Abstract
Inherited cardiomyopathies are frequent causes of sudden cardiac death (SCD), especially in young patients. Despite at the autopsy they usually have distinctive microscopic and/or macroscopic diagnostic features, their phenotypes may be mild or ambiguous, possibly leading to misdiagnoses or missed diagnoses. In this review, the main differential diagnoses of hypertrophic cardiomyopathy (e.g., athlete’s heart, idiopathic left ventricular hypertrophy), arrhythmogenic cardiomyopathy (e.g., adipositas cordis, myocarditis) and dilated cardiomyopathy (e.g., acquired forms of dilated cardiomyopathy, left ventricular noncompaction) are discussed. Moreover, the diagnostic issues in SCD victims affected by phenotype-negative hypertrophic cardiomyopathy and the relationship between myocardial bridging and hypertrophic cardiomyopathy are analyzed. Finally, the applications/limits of virtopsy and post-mortem genetic testing in this field are discussed, with particular attention to the issues related to the assessment of the significance of the genetic variants.
1. Introduction
Sudden unexplained death (SUD) is a fatal event that encompasses several heart disorders which lead to abrupt and unpredicted death. Normally, the victim has no known history of heart disease. In adult population (16–64 years) the SUD rate is 11/100,000 per year, while, in the young population (<16 years of age), it is 7.5/100,000 [1]. According to the evidence of the last 15 years, most of the SUD cases (at least the 5–20% of them) are of cardiac origin [2]. It is well known that Sudden Cardiac Death (SCD) is one of the most common causes of death in developed countries, with a yearly incidence of 30–200/100,000. In young population, SCD is a rare event, having an incidence of approximately 2–5/100,000 patients per year [3]. Coronary artery disease and acquired cardiomyopathies are the most frequent causes of SCD in the adults, while in those younger than 35 years the main cause of SCD is represented by non-ischemic diseases [4,5]. Cardiomyopathies are the main cause of SCD in those younger than 35 years, while up to 40% of young cases of SCD are caused by pathogenic alterations in the genes that code for ion channels or proteins associated with their proper functioning [6,7]. Currently, nearly the 20% of total deaths in young population remain without a conclusive explanation after a complete autopsy [8,9,10,11]. Inherited arrhythmogenic syndromes—channelopathies—account for most of the autopsy-negative cases (if acute intoxications are excluded). On the other hand, cardiomyopathies are generally thought to have distinctive macroscopic and microscopic features. However, in the forensic field cardiomyopathies are often extremely challenging, mainly because of two factors: (i) the phenotypes of cardiomyopathies gradually develop, and some of cases of SCD occur in young victims, with only mild microscopic and/or macroscopic signs of disease; (ii) when the diagnosis has not been made before the death, at the autopsy it is often difficult to distinguish a cardiomyopathy from another pathological (or from a physiological) condition.
In this paper, we review the main issues that the pathologist can encounter at the macroscopic and microscopic examination of cardiomyopathies cases and we discuss the contributions that can be given by virtopsy and post-mortem genetic testing, focusing on the issues of medico-legal interest.
2. Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) has a prevalence of 1/500 [12]. It is macroscopically characterized by abnormal left ventricular (LV) wall thickness and/or heart weight. In HCM the LV hypertrophy is typically asymmetrical, but it can also be symmetrical and can involve only delimited regions, like the apex. The LV wall thickness is considered a strong predictor of sudden death [13]. However, the predisposition to SCD is multifactorial, since it also depends on anamnestic factors (familiarity for SCD and history of syncope and/or non-sustained ventricular tachycardia) [14]. At the autopsy, as said, an abnormal heart weight can be suggestive of HCM, but there is little evidence on what the normal range of this parameter is [15]. However, it is usually assumed that a weight > 500 g is of pathological significance [16]. The diagnostic microscopic features are hypertrophy and disarray (i.e., the loss of the normal parallel alignment of the myocytes), associated with interstitial fibrosis (Figure 1). LV hypertrophy and/or myocardial fibrosis without myocardial disarray are features of uncertain significance [17].
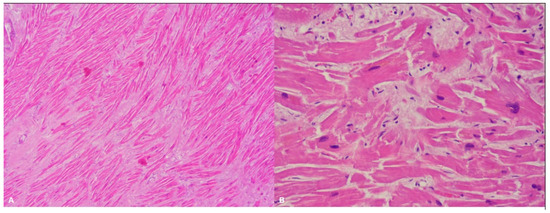
Figure 1.
Left ventricle: diffuse disarray and mild fibrosis (A). Hematoxylin and eosin stain, 4× magnification), associated with focal and mild myocardial hypertrophy (B). Hematoxylin and eosin stain, 40× magnification) in a 48-year-old man who suddenly died while playing sport.
2.1. Differential Diagnosis
At the autopsy, the hypertrophy of the ventricular walls is the most misleading feature: ventricular hypertrophy is an extremely common finding, but it must be carefully evaluated to infer on its origin. In detail, the hypertrophy can be concentric or eccentric. Concentric hypertrophy may be associated with both congenital (HCM) (Figure 2) and acquired (hypertension, aortic stenosis, chronic abuse of anabolic steroids) conditions. Eccentric hypertrophy is rarely found at the autopsy, since it does not reduce the volumes of ventricular chambers, and thus the diagnosis can be easily missed. Eccentric hypertrophy is usually caused by valvular anomalies, like aortic or mitral insufficiency.
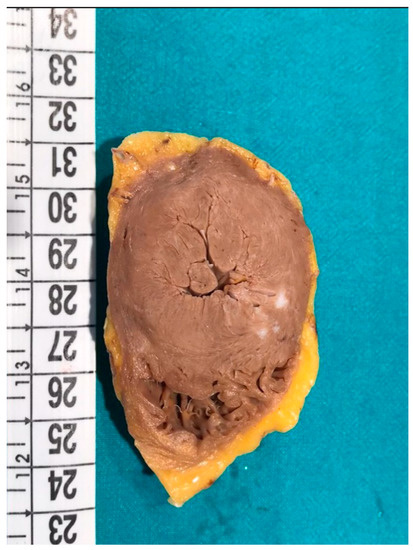
Figure 2.
Macroscopic transversal section of the heart of a 48-year-old man affected by hypertrophic cardiomyopathy (HCM) (the same case of Figure 1). Concentric hypertrophy is clearly visible.
Furthermore, several adaptive or idiopathic conditions can be misdiagnosed as HCM. For instance, in obese individuals a state of chronically high cardiac output can cause the so-called “obesity associated heart disease”, that can mimic HCM [18]. Moreover, competitive athletes (in particular endurance athletes) frequently show an adaptive ventricular remodelling known as athletic heart syndrome (“athlete’s heart”). Because of the small increases in the thickness of the ventricular walls, athlete’s heart can be easily misdiagnosed as a mild HCM (a LV wall of 13–15 mm is typically considered a diagnostic grey zone) [19]. However, differently from HCM, athlete’s heart is usually associated with left atrium enlargement and, according to current evidence, does not increase the risk of supraventricular tachyarrhythmias [20,21]. Finally, in the young population (in both athletes and non-athletes) an idiopathic left ventricular hypertrophy (ILVH) can be reported [22]. The ILVH is a condition of uncertain significance, despite it has been associated with an increased risk of sudden death, especially in presence of myocardial fibrosis [17].
2.2. Diagnostic Issues in Young SCD Victims
In HCM the impaired mechanical function of myocytes progressively causes compensatory disarray, hypertrophy, and fibrosis. Therefore, in children this cardiomyopathy often has no or extremely mild phenotype, with most of the patients developing clear hypertrophy only during or after the adolescence [22,23]. In case of young victims, since hypertrophy is rarely found, it is very important to look for the myocyte disarray. This finding can be physiological in the parts of the anterior and posterior walls of the right ventricle that are near the septum, and thus it is very important to collect several samples in different areas of the heart [23].
