
JDP2, a Novel Molecular Key in Heart Failure and Atrial Fibrillation?



Abstract
1. Introduction
2. Transcriptional Control by JDP2
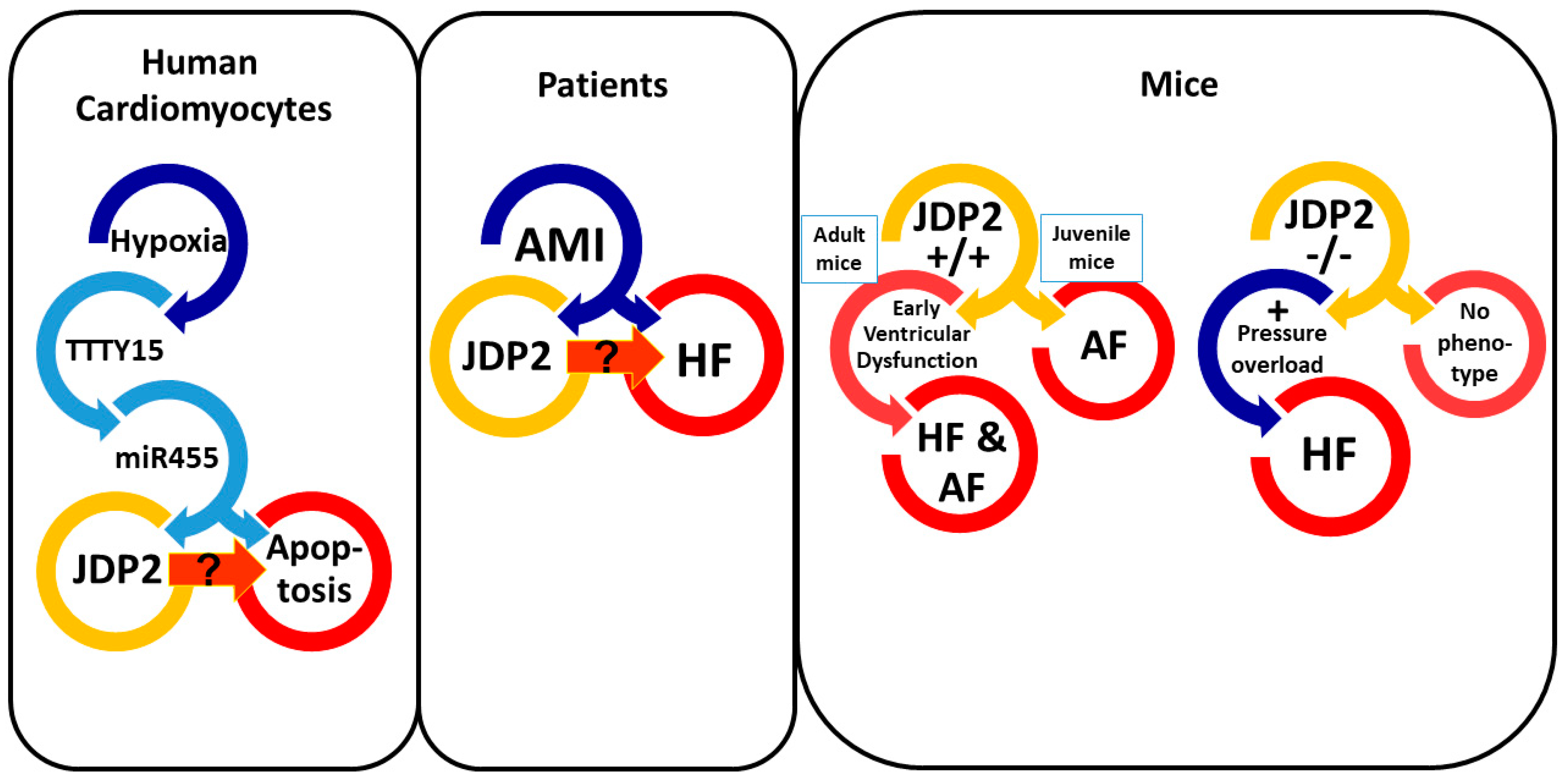
3. Correlation between JDP2 Expression and Heart Failure
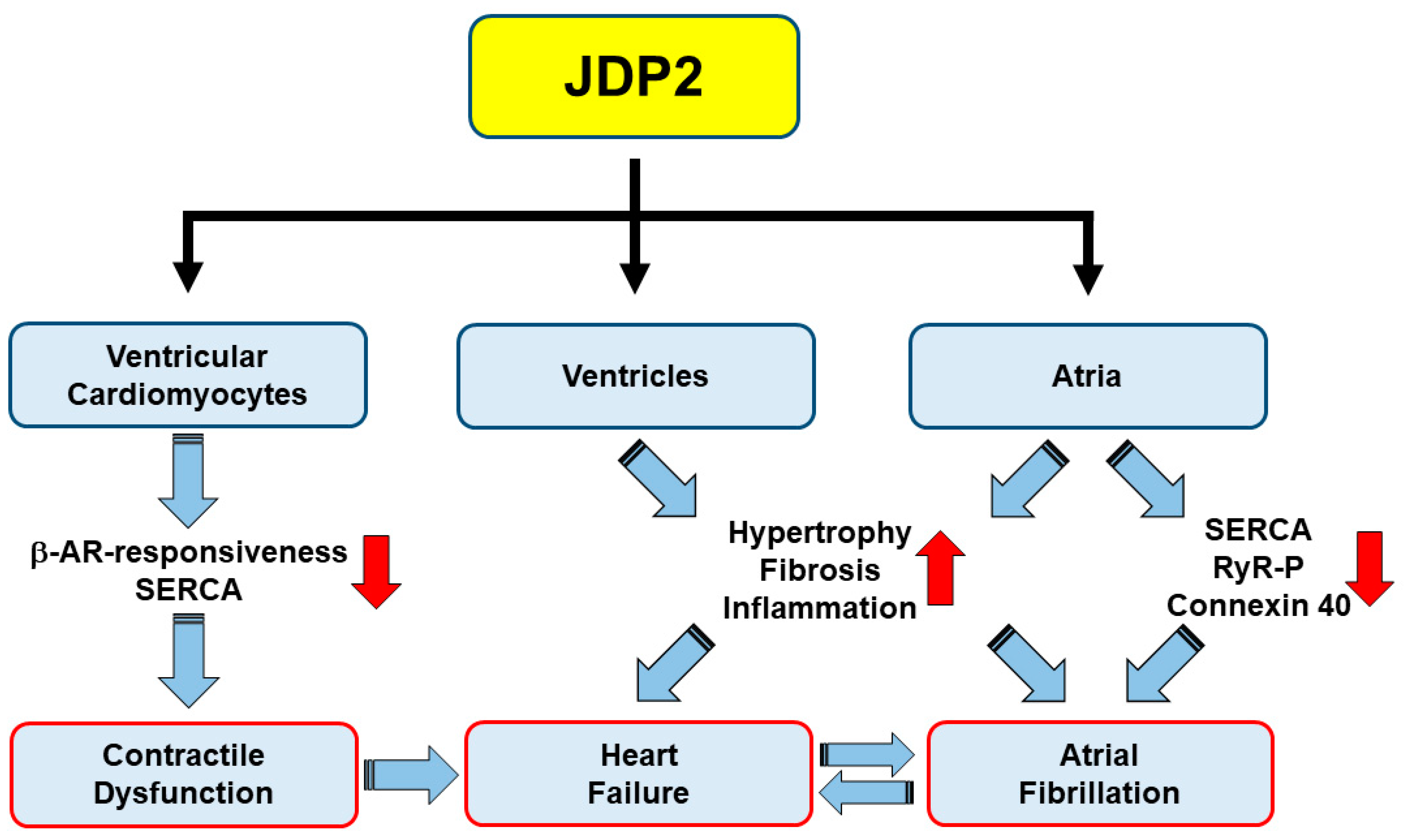
4. Influence of JDP2 on Ventricular Remodeling and Function
5. JDP2 Promotes Atrial Remodeling and Arrhythmias
6. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bragazzi, N.; Zhong, W.; Shu, J.; Abu Much, A.; Lotan, D.; Grupper, A.; Younis, A.; Dai, H. Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur. J. Prev. Cardiol. 2021, zwaa147. [Google Scholar] [CrossRef]
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.J.; Helm, R.H. Atrial Fibrillation: Epidemiology, Pathophysiology, and Clinical Outcomes. Circ. Res. 2017, 120, 1501–1517. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. ESC Scientific Document Group. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Wuputra, K.; Lin, Y.-C.; Lin, C.-S.; Yokoyama, K.K. Multiple functions of the histone chaperone Jun dimerization protein 2. Gene 2016, 590, 193–200. [Google Scholar] [CrossRef]
