol. 2009, 58, 145–151. [Google Scholar] [CrossRef]
- Słomnicka, R.; Olczak-Woltman, H.; Korzeniewska, A.; Gozdowski, D.; Niemirowicz-Szczytt, K.; Bartoszewski, G. Genetic mapping of psl locus and quantitative trait loci for angular leaf spot resistance in cucumber (Cucumis sativus L.). Mol. Breed. 2018, 38, 111. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, J.; Wu, Z.; VandenLangenberg, K.; Wehner, T.C.; Wen, C.L.; Zheng, X.Y.; Owens, K.; Thornton, A.; Bang, H.H.; et al. STAYGREEN, STAY HEALTHY: A loss-of-susceptibility mutation in the STAYGREEN gene provides durable, broad-spectrum disease resistances for over 50 years of US cucumber production. New Phytol. 2019, 221, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wei, Y.; Njogu, M.K.; Wang, X.; Lou, Q.; Li, J.; Chen, J. Genetic mapping of angular leaf spot resistance to Pseudomonas syringae pv. lachrymans in a Cucumis hystrix introgression line of cucumber. Euphytica 2019, 215, 176. [Google Scholar] [CrossRef]
- Kong, W.; Chen, N.; Liu, T.; Zhu, J.; Wang, J.; He, X.; Jin, Y. Large-scale transcriptome analysis of cucumber and Botrytis cinerea during infection. PLoS ONE 2015, 10, e0142221. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Z.; Gu, Q.; Li, H.; Han, W.; Shi, Y. Transcriptome analysis of Cucumis sativus infected by Cucurbit chlorotic yellows virus. Virol. J. 2017, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Šubr, Z.; Predajna, L.; Šoltys, K.; Bokor, B.; Budiš, J.; Glasa, M. Comparative transcriptome analysis of two cucumber cultivars with different sensitivity to Cucumber Mosaic Virus infection. Pathogens 2020, 9, 145. [Google Scholar] [CrossRef]
- Savory, E.A.; Adhikari, B.N.; Hamilton, J.P.; Vaillancourt, B.; Buell, C.R.; Day, B. mRNA-Seq analysis of the Pseudoperonospora cubensis transcriptome during cucumber (Cucumis sativus L.) infection. PLoS ONE 2012, 7, 35796. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, A.; Day, B. Transcriptome and small RNAome dynamics during a resistant and susceptible interaction between cucumber and downy mildew. Plant Genome 2016, 9, 1–19. [Google Scholar] [CrossRef]
- Xu, X.; Wang, R.; Chao, J.; Lin, Y.; Jin, Q.; He, X.; Luo, S.; Wu, T. The expression patterns of Cucumis sativus WRKY (CsWRKY) family under the condition of inoculation with Phytophthora melonis in disease resistant and susceptible cucumber cultivars. Can. J. Plant Sci. 2015, 95, 1121–1131. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, D.; Cui, N.; Yu, Y.; Yu, G.; Fan, H. Transcriptome and miRNA analyses of the response to Corynespora cassiicola in cucumber. Sci. Rep. 2018, 8, 7798. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, Y.; Xian, Q.; Chen, X.; Xu, J. Transcriptome analysis reveals ethylene-mediated defense responses to Fusarium oxysporum f. sp. cucumerinum infection in Cucumis sativus L. BMC Plant Biol. 2020, 20, 334. [Google Scholar] [CrossRef]
- Li, Y.; Tian, S.; Yang, X.; Wang, X.; Guo, Y.; Ni, H. Transcriptomic analysis reveals distinct resistant response by physcion and chrysophanol against cucumber powdery mildew. PeerJ 2016, 4, e1991. [Google Scholar] [CrossRef]
- Sa, R.; Liu, D.; Chen, L.; Liu, L.; Zhang, Y.; Zhang, X.; Xu, C. Transcriptome analysis of mechanisms and candidate genes associated with cucumber response to cucumber alternaria leaf spot infection. Physiol. Mol. Plant Pathol. 2020, 111, 101490. [Google Scholar] [CrossRef]
- Liang, D.; Liu, M.; Hu, Q.; Qi, X.; Xu, Q.; Zhou, F.; Chen, X. Identification of differentially expressed genes related to aphid resistance in cucumber (Cucumis sativus L.). Sci. Rep. 2015, 5, 9645. [Google Scholar] [CrossRef]
- He, J.; Bouwmeester, H.J.; Dicke, M.; Kappers, I. Transcriptional and metabolite analysis reveal a shift in direct and indirect defenses in response to spider-mite infestation in cucumber (Cucumis sativus). Plant Mol. Biol. 2020, 103, 489–505. [Google Scholar] [CrossRef]
- Howard, B.E.; Hu, Q.; Babaoglu, A.C.; Chandra, M.; Borghi, M.; Tan, X.; He, L.; Winter-Sederoff, H.; Gassmann, W.; Veronese, P. High-throughput RNA sequencing of Pseudomonas-infected Arabidopsis reveals hidden transcriptome complexity and novel splice variants. PLoS ONE 2013, 8, e74183. [Google Scholar] [CrossRef] [PubMed]