HCM is characterized by variable expressivity, and thus a carrier of a pathogenic variant could never develop macroscopic or microscopic signs of disease. However, genotype-positive patients without hypertrophy of the LV walls still have altered cardiac dimensions/function and a higher burden of early phenotypes [22].
2.3. HCM and Myocardial Bridging
Myocardial bridging (MB) (Figure 3) is a common congenital anomaly, that can be found in up to the 85% forensic autopsies. It usually affects the middle trait of the left anterior descending coronary artery [24,25].
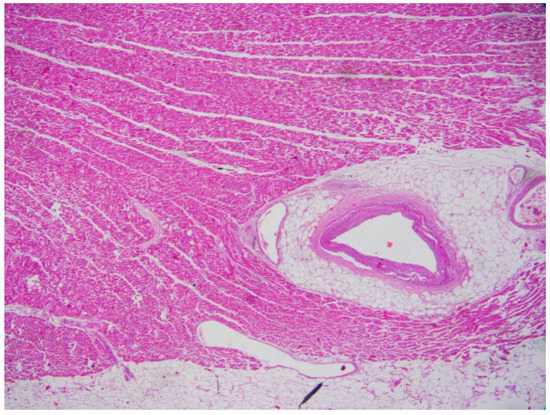
Figure 3.
Myocardial bridging in the heart of a 32-year-old man who suddenly died (Hematoxylin and eosin stain, 20× magnification).
Brodsky et al. found that variables like age, sex, body mass index, heart weight, left ventricle wall thickness, and circumference of pulmonary/aortic valves do not have a statistically significant relationship with MB [26].
MB is more frequently reported in forensic rather than in clinical scientific literature: according to current evidence, it is found in up to the 16% of coronary angiographies and in up to the 85% of forensic autopsies [26].
It is a condition of uncertain clinical significance. However, despite very common, some authors have reported associations between MB and myocardial ischemia, left ventricle dysfunction, arrhythmias, and sudden cardiac death [6,27,28]. In particular, Schwarz et al. found that MB can cause a decrease in coronary flow reserve, which may be responsible for myocardial ischemia [29]. Moreover, Mohiddin et al., found that thallium perfusion is reduced by the LV hypertrophy of the area where the MB is, but they did not find any significant association between this vascular anomaly and ventricular arrhythmias or sudden death [30]. Despite some cases of SCD in young patients with MB have been reported, the first clinical manifestation generally occurs in the adult age, and it is thought to be related to a loss of elasticity of the coronary arteries (and thus to a reduction of diastolic lumen) [6,23,31].
From a forensic point of view, one of the most interesting issues regarding MB is its possible association with some features of HCM or with HCM itself [6,32,33,34,35]. Several authors reported that the depth of the intramyocardial course (≥2 mm) is significatively associated with a greater amount of myocardial fibrosis [28,32,36]. The fibrotic tissue is due to an ischemic process in the territory of the MB, which consequently could predispose the individual to an increased risk of sudden death [36,37]. As noted by Yetman et al., chronic ischemia can cause a myocardial damage characterized by disarray and diffuse fibrosis, which can consequently predispose the subject to develop ventricular arrhythmias and sudden death [32]. In general, the prevalence of MB among patients with HCM has been reported to be of 21–41% [33]. This association is considered important by many authors because the pathological substrate of HCM can increase the risk of MB clinical manifestation. For example, Sharzhee et al. observed that the structure of MB is essentially the same in HCM and non-HCM individuals, but the heart hypertrophy causes a greater compression of the bridging, which is responsible for the decrease in the flow rate and for a higher pressure drop coefficient [38].
2.4. Genetics
HCM normally has an autosomal dominant inheritance pattern, and it is usually caused by rare variants in genes encoding cardiac sarcomeric proteins [39,40,41]. Variants in two genes (MYH7 and MYBPC3) account for 60–70% of HCM patients with known pathogenic variants, while pathogenic variants of TNNI3, TNNT3, TPM1 and MYL3 are rarer [42,43,44]. Rare variants in the MYH7 gene are predominantly missense, so single nucleotide-base variants result in a non-synonymous single amino acid substitution [45,46]. The majority of rare alterations in the MYBPC3 gene are caused by the introduction of a premature stop codon (PTC), that results in a truncated protein transcript. This PTC may be caused by nonsense mutations and insertions/deletions that alter the reading frame (known as frameshifts) or splice-site variants [43,47].
HCM can also (rarely) be caused by mutations of non-sarcomeric genes, like CSRP3 (coding for a Z-disk protein), FHL-1 (coding for a sarcomere-associated protein) and PLN (coding for a regulator of sarcoplasmic reticulum calcium) [48,49].
The penetrance of the pathogenic variants is variable, being relatively high for some of them (in particular, MYH7 and MYBPC3) and low to moderate for many others [49]. Moreover, as said, HCM shows a significant variability in its phenotypic expression. Indeed, the severity of the phenotype (and thus the risk of SCD) is thought to be increased, for example, by an insertion/deletion variant in the angiotensin-1 converting enzyme gene (ACE) and by relevant changes in loading conditions (like systemic arterial hypertension) [48]. Moreover, in the carriers of pathogenic sarcomere protein mutations, hypertrophy and myocardial fibrosis tend to be more severe and the prognosis is generally poorer than in patients in which no variant is found [50]. In particular, the phenotype is more severe in those who carry multiple variants (about the 5% of the total carriers) [50].
Finally, it should be noted that mutations of sarcomeric genes like MYH7 or TNNT2 may have pleiotropic effects, causing different cardiomyopathies within the same families [48].
3. Arrhythmogenic Cardiomyopathy
Arrhythmogenic Cardiomyopathy (ACM) has a prevalence up to 1/2000 [51], being particularly frequent in some geographical areas like Veneto region (Italy). It is characterized by the fibrofatty replacement of the myocardium, that progresses from the epicardium towards endocardium and tends to be in the “triangle of dysplasia” (outflow tract of right ventricle, inferior wall beneath the posterior tricuspid leaflet and the ventricular apex) [52]. Epicardial adiposity is supposed to have arrhythmogenic effects through different patterns of action: structural barrier to electric impulse propagation, adipogenesis/fibrosis, increased oxidative stress, and formation of cytokines [53]. However, the clinical significance of this recurrent finding is still to be determined.
3.1. Differential Diagnosis
Fibrofatty replacement is not pathognomonic of ACM, since it can also be found, for example, in PRKAG2 cardiac syndrome, Uhl’s anomaly, and HCM [54,55]. However, the most important differential diagnosis is the normal (and sporadic) fatty infiltration of the ventricles (i.e., “adipositas cordis”), that is often found (up to the 85% of the cases), especially in the elderlies and in the obese patients (Figure 4) [56,57]. Corradi et al., observed that a normal heart contains an average amount of 205 g of ventricular myocardium and 54 g of ventricular fat [58]. In normal hearts, a certain amount of subepicardial fatty tissue is often found in the ventricular walls (especially in the antero-lateral and apical areas), but it is clearly separated from the inner myocardium [33,59]. Some authors, like Anumonwo et al., reported that the volume and the thickness of epicardial fat are possible markers of ACM, considering pathological a volume greater than 125 mL and a thickness greater than 5 mm [53]. However, normal fatty infiltration can be easily distinguished from ACM through the observation of myocytes atrophy, that gives the ventricular wall a translucent appearance at the autopsy. Another important differential diagnosis is myocarditis, since in ACM signs of recurrent, chronic myocarditis (inflammatory cells infiltrates with focal myocyte necrosis) can often be observed [33].
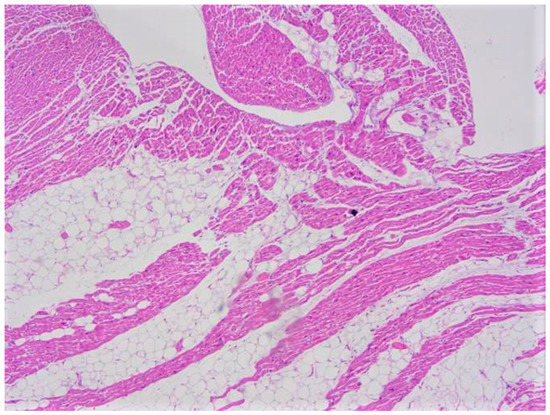
Figure 4.