- Aronheim, A.; Zandi, E.; Hennemann, H.; Elledge, S.J.; Karin, M. Isolation of an AP-1 repressor by a novel method for detecting protein–protein interactions. Mol. Cell. Biol. 1997, 17, 3094–3102. [Google Scholar] [CrossRef]
- Jin, C.; Ugai, H.; Song, J.; Murata, T.; Nili, F.; Sun, K.; Horikoshi, M.; Yokoyama, K.K. Identification of mouse Jun dimerization protein 2 as a novel repressor of ATF-2. FEBS Lett. 2001, 489, 34–41. [Google Scholar] [CrossRef]
- Jin, C.; Kato, K.; Chimura, T.; Yamasaki, T.; Nakade, K.; Murata, T.; Li, H.; Pan, J.; Zhao, M.; Sun, K.; et al. Regulation of histone acetylation and nucleosome assembly by transcription factor JDP2. Nat. Struct. Mol. Biol. 2006, 13, 331–338. [Google Scholar] [CrossRef]
- Huang, Y.C.; Saito, S.; Yokoyama, K.K. Histone chaperone Jun dimerization protein 2 (JDP2): Role in cellular senescence and aging. Kaohsiung J. Med. Sci. 2010, 26, 515–531. [Google Scholar] [CrossRef]