- Nemchinov, L.G.; Shao, J.; Lee, M.N.; Postnikova, O.A.; Samac, D.A. Resistant and susceptible responses in alfalfa (Medicago sativa) to bacterial stem blight caused by Pseudomonas syringae pv. syringae. PLoS ONE 2017, 12, e0189781. [Google Scholar] [CrossRef]
- Song, Y.; Sun, L.; Lin, M.; Chen, J.; Qi, X.; Hu, C.; Fang, J. Comparative transcriptome analysis of resistant and susceptible kiwifruits in response to Pseudomonas syringae pv. actinidiae during early infection. PLoS ONE 2019, 14, e0211913. [Google Scholar] [CrossRef]
- Pérez-Rodriguez, P.; Riaño-Pachón, D.M.; Corrêa, L.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, D822–D827. [Google Scholar] [CrossRef] [PubMed]
- Harighi, B. Angular leaf spot of cucumber caused by Pseudomonas syringae pv. lachrymans in Kurdistan. Plant Dis. 2007, 91, 769. [Google Scholar] [CrossRef]
- Meng, X.L.; Xie, X.W.; Shi, Y.X.; Chai, A.L.; Ma, Z.H.; Li, B.J. Evaluation of a loop-mediated isothermal amplification assay based on hrpZ gene for rapid detection and identification of Pseudomonas syringae pv. lachrymans in cucumber leaves. J. Appl. Microbiol. 2016, 122, 441–449. [Google Scholar] [CrossRef]
- Olczak-Woltman, H.; Marcinkowska, J.; Niemirowicz-Szczytt, K. The genetic basis of resistance to downy mildew in Cucumis spp.—latest developments and prospects. J. Appl. Genet. 2011, 52, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Newberry, E.A.; Jardini, T.M.; Rubio, I.; Roberts, P.D.; Babu, B.; Koike, S.T.; Bouzar, H.; Goss, E.M.; Jones, J.B.; Bull, C.T.; et al. Angular leaf spot of cucurbits is associated with genetically diverse Pseudomonas syringae strains. Plant Dis. 2016, 100, 1397–1404. [Google Scholar] [CrossRef]
- Newberry, E.A.; Babu, B.; Roberts, P.D.; Dufault, N.S.; Goss, E.M.; Jones, J.B.; Paret, M.L. Molecular epidemiology of Pseudomonas syringae pv. syringae causing bacterial leaf spot of watermelon and squash in Florida. Plant Dis. 2018, 102, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Gardan, L.; Shafik, H.; Belouin, S.; Broch, R.; Grimont, F.; Grimont, P.A.D. DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas cannabina sp. nov. (ex Sutic and Dowson, 1959). Int. J. Syst. Bacteriol. 1999, 49, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Olczak-Woltman, H.; Schollenberger, M.; Mądry, W.; Niemirowicz-Szczytt, K. Evaluation of cucumber (Cucumis sativus L.) cultivars grown in Eastern Europe and progress in breeding for resistance to angular leaf spot (Pseudomonas syringae pv. lachrymans). Eur. J. Plant Pathol. 2008, 122, 385–393. [Google Scholar] [CrossRef]
- Xu, X.; Liu, X.; Yan, Y.; Wang, W.; Gebretsadik, K.; Qi, X.; Xu, Q.; Chen, X. Comparative proteomic analysis of cucumber powdery mildew resistance between a single-segment substitution line and its recurrent parent. Horticult. Res. 2019, 6, 115. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Bhuyan, M.H.M.B.; Zulfiqar, F.; Raza, A.; Mohsin, S.M.; Mahmud, J.A.; Fujita, M.; Fotopoulos, V. Reactive oxygen species and antioxidant defense in plants under abiotic stress: Revisiting the crucial role of a universal defense regulator. Antioxidants 2020, 9, 681. [Google Scholar] [CrossRef]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Chisholm, S.T.; Coaker, G.; Day, B.; Staskawicz, B.J. Host-microbe interactions: Shaping the evolution of the plant immune response. Cell 2006, 124, 803–814. [Google Scholar] [CrossRef]
- Monaghan, J.; Zipfel, C. Plant pattern recognition receptor complexes at the plasma membrane. Curr. Opin. Plant Biol. 2012, 15, 1–9. [Google Scholar]
- Yang, Y.; Wang, X.; Chen, P.; Zhou, K.; Xue, W.; Abid, K.; Chen, S. Redox status, JA and ET signaling pathway regulating responses to Botrytis cinerea infection between the resistant cucumber genotype and its susceptible mutant. Front. Plant Sci. 2020, 11, 559070. [Google Scholar] [CrossRef]