Fatty infiltration with an epicardial-endocardial gradient clearly separated by the myocardium and without fibrosis (adipositas cordis). This case regards an obese 22-year-old woman who suddenly died at home (hematoxylin and eosin stain, 10× magnification).
3.2. Genetics
ACM can be caused by deleterious alterations located in genes encoding mainly desmosomal proteins but also proteins involved in electric signal transmission [60,61,62,63,64]. Currently, more than 1000 rare genetic variants have been identified in more than 15 genes (ANK2, CTNNA3, DES, DSC2, DSG2, DSP, FLNC, ILK, JUP, LMNA, PKP2, PLN, PNPLA2, SCN5A, TGFB3, TJP1, TMEM43, TP63, and TTN), but only about 400 rare genetic alterations have been classified as certainly pathogenic [65]. All the other rare variants have an ambiguous role and further data are needed to determine whether they are pathogenic for ACM or not [66]. Most of the pathogenic variants affect desmosomal genes, like PKP2 (41.6% of the pathogenic variants), DSP (21.2%), DSG2 (12.2%), DSC2 (9.7%), and JUP (3.6%) [65]. In up to the 16% of the cases, a single patient carries multiple mutations, that can affect the same gene (compound heterozygosity) or different genes (digenic heterozygosity) [65,67,68]. Furthermore, ACM can be caused by variants of extradesmosomal genes, like LMNA, DES and TTN [65].
When a carrier of a pathogenic variant is found, his relatives should be carefully evaluated [69]. ACM mainly follows an autosomal dominant pattern of inheritance, with incomplete and age-related penetrance as well as polymorphic phenotypic expression [70,71]. Autosomal recessive forms have also been reported, although in a smaller number of cases (Naxos disease, caused by a deletion in the JUP gene, and Carvajal syndrome, caused by mutations in the DSP gene) [72,73,74]. In addition, alterations in number of copies (Copy Number Variation, CNV) were also associated with ACM [75].
Despite these recent advances, only in about the 50% of ACM patients a pathogenic variant is found [76,77]. However, even when it is found, the patient could show no phenotype because of the variable expressivity and incomplete penetrance [78]. Therefore, clinical translation should be done carefully, after a comprehensive personalized interpretation of all the obtained data.
4. Dilated Cardiomyopathy
Dilated Cardiomyopathy (DCM) has a prevalence of 1/2500 [79]. Its mortality rate is up to 20% [80]. Death is usually due to heart failure or ventricular arrhythmias. SCD has an incidence of about 2–3%, can be the first manifestation of disease and is caused by electromechanical dissociation or arrhythmias [81]. LV dilatation and contractile impairment are the main risk factors for sudden death [82]. Greater left atrial volume also increases the risk of adverse outcomes [83]. In the paediatric population, the predictors of SD are the age at diagnosis, familiarity, and severe LV systolic disfunction [81]. At the forensic autopsies, DCM is macroscopically characterised by the dilatation of the cardiac chambers (greater in the ventricles than in the atria). These signs can be associated with other common findings, like intracavitary thrombi and, in case of right heart failure, hepatomegaly, ascites and peripheral oedema [79]. At the histopathologic examination, diffuse fibrosis with some areas of necrosis and atrophied and/or hypertrophied cardiomyocytes is usually found (Figure 5). Fibrosis plays an important role in this disease since it causes contractile impairment and ventricular re-entrant arrhythmias [84]. In DCM, two kinds of fibrosis can be found: interstitial and replacement fibrosis. Fibrosis results from the so-called “replacement”, which consists of myocyte cell death and scarring formation or directly from an expansion of interstitial collagen [85]. Replacement fibrosis is of great clinical significance because it is associated with sustained or inducible VT [86]. On the other hand, interstitial fibrosis, that is almost always found in DCM cases, is thought to cause focal tachycardias and to be involved in the maintenance of re-entry circuits [87].
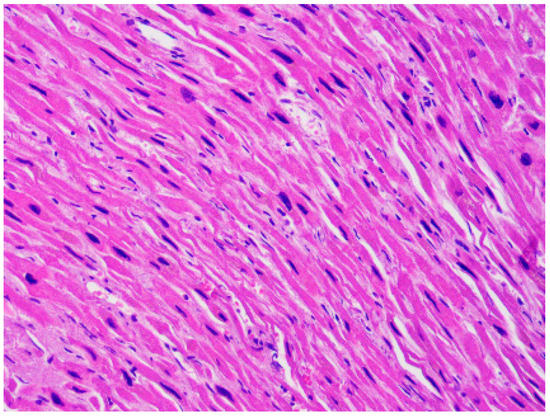
Figure 5.
Mild fibrosis and increased nuclear-cytoplasmic ratio in the cardiomyocytes of the heart of a 45-year-old woman affected by dilated cardiomyopathy (hematoxylin and eosin stain, 40× magnification).
4.1. Differential Diagnosis
DCM has many possible causes. It is usually distinguished in primary (congenital) and secondary (acquired). Secondary DCM can be caused by many factors, like toxic substances (e.g., cocaine or a chronic alcohol intake > 80 g/day), pathogens (virus, bacteria, fungi, spirochete, protozoans, rickettsia), endocrine or metabolic disfunctions (electrolyte disturbances, Cushing’s disease), inflammatory conditions, and autoimmune or neuromuscular diseases [81]. It is important to note that DCM can be also a long-term toxic manifestation: for example, it can occur even 10 years after chronic anthracycline exposure [81]. Therefore, when DCM is found at the autopsy, the main tool for differential diagnosis is a complete and accurate anamnesis. From a macroscopic point of view, at the autopsy it can be difficult to differentiate DCM from ischemic cardiomyopathy, hypertensive heart disease, athlete’s heart, and other cardiomyopathies [82]. In particular, the presence of significant coronary stenosis allows to distinguish ischemic cardiomyopathy from DCM. Instead, the differential diagnosis between DCM and left ventricular noncompaction is performed through the identification of LV trabeculations, deep intertrabecular recesses, and a thin layer of normal myocardium. Histopathological examination can be of great help to distinguish a DCM from a viral or immune-mediated myocarditis thanks to the identification of a lymphocytic infiltrate (Figure 6) and, in case of infective myocarditis, the post-mortem microbiological testing (PCR) [81].
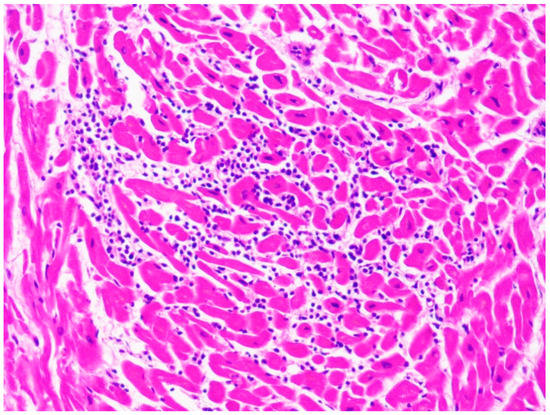
Figure 6.
Lymphocytic infiltrate in the case of a 10-year-old child affected by myocarditis (hematoxylin and eosin stain, 40× magnification).
4.2. Genetics
As already said, nearly the 60% of familial DCM cases show genetic alteration in one of over 60 genes associated with DCM [88]. Most of the familial DCM cases are due to pathogenic variants that follow an autosomal pattern of inheritance. These pathogenic variants have been identified in several genes encoding proteins with different functions, such as ion channels, transcription factors, and sarcomeric/desmosomal/nuclear proteins. Currently, few DCM cases following an autosomal recessive pattern of inheritance have been reported [89]. Recent studies reported pathogenic variants in the TTN gene as the main causes of familial DCM [90]. Despite nearly the 30–35% of families affected by DCM show alterations in this gene, most of the found variants are classified as of unknown significance [90]. The second most prevalent gene in familial DCM is LMNA, responsible for nearly 10–15% of cases [6]. Several other genes have been associated with this disease, being responsible for nearly the 5–10% of all the familial DCM cases. Finally, other genetic alterations such as Copy Number Variation (CNV) have been reported to (rarely) be causes of DCM.
5. Molecular Autopsy
Molecular autopsy has been recommended as part of the autopsy process, for example, by the Heart Rhythm Society and the European Heart Rhythm Association (HRS/EHRA) [91], the European Society of Cardiology [92], the Canadian Cardiovascular Society/Canadian Heart Rhythm Society [93], and the Swiss Society of Legal Medicine [94]. Recently, the Trans-Tasman Response Against Sudden Death in the Young (TRAGADY), together with the Royal College of Pathologists of Australasia and the National Heart Foundation of New Zealand, have proposed a guide to standardize the autopsies in young cases of SCD (http://www.rcpa.edu.au/Library/Publications/Joint-and-Third-Party-Guidelines) (accessed on 3 April 2021). Despite these recent advances, these guidelines still only recommend the analysis of the main genes associated with arrhythmogenic syndromes, mainly because of economic reasons. Thanks to the recent advances in the field of genetics, more than 100 genes (about 60 genes associated with cardiomyopathies and about 40 genes associated with channelopathies) are currently known (Figure 7) [95]. The pathogenic genetic variants have been discovered thanks to the Next Generation Sequencing (NGS), that allows massive analysis of genes in a short time and in a cost-effective way [96]. Recent studies reported that the yield of genetic testing in SCD cases with autopsy findings suggestive of a cardiomyopathy is comparable with the yield in alive patients affected by cardiomyopathies [96,97]. Despite this evidence, as said, performing molecular autopsy (when indicated) is still discretionary.
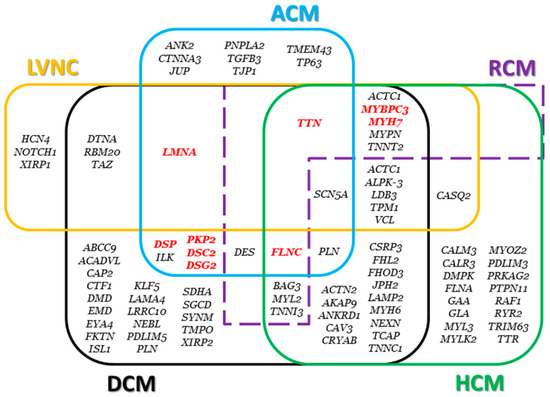
Figure 7.
Genes associated with cardiomyopathies. The main genes are highlighted in red. Genes associated with restrictive cardiomyopathy (RCM) and left ventricular noncompaction (LVNC) are also displayed.
6. Discussion
Inherited cardiomyopathies (Table 1) are relatively common and SCD is often the first manifestation of disease. When a correct diagnosis has not been made before the death, identifying a cardiomyopathy at the autopsy is extremely important for forensic and public health issues. From a public health point of view, diagnosing inherited cardiomyopathies is essential to identify other carriers of the pathogenic variants within the family and promptly adopt preventive measures. Indeed, it is very common that in the family of the victim a post-mortem diagnosis of cardiomyopathy allows new diagnoses—that would not have been otherwise made (since, as said, cardiomyopathies are generally autosomal dominant and have incomplete penetrance and variable expressivity). From a medico-legal point of view, the pathologist is often asked to determine whether the cause of the death was a condition that, for example, the cardiologist of the victim should have diagnosed. This issue is particularly relevant in countries, like Italy, where athletes have to regularly undergo cardiologic evaluation to exclude diseases, like cardiomyopathies, that contraindicate physical activity [98]. In these cases, especially when ECGs of uncertain significance were obtained and no radiological procedures were indicated, finding that the death was caused, for example, by a phenotype-positive cardiomyopathy is essential to prove the liability of the physician. Moreover, as said, it is important to distinguish inherited cardiomyopathies from myocarditis and infective cardiomyopathies. This issue is of great medico-legal relevance because, in case of death of an inpatient, if a hospital-acquired infection is suspected, it is important to assess whether the cause of the death was a primary cardiomyopathy or a myocarditis/secondary cardiomyopathy. Currently, this problem is particularly relevant, since in about one third of the critically ill COVID-19 patients a cardiomyopathy is found [99,100].

Table 1.
The main features of inherited HCM, Arrhythmogenic Cardiomyopathy (ACM) and Dilated Cardiomyopathy (DCM).
6.1. Importance of Good Practice
It is fundamental to carefully investigate any aspect of a SCD case (Table 1). Complete autopsies must be performed. Dissection and sampling of the heart must be done by well-trained pathologists in order to collect all the data useful for a correct diagnosis [48,54,101]. Inadequate sampling or interpretation of the microscopic features [54,82,102,103] can easily lead to missed diagnosis or misdiagnosis, especially in cases with mild phenotypes (children). Acquiring information on the clinical history of both the SD victim and his relatives is often necessary to distinguish cardiomyopathies from their many differential diagnoses [54,81,82,104] or, in case of negative autopsy, to correctly assess the significance of a variant of a gene involved in a cardiomyopathy. It is important to carefully study the clinical history of the victim and the circumstances of his death [102,104,105,106] also because, in case of negative autopsy, the pathologist can promptly suspect an IAS and thus collect fresh blood and samples of tissues for genetic testing [6,12,50,54,88,101]. This procedure is important because the DNA extracted from formalin-fixed paraffin-embedded samples is often low-template.
6.2. Virtopsy
In forensic practice, virtopsy (post-mortem CT and MR) has been proposed in combination with complete (or minimally invasive) autopsy, or even to replace it when the family opposes it for psycho-emotional or religious reasons [108,109]. As stated by Femia et al., in cases of SCD, virtopsy can play a strategical role in identifying the phenotype of a cardiomyopathy, in differentiating cardiomyopathies from other conditions (e.g., the asymmetric hypertrophy of HCM from a post-mortem myocardial oedema) and in reliably excluding any heart anomaly (thus orienting towards the indication to molecular autopsy) [107]. According to current evidence, both post-mortem CT and MR are accurate for the diagnosis of cardiomyopathies, but in those younger than 35 years MR is superior to CT, while, according to the current (few) evidence, in older SCD cases the concordance of post-mortem CT to conventional autopsy is higher [107]. Since ACM is characterized by a tissue replacement, virtopsy can be particularly useful when this cardiomyopathy is suspected. Kimura et al. proposed some criteria for the diagnosis of clinical cases of ACM at CT: “dilatation of the RV (RV body and outflow tract), abundant epicardial fat, myocardial fat in the RV trabeculae and moderator band, conspicuous trabeculae, and scalloped or bulging appearance of the RV free wall” [52]. Virtopsy is not only important for the diagnosis of ACM but also for distinguishing it from other conditions. For example, in up to the 62% of the CT-scans of patients with a history of myocardial infarction myocardial fat can be observed in the left ventricle [52]. This fatty infiltration tends to be thin and located in the subendocardial areas, near the occluded coronary artery, thus helping to distinguish a post-ischemic modification from a sign of ACM [52].