- Shang, Q.M.; Li, L.; Dong, C.J. Multiple tandem duplication of the phenylalanine ammonia-lyase genes in Cucumis sativus L. Planta 2012, 236, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.J.; Cao, N.; Shang, Q.M.; Zhang, Z.G. Phenylalanine ammonia-lyase gene families in cucurbit species: Structure, evolution, and expression. J. Integr. Agr. 2016, 15, 1239–1255. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, Y.; Luo, X.; Zhou, S. Comparative proteomic analysis provides insights into the complex responses to Pseudoperonospora cubensis infection of cucumber (Cucumis sativus L.). Sci. Rep. 2019, 9, 433. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, L.; Xu, S.; Jiang, L.; Liu, S. Identification and transcriptional analysis of dehydrin gene family in cucumber (Cucumis sativus). Acta Physiol. Plant. 2018, 40, 144. [Google Scholar] [CrossRef]
- Rorat, T. Plant dehydrins. Tissue location, structure and function. Cell. Mol. Biol. Lett. 2006, 11, 536–556. [Google Scholar] [CrossRef]
- Olczak-Woltman, H.; Masny, A.; Bartoszewski, G.; Płucienniczak, A.; Niemirowicz-Szczytt, K. Genetic diversity of Pseudomonas syringae pv. lachrymans strains isolated from cucumber leaves collected in Poland. Plant Pathol. 2007, 56, 373–382. [Google Scholar] [CrossRef]
- Słomnicka, R.; Olczak-Woltman, H.; Oskiera, M.; Schollenberger, M.; Niemirowicz-Szczytt, K.; Bartoszewski, G. Genome analysis of Pseudomonas syringae pv. lachrymans strain 814/98 indicates diversity within the pathovar. Eur. J. Plant Pathol. 2018, 151, 663–676. [Google Scholar] [CrossRef]
- Thordal-Christensen, H.; Zhang, Z.; Wei, Y.; Collinge, D.B. Subcellular localization of H2O2 in plants: H2O2 accumulation in papillae and hypersensitive response during the barley-powdery mildew interaction. Plant J. 1997, 11, 1187–1194. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Piszczek, E.; Dudkiewicz, M.; Sobczak, M. Molecular cloning and phylogenetic analysis of cereal type II metacaspase cDNA from wheat (Triticum aestivum). Biol. Plant. 2011, 55, 614–624. [Google Scholar] [CrossRef]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.; Antonescu, C.; Chang, T.-S.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.T.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef]
- Oliveros, J.C. VENNY. An interactive tool for comparing lists with Venn Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 15 March 2021).
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Osipowski, P.; Pawełkowicz, M.; Wojcieszek, M.; Skarzyńska, A.; Przybecki, Z.; Pląder, W. A high-quality cucumber genome assembly enhances computational comparative genomics. Mol. Genet. Genomics 2020, 295, 177–193. [Google Scholar] [CrossRef]
- Mróz, T.L.; Havey, M.J.; Bartoszewski, G. Cucumber possesses a single terminal alternative oxidase gene that is upregulated by cold stress and in the mosaic (MSC) mitochondrial mutants. Plant Mol. Biol. Rep. 2015, 33, 1893–1906. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 0034.1. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopád, A.; Prgomet, C.; Neuvins, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Słomnicka, R.; Olczak-Woltman, H.; Sobczak, M.; Bartoszewski, G. Transcriptome Profiling of Cucumber (Cucumis sativus L.) Early Response to Pseudomonas syringae pv. lachrymans. Int. J. Mol. Sci. 2021, 22, 4192. https://doi.org/10.3390/ijms22084192
Słomnicka R, Olczak-Woltman H, Sobczak M, Bartoszewski G. Transcriptome Profiling of Cucumber (Cucumis sativus L.) Early Response to Pseudomonas syringae pv. lachrymans. International Journal of Molecular Sciences. 2021; 22(8):4192. https://doi.org/10.3390/ijms22084192
Chicago/Turabian StyleSłomnicka, Renata, Helena Olczak-Woltman, Mirosław Sobczak, and Grzegorz Bartoszewski. 2021. "Transcriptome Profiling of Cucumber (Cucumis sativus L.) Early Response to Pseudomonas syringae pv. lachrymans" International Journal of Molecular Sciences 22, no. 8: 4192. https://doi.org/10.3390/ijms22084192
APA StyleSłomnicka, R., Olczak-Woltman, H., Sobczak, M., & Bartoszewski, G. (2021). Transcriptome Profiling of Cucumber (Cucumis sativus L.) Early Response to Pseudomonas syringae pv. lachrymans. International Journal of Molecular Sciences, 22(8), 4192. https://doi.org/10.3390/ijms22084192