6.3. Molecular Autopsy
As said, cardiomyopathies often show mild or ambiguous macroscopic/microscopic signs of disease. This issue mainly affects young population, in which, at the same time, most of the SCDs are caused by cardiomyopathies. When, after the autopsy, the pathologist cannot make inference on the cause of the death, a molecular autopsy (post-mortem genetic testing) should be performed. This procedure is not mandatory, but it is highly recommended, since in cardiomyopathies the diagnostic rate is relatively good (80% for HCM, 50% for ACM, 30–40% for DCM) [6,12,88]. It is important to remark that in the last years rare variants in genes that code for structural proteins have been described as possible inducers of heart arrhythmias in absence of structural alterations [110]. This is because genes can affect the expression of nearby or distant genes, and some of these variants have recently been associated with the development of cardiac pathologies [111]. Moreover, as said, structural anomalies tend to be progressive and may cause arrhythmias in the young even before the complete development of the phenotype [112,113,114]. Proper sampling and storage of the blood and/or of the tissues (liver, spleen and/or heart) for the genetic testing is crucial for optimizing the results of the procedure. The choice of the source of DNA (blood vs tissues) depends on the protocol that the forensic geneticist wants to adopt. At the same time, high-throughput techniques (NGS) are particularly recommended, since variants of many different genes can cause inherited cardiomyopathies [6].
Currently, the main challenge in this field is represented by the clinical interpretation of the NGS findings. The significance of the variants is assessed following clinical guidelines, in particular those proposed by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) [115,116,117]. In the forensic field, the assessment of the significance of the variants is mainly limited by two issues: the absence of forensic guidelines (that take into account the peculiarities and the limits recurring in forensic practice) and the fact that most of the variants cannot be interpreted because of the lack of the necessary data. Indeed, in a forensic investigation acquiring complete information on the clinical history of the victim (and of his family) and performing a segregation analysis must be authorized by the competent authorities and can be extremely time-consuming (because of the bureaucratic procedures) [6]. Moreover, since relatively few molecular autopsies are performed, the lack of shared data can limit the interpretation of a variant.
Another important issue is represented by the fact that, since the significance of a variant depends on the data known at the time of the investigation, it can change over time.
In 2020, Campuzano et al. analyzed how many variants of genes involved in IAS had to be reclassified after 10 years [118]. The authors found that the 69.23% and the 94.11% of the variants found in, respectively, clinical and forensic cases had to be reclassified and that many of the variants previously classified as likely pathogenic were re-classified as variants of uncertain significance (VUS). In total, the prevalence of VUS was 18.75% in 2010 and 60.15% in 2020. Regarding the cardiomyopathies, while the prevalence of VUS in HCM and DCM cases did not change after reclassification, in ACM cases it significantly increased. The authors noted that the increase of the VUS prevalence in IAS cases depended on the higher stringency of the diagnostic criteria of the ACMG/AMP guidelines.
In 2021 Vallverdù-Prats et al. reevaluated 39 rare variants of genes associated with ACM that had already been classified according to the ACMG/AMP guidelines in 2016 [119]. In particular, the authors reported a 17.95% decrease in VUS and a 5.12% increase in likely pathogenic variants [119]. They underlined that these variations were mainly due to the updated global frequencies, thus underlying the importance of periodic reevaluations.
Hence, the assessment of the significance of a variant is an extremely critical step in the forensic evaluation of a SCD case. For instance, in forensic field, finding only a variant of uncertain significance means failing to find the cause of the death and thus making impossible to draw medico-legal conclusions on a specific case, while, on the other hand, misinterpreting a variant (e.g., a variant is classified as likely pathogenic and then, after ten years, is re-classified as VUS) can severely jeopardize the reliability of a medico-legal report. The communication of the results of a molecular autopsy to the competent authorities and, if requested by the local law, to the family of the victim should stress that the significance of a variant can be “dynamic”, largely depending on the data that are available at the time of the investigation, and that, if not certain, should be periodically reevaluated. As said, it can be particularly complex to explain the (absence of) diagnostic value of a VUS, especially when it is the only finding in an autopsy-negative forensic case.
7. Conclusions
Autopsies of forensic cases of inherited cardiomyopathies are extremely challenging when the phenotypes of these diseases are mild or difficult to be distinguished from physiological or other pathological conditions. In these cases, if accurate microscopic analysis is not diriment, virtopsy and (in particular) post-mortem genetic testing can be of great help. The results of molecular autopsies should always be interpreted by a team composed by (at least) a forensic pathologist and a forensic geneticist and, if uncertain, should be always communicated stressing that the significance of a variant can be dynamic.
Author Contributions
Conceptualization, A.O. and R.B.; Review of the literature, S.G., O.C., M.C. and F.C.; Writing–Original draft preparation, review & editing, all the authors; Figures acquisition and preparation, V.A.; Funding acquisition, A.O. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by Fondi di Ateneo, Linea D3.2—Project “Funzioni pubbliche, controllo privato. Profili interdisciplinari sulla governance senza governo della società algoritmica”, Università Cattolica del Sacro Cuore. This work was also supported by Obra Social “La Caixa Foundation” (LCF/PR/GN16/50290001 and LCF/PR/GN19/50320002). CIBERCV is an initiative of the ISCIII, Spanish Ministry of Economy and Competitiveness. Funders had no role in study design, data collection, data analysis, interpretation, or writing of the report.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not required, since no data that regard or allow to identify specific persons are discussed.
Data Availability Statement
Not applicable.
Acknowledgments
The authors deeply thank Gabriele Della Morte (P.I. of the project “Funzioni pubbliche, controllo privato. Profili interdisciplinari sulla governance senza governo della società algoritmica”) and Giovanna Mascheroni for their support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vos, A.; van der Wal, A.C.; Teeuw, A.H.; Bras, J.; Vink, A.; Nikkels, P.G.J.; Dutch NODO group. Cardiovascular causes of sudden unexpected death in children and adolescents (0–17 years): A nationwide autopsy study in the Netherlands. Neth. Heart J. 2018, 26, 500–505. [Google Scholar] [CrossRef]
- Tan, H.L.; Hofman, N.; van Langen, I.M.; van der Wal, A.C.; Wilde, A.A. Sudden unexplained death: Heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 2005, 112, 207–213. [Google Scholar] [CrossRef]
- Driscoll, D.J.; Edwards, W.D. Sudden unexpected death in children and adolescents. J. Am. Coll. Cardiol. 1985, 5, 118B–121B. [Google Scholar] [CrossRef]
- Arzamendi, D.; Benito, B.; Tizon-Marcos, H.; Flores, J.; Tanguay, J.F.; Ly, H.; Doucet, S.; Leduc, L.; Leung, T.K.; Campuzano, O.; et al. Increase in sudden death from coronary artery disease in young adults. Am. Heart J. 2011, 161, 574–580. [Google Scholar] [CrossRef]
- Rodríguez-Calvo, M.S.; Brion, M.; Allegue, C.; Concheiro, L.; Carracedo, A. Molecular genetics of sudden cardiac death. Forensic Sci. Int. 2008, 182, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Campuzano, O.; Coll, M.; Brión, M.; Arena, V.; Iglesias, A.; Carracedo, Á.; Brugada, R.; Oliva, A. Genetic variants of uncertain significance: How to match scientific rigour and standard of proof in sudden cardiac death? Leg. Med. 2020, 45, 101712. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Medeiros-Domingo, A.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Cardiac channel molecular autopsy: Insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin. Proc. 2012, 87, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.S.; Senashova, O.; Watts, A.; Tran, P.T.; Zhou, Z.; Gong, Q.; Titus, J.L.; Hayflick, S.J. Postmortem molecular screening in unexplained sudden death. J. Am. Coll. Cardiol. 2004, 43, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Allegue, C.; Partemi, S.; Mates, J.; Del Olmo, B.; Campuzano, O.; Pascali, V.; Iglesias, A.; Striano, P.; Oliva, A.; et al. Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int. J. Leg. Med. 2016, 130, 331–339. [Google Scholar]
- Gurrieri, F.; Zollino, M.; Oliva, A.; Pascali, V.; Orteschi, D.; Pietrobono, R.; Camporeale, A.; Coll Vidal, M.; Partemi, S.; Brugada, R.; et al. Mild Beckwith-Wiedemann and severe long-QT syndrome due to deletion of the imprinting center 2 on chromosome 11p. Eur. J. Hum. Genet. 2013, 21, 965–969. [Google Scholar] [CrossRef]
- Sanchez, O.; Campuzano, O.; Fernández-Falgueras, A.; Sarquella-Brugada, G.; Cesar, S.; Mademont, I.; Mates, J.; Pérez-Serra, A.; Coll, M.; Pico, F.; et al. Natural and Undetermined Sudden Death: Value of Post-Mortem Genetic Investigation. PLoS ONE 2016, 11, e0167358. [Google Scholar] [CrossRef]
- Atteya, G.; Lampert, R. Sudden Cardiac Death in Genetic Cardiomyopathies. Card. Electrophysiol. Clin. 2017, 9, 581–603. [Google Scholar] [CrossRef] [PubMed]
- Spirito, P.; Bellone, P.; Harris, K.M.; Bernabo, P.; Bruzzi, P.; Maron, B.J. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N. Engl. J. Med. 2000, 342, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Gimeno Blanes, J.R.; Mahon, N.G.; Poloniecki, J.D.; McKenna, W.J. Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet 2001, 357, 420–424. [Google Scholar] [CrossRef]
- Scholz, D.G.; Kitzman, D.W.; Hagen, P.T.; Ilstrup, D.M.; Edwards, W.D. Age-related changes in normal human hearts during the first 10 decades of life. Part I (Growth): A quantitative anatomic study of 200 specimens from subjects from birth to 19 years old. Mayo Clin. Proc. 1988, 63, 126–136. [Google Scholar] [CrossRef]
- Sheppard, M.N. Practical Cardiovascular Pathology, 2nd ed.; Hodder Arnold: London, UK, 2011. [Google Scholar]
- Papadakis, M.; Raju, H.; Behr, E.R.; De Noronha, S.V.; Spath, N.; Kouloubinis, A.; Sheppard, M.N.; Sharma, S. Sudden cardiac death with autopsy findings of uncertain significance: Potential for erroneous interpretation. Circ. Arrhythm. Electrophysiol. 2013, 6, 588–596. [Google Scholar] [CrossRef]
- Cunningham, K.S.; Spears, D.A.; Care, M. Evaluation of cardiac hypertrophy in the setting of sudden cardiac death. Forensic Sci. Res. 2019, 4, 223–240. [Google Scholar] [CrossRef]
- Caselli, S.; Maron, M.S.; Urbano-Moral, J.A.; Pandian, N.G.; Maron, B.J.; Pelliccia, A. Differentiating left ventricular hypertrophy in athletes from that in patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 2014, 114, 1383–1389. [Google Scholar] [CrossRef]
- Pelliccia, A.; Maron, M.S.; Maron, B.J. Assessment of left ventricular hypertrophy in a trained athlete: Differential diagnosis of physiologic athlete’s heart from pathologic hypertrophy. Prog. Cardiovasc. Dis. 2012, 54, 387–396. [Google Scholar] [CrossRef]
- Pelliccia, A.; Maron, B.J.; Di Paolo, F.M.; Biffi, A.; Quattrini, F.M.; Pisicchio, C.; Roselli, A.; Caselli, S.; Culasso, F. Prevalence and clinical significance of left atrial remodeling in competitive athletes. J. Am. Coll. Cardiol. 2005, 46, 690–696. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Colan, S.D.; Russell, M.W.; Towbin, J.A.; Sherrid, M.V.; Canter, C.E.; Jefferies, J.L.; Murphy, A.M.; Cirino, A.L.; et al. HCMNet Investigators. The Burden of Early Phenotypes and the Influence of Wall Thickness in Hypertrophic Cardiomyopathy Mutation Carriers: Findings from the HCMNet Study. JAMA Cardiol. 2017, 2, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.E. The pathology of hypertrophic cardiomyopathy. Histopathology 2004, 44, 412–427. [Google Scholar] [CrossRef]
- Corban, M.T.; Hung, O.Y.; Eshtehardi, P.; Rasoul-Arzrumly, E.; McDaniel, M.; Mekonnen, G.; Timmins, L.H.; Lutz, J.; Guyton, R.A.; Samady, H. Myocardial bridging: Contemporary understanding of pathophysiology with implications for diagnostic and therapeutic strategies. J. Am. Coll. Cardiol. 2014, 63, 2346–2355. [Google Scholar] [CrossRef]
- Yuan, S.M. Myocardial Bridging. Braz. J. Cardiovasc. Surg. 2016, 31, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.V.; Roh, L.; Ashar, K.; Braun, A.; Ramaswamy, G. Myocardial bridging of coronary arteries: A risk factor for myocardial fibrosis? Int. J. Cardiol. 2008, 124, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, R.B.; Costa, R.S.; Zucolotto, S.; Oliveira, J.S. Fatal outcome associated with autopsy proven myocardial bridging of the left anterior descending coronary artery. Eur. Heart J. 1989, 10, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Hostiuc, S.; Curca, G.C.; Dermengiu, D.; Dermengiu, S.; Hostiuc, M.; Rusu, M.C. Morphological changes associated with hemodynamically significant myocardial bridges in sudden cardiac death. Thorac. Cardiovasc. Surg. 2011, 59, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.R.; Klues, H.G.; vom Dahl, J.; Klein, I.; Krebs, W.; Hanrath, P. Functional, angiographic and intracoronary Doppler flow characteristics in symptomatic patients with myocardial bridging: Effect of short-term intravenous beta-blocker medication. J. Am. Coll. Cardiol. 1996, 27, 1637–1645. [Google Scholar] [CrossRef]
- Mohiddin, S.A.; Begley, D.; Shih, J.; Fananapazir, L. Myocardial bridging does not predict sudden death in children with hypertrophic cardiomyopathy but is associated with more severe cardiac disease. J. Am. Coll. Cardiol. 2000, 36, 2270–2278. [Google Scholar] [CrossRef]
- Berry, J.F.; von Mering, G.O.; Schmalfuss, C.; Hill, J.A.; Kerensky, R.A. Systolic compression of the left anterior descending coronary artery: A case series, review of the literature, and therapeutic options including stenting. Catheter. Cardiovasc. Interv. 2002, 56, 58–63. [Google Scholar] [CrossRef]
- Yetman, A.T.; McCrindle, B.W.; MacDonald, C.; Freedom, R.M.; Gow, R. Myocardial bridging in children with hypertrophic cardiomyopathy—A risk factor for sudden death. N. Engl. J. Med. 1998, 339, 1201–1209. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Mackey-Bojack, S.; Frigo, A.C.; Corrado, D.; Maron, B.J. Myocardial bridging, a frequent component of the hypertrophic cardiomyopathy phenotype, lacks systematic association with sudden cardiac death. Eur. Heart J. 2009, 30, 1627–1634. [Google Scholar] [CrossRef]
- Gori, F.; Basso, C.; Thiene, G. Myocardial infarction in a patient with hypertrophic cardiomyopathy. N. Engl. J. Med. 2000, 342, 593–594. [Google Scholar] [CrossRef]
- Thiene, G. Sudden cardiac death in the young: A genetic destiny? Clin. Med. 2018, 18, 17–23. [Google Scholar] [CrossRef]
- Morales, A.R.; Romanelli, R.; Tate, L.G.; Boucek, R.J.; de Marchena, E. Intramural left anterior descending coronary artery: Significance of the depth of the muscular tunnel. Hum. Pathol. 1993, 24, 693–701. [Google Scholar] [CrossRef]
- Virmani, R.; Farb, A.; Burke, A.P. Ischemia from myocardial coronary bridging: Fact or fancy? Hum. Pathol. 1993, 24, 687–688. [Google Scholar] [CrossRef]
- Sharzehee, M.; Chang, Y.; Song, J.P.; Han, H.C. Hemodynamic effects of myocardial bridging in patients with hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1282–H1291. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Rahman, M.S.; Elliott, P.M. A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013, 99, 1800–1811. [Google Scholar] [CrossRef]
- Erdmann, J.; Daehmlow, S.; Wischke, S.; Senyuva, M.; Werner, U.; Raible, J.; Tanis, N.; Dyachenko, S.; Hummel, M.; Hetzer, R.; et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin. Genet. 2003, 64, 339–349. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.P.; Lyons, R.G.; Bezold, K.L. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 2011, 108, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Rehm, H.L.; Menesses, A.; McDonough, B.; Roberts, A.E.; Kucherlapati, R.; Towbin, J.A.; Seidman, J.G.; Seidman, C.E. Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 2008, 358, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Watkins, H.; Rosenzweig, A.; Hwang, D.S.; Levi, T.; McKenna, W.; Seidman, C.E.; Seidman, J.G. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N. Engl. J. Med. 1992, 326, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Carrier, L.; Bonne, G.; Bährend, E.; Yu, B.; Richard, P.; Niel, F.; Hainque, B.; Cruaud, C.; Gary, F.; Labeit, S.; et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ. Res. 1997, 80, 427–434. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- Corrado, D.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Clinical impact of molecular genetic studies. Circulation 2006, 113, 1634–1637. [Google Scholar] [CrossRef]
- Kimura, F.; Matsuo, Y.; Nakajima, T.; Nishikawa, T.; Kawamura, S.; Sannohe, S.; Hagiwara, N.; Sakai, F. Myocardial fat at cardiac imaging: How can we differentiate pathologic from physiologic fatty infiltration? Radiographics 2010, 30, 1587–1602. [Google Scholar] [CrossRef]
- Anumonwo, J.M.B.; Herron, T. Fatty Infiltration of the Myocardium and Arrhythmogenesis: Potential Cellular and Molecular Mechanisms. Front. Physiol. 2018, 9, 2. [Google Scholar] [CrossRef]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. International Experts. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar]
- Roberts, J.D.; Veinot, J.P.; Rutberg, J.; Gollob, M.H. Inherited cardiomyopathies mimicking arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 2010, 19, 316–320. [Google Scholar] [CrossRef]
- Tansey, D.K.; Aly, Z.; Sheppard, M.N. Fat in the right ventricle of the normal heart. Histopathology 2005, 46, 98–104. [Google Scholar] [CrossRef]
- Liang, Y.H.; Zhu, J.; Zhong, D.R.; Hou, D.Y.; Ma, G.L.; Zhang, Z.Y.; Zhang, L. Adipositas cordis sudden death: A series of 79 patients. Int. J. Clin. Exp. Pathol. 2015, 8, 10861–10867. [Google Scholar]
- Corradi, D.; Maestri, R.; Callegari, S.; Pastori, P.; Goldoni, M.; Luong, T.V.; Bordi, C. The ventricular epicardial fat is related to the myocardial mass in normal, ischemic and hypertrophic hearts. Cardiovasc. Pathol. 2004, 13, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Farb, A.; Tashko, G.; Virmani, R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: Are they different diseases? Circulation 1998, 97, 1571–1580. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brooke, M.A.; Calkins, H.; Corrado, D.; Duru, F.; Green, K.J.; et al. Definition and treatment of arrhythmogenic cardiomyopathy: An updated expert panel report. Eur. J. Heart Fail. 2019, 21, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Chen, S.N. Editorial of Special Issue “Genetics and Molecular Pathogenesis of Non-Ischemic Cardiomyopathies”. Int. J. Mol. Sci. 2020, 21, 9398. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Taylor, M.R.G.; Mestroni, L. Unraveling Missing Genes and Missing Inheritance in Arrhythmogenic Cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2017, 10, e005813. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, E.; Jongbloed, J.D.; Pilichou, K.; Thiene, G.; Basso, C.; Bikker, H.; Charbon, B.; Swertz, M.; van Tintelen, J.P.; van der Zwaag, P.A. The ARVD/C genetic variants database: 2014 Update. Hum. Mutat. 2015, 36, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Riele, A.S.T.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.P.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef]
- Nakajima, T.; Kaneko, Y.; Irie, T.; Takahashi, R.; Kato, T.; Iijima, T.; Iso, T.; Kurabayashi, M. Compound and digenic heterozygosity in desmosome genes as a cause of arrhythmogenic right ventricular cardiomyopathy in Japanese patients. Circ. J. 2012, 76, 737–743. [Google Scholar] [CrossRef]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef]
- Oliva, A.; Brugada, R.; D’Aloja, E.; Boschi, I.; Partemi, S.; Brugada, J.; Pascali, V.L. State of the art in forensic investigation of sudden cardiac death. Am. J. Forensic Med. Pathol. 2011, 32, 1–16. [Google Scholar] [CrossRef]
- Quarta, G.; Muir, A.; Pantazis, A.; Syrris, P.; Gehmlich, K.; Garcia-Pavia, P.; Ward, D.; Sen-Chowdhry, S.; Elliott, P.M.; McKenna, W.J. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: Impact of genetics and revised task force criteria. Circulation 2011, 123, 2701–2709. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Pantazis, A.; Quarta, G.; McKenna, W.J.; Chambers, J.C. Mutational heterogeneity, modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ. Cardiovasc Genet. 2010, 3, 323–330. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Awad, M.M.; Dalal, D.; Tichnell, C.; James, C.; Tucker, A.; Abraham, T.; Spevak, P.J.; Calkins, H.; Judge, D.P. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum. Mutat. 2006, 27, 1157. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- Roberts, J.D.; Herkert, J.C.; Rutberg, J.; Nikkel, S.M.; Wiesfeld, A.C.; Dooijes, D.; Gow, R.M.; van Tintelen, J.P.; Gollob, M.H. Detection of genomic deletions of PKP2 in arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 2013, 83, 452–456. [Google Scholar] [CrossRef]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.D.; Landstrom, A.P.; Salisbury, B.A.; Callis, T.E.; Pollevick, G.D.; Tester, D.J.; Cox, M.G.; Bhuiyan, Z.; Bikker, H.; Wiesfeld, A.C.; et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J. Am. Coll. Cardiol. 2011, 57, 2317–2327. [Google Scholar] [CrossRef]
- Patel, V.; Asatryan, B.; Siripanthong, B.; Munroe, P.B.; Tiku-Owens, A.; Lopes, L.R.; Khanji, M.Y.; Protonotarios, A.; Santangeli, P.; Muser, D.; et al. State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 6615. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Towbin, J.A. Dilated cardiomyopathy. Lancet 2010, 375, 752–762. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator implantation in patients with nonischemic systolic heart failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.; Jabbour, A.; Ismail, T.F.; Guha, K.; Khwaja, J.; Raza, S.; Morarji, K.; Brown, T.D.; Ismail, N.A.; Dweck, M.R.; et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013, 309, 896–908. [Google Scholar] [CrossRef]
- Narayanan, K.; Reinier, K.; Teodorescu, C.; Uy-Evanado, A.; Aleong, R.; Chugh, H.; Nichols, G.A.; Gunson, K.; London, B.; Jui, J.; et al. Left ventricular diameter and risk stratification for sudden cardiac death. J. Am. Heart Assoc. 2014, 3, e001193. [Google Scholar] [CrossRef]
- Beltrami, C.A.; Finato, N.; Rocco, M.; Feruglio, G.A.; Puricelli, C.; Cigola, E.; Son-nenblick, E.H.; Olivetti, G.; Anversa, P. The cellular basis of dilated cardiomyopathy in humans. J. Mol. Cell. Cardiol. 1995, 27, 291–305. [Google Scholar] [CrossRef]
- Bogun, F.M.; Desjardins, B.; Good, E.; Gupta, S.; Crawford, T.; Oral, H.; Ebinger, M.; Pelosi, F.; Chugh, A.; Jongnarangsin, K.; et al. Delayed-enhanced magnetic resonance imaging in nonischemic cardiomyopathy: Utility for identifying the ventricular arrhythmia substrate. J. Am. Coll. Cardiol. 2009, 53, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Halliday, B.P.; Cleland, J.G.F.; Goldberger, J.J.; Prasad, S.K. Personalizing Risk Stratification for Sudden Death in Dilated Cardiomyopathy: The Past, Present, and Future. Circulation 2017, 136, 215–231. [Google Scholar] [CrossRef]
- García-Pavía, P.; Cobo-Marcos, M.; Guzzo-Merello, G.; Gómez-Bueno, M.; Bornstein, B.; Lara-Pezzi, E.; Segovia, J.; Alonso-Pulpón, L. Genetics in Dilated Cardiomyopathy. Biomark. Med. 2013, 7, 517–533. [Google Scholar] [CrossRef]
- Murphy, R.T.; Mogensen, J.; Shaw, A.; Kubo, T.; Hughes, S.; McKenna, W.J. Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet 2004, 363, 371–372. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135a. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.H.; Hamilton, R.M.; et al. Hrs/ehra expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the heart rhythm society (hrs) and the european heart rhythm association (ehra)>. Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. ESC Scientific Document Group. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar]
- Gollob, M.H.; Blier, L.; Brugada, R.; Champagne, J.; Chauhan, V.; Connors, S.; Gardner, M.; Green, M.S.; Gow, R.; Hamilton, R.; et al. Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian cardiovascular society/canadian heart rhythm society joint position paper. Can. J. Cardiol. 2011, 27, 232–245. [Google Scholar] [CrossRef]
- Wilhelm, M.; Bolliger, S.A.; Bartsch, C.; Fokstuen, S.; Gräni, C.; Martos, V.; Medeiros Domingo, A.; Osculati, A.; Rieubland, C.; Sabatasso, S.; et al. Sudden cardiac death in forensic medicine—Swiss recommendations for a multidisciplinary approach. Swiss Med. Wkly. 2015, 145, w14129. [Google Scholar] [CrossRef]
- Fernández-Falgueras, A.; Sarquella-Brugada, G.; Brugada, J.; Brugada, R.; Campuzano, O. Cardiac Channelopathies and Sudden Death: Recent Clinical and Genetic Advances. Biology 2017, 6, 7. [Google Scholar] [CrossRef]
- Hertz, C.L.; Christiansen, S.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; LuCamp; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Next-generation sequencing of 100 candidate genes in young victims of suspected sudden cardiac death with structural abnormalities of the heart. Int. J. Leg. Med. 2016, 130, 91–102. [Google Scholar] [CrossRef]
- Lahrouchi, N.; Raju, H.; Lodder, E.M.; Papatheodorou, S.; Miles, C.; Ware, J.S.; Papadakis, M.; Tadros, R.; Cole, D.; Skinner, J.R.; et al. The yield of postmortem genetic testing in sudden death cases with structural findings at autopsy. Eur. J. Hum. Genet. 2020, 28, 17–22. [Google Scholar] [CrossRef]
- Oliva, A.; Grassi, V.M.; Campuzano, O.; Brion, M.; Arena, V.; Partemi, S.; Coll, M.; Pascali, V.L.; Brugada, J.; Carracedo, A.; et al. Medico-legal perspectives on sudden cardiac death in young athletes. Int. J. Leg. Med. 2017, 131, 393–409. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef]
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington State. JAMA 2020, 323, 1612–1614. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef]
- Maron, B.J.; Mackey-Bojack, S.; Facile, E.; Duncanson, E.; Rowin, E.J.; Maron, M.S. Hypertrophic Cardiomyopathy and Sudden Death Initially Identified at Autopsy. Am. J. Cardiol. 2020, 127, 139–141. [Google Scholar] [CrossRef]
- Niimura, H.; Patton, K.K.; McKenna, W.J.; Soults, J.; Maron, B.J.; Seidman, J.G.; Seidman, C.E. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002, 105, 446–451. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e533–e557. [Google Scholar] [PubMed]
- Cadrin-Tourigny, J.; Bosman, L.P.; Wang, W.; Tadros, R.; Bhonsale, A.; Bourfiss, M.; Lie, Ø.H.; Saguner, A.M.; Svensson, A.; Andorin, A.; et al. Sudden Cardiac Death Prediction in Arrhythmogenic Right Ventricular Cardiomyopathy: A Multinational Collaboration. Circ. Arrhythm Electrophysiol. 2021, 14, e008509. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Femia, G.; Semsarian, C.; Langlois, N.; McGuire, M.; Raleigh, J.; Taylor, A.; Puranik, R. Post-Mortem Imaging Adjudicated Sudden Death: Causes and Controversies. Heart Lung Circ. 2019, 28, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Thali, M.J.; Yen, K.; Schweitzer, W.; Vock, P.; Boesch, C.; Ozdoba, C.; Schroth, G.; Ith, M.; Sonnenschein, M.; Doernhoefer, T.; et al. Virtopsy, a new imaging horizon in forensic pathology: Virtual autopsy by postmortem multislice computed tomography (MSCT) and magnetic resonance imaging (MRI)—A feasibility study. J. Forensic Sci. 2003, 48, 386–403. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.; Grassi, S.; Grassi, V.M.; Pinchi, V.; Floris, R.; Manenti, G.; Colosimo, C.; Filograna, L.; Pascali, V.L. Postmortem CT and autopsy findings in nine victims of terrorist attack. Int. J. Leg. Med. 2021, 135, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Barc, J.; Briec, F.; Schmitt, S.; Kyndt, F.; Le Cunff, M.; Baron, E.; Vieyres, C.; Sacher, F.; Redon, R.; Le Caignec, C.; et al. Screening for copy number variation in genes associated with the long QT syndrome: Clinical relevance. J. Am. Coll. Cardiol. 2011, 57, 40–47. [Google Scholar] [CrossRef]
- Campuzano, O.; Sanchez-Molero, O.; Allegue, C.; Coll, M.; Mademont-Soler, I.; Selga, E.; Ferrer-Costa, C.; Mates, J.; Iglesias, A.; Sarquella-Brugada, G.; et al. Post-mortem genetic analysis in juvenile cases of sudden cardiac death. Forensic Sci. Int. 2014, 245, 30–37. [Google Scholar] [CrossRef]
- Mori, A.A.; Castro, L.R.; Bortolin, R.H.; Bastos, G.M.; Oliveira, V.F.; Ferreira, G.M.; Hirata, T.D.C.; Fajardo, C.M.; Sampaio, M.F.; Moreira, D.A.R.; et al. Association of variants in MYH7, MYBPC3 and TNNT2 with sudden cardiac death-related risk factors in Brazilian patients with hypertrophic cardiomyopathy. Forensic Sci. Int. Genet. 2021, 52, 102478. [Google Scholar] [CrossRef]
- Sarquella-Brugada, G.; Campuzano, O.; Cesar, S.; Iglesias, A.; Fernandez, A.; Brugada, J.; Brugada, R. Sudden infant death syndrome caused by cardiac arrhythmias: Only a matter of genes encoding ion channels? Int. J. Legal Med. 2016, 130, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Allegue, C.; Fernandez, A.; Iglesias, A.; Brugada, R. Determining the pathogenicity of genetic variants associated with cardiac channelopathies. Sci. Rep. 2015, 5, 7953. [Google Scholar] [CrossRef] [PubMed]
- Amendola, L.M.; Jarvik, G.P.; Leo, M.C.; McLaughlin, H.M.; Akkari, Y.; Amaral, M.D.; Berg, J.S.; Biswas, S.; Bowling, K.M.; Conlin, L.K.; et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am. J. Hum. Genet. 2016, 98, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Coll, M.; Iglesias, A.; Ferrer-Costa, C.; Cesar, S.; Arbelo, E.; García-Álvarez, A.; Jordà, P.; et al. Reanalysis and reclassification of rare genetic variants associated with inherited arrhythmogenic syndromes. EBioMedicine 2020, 54, 102732. [Google Scholar] [CrossRef] [PubMed]
- Vallverdú-Prats, M.; Alcalde, M.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Fernandez-Falgueras, A.; Coll, M.; Pérez-Serra, A.; Puigmulé, M.; Iglesias, A.; et al. Rare Variants Associated with Arrhythmogenic Cardiomyopathy: Reclassification Five Years Later. J. Pers. Med. 2021, 11, 162. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).