- Darlyuk-Saadon, I.; Weidenfeld-Baranboim, K.; Yokoyama, K.K.; Hai, T.; Aronheim, A. The bZIP repressor proteins, c-Jun dimerization protein 2 and activating transcription factor 3, recruit multiple HDAC members to the ATF3 promoter. Biochim. Biophys. Acta 2012, 1819, 1142–1153. [Google Scholar] [CrossRef]
- Maruyama, K.; Fukasaka, M.; Vandenbon, A.; Saitoh, T.; Kawasaki, T.; Kondo, T.; Yokoyama, K.K.; Kidoya, H.; Takakura, N.; Standley, D.; et al. The transcription factor Jdp2 controls bone homeostasis and antibacterial immunity by regulating osteoclast and neutrophil differentiation. Immunity 2012, 37, 1024–1036. [Google Scholar] [CrossRef] [PubMed]
- Nakade, K.; Pan, J.; Yoshiki, A.; Ugai, H.; Kimura, M.; Liu, B.; Li, H.; Obata, Y.; Iwama, M.; Itohara, S.; et al. JDP2 suppresses adipocyte differentiation by regulating histone acetylation. Cell. Death. Differ. 2007, 14, 1398–1405. [Google Scholar] [CrossRef]
- Edwards, D.P.; Wardell, S.E.; Boonyaratanakornkit, V. Progesterone receptor interacting coregulatory proteins and cross talk with cell signaling pathways. J. Steroid Biochem. Mol. Biol. 2002, 83, 173–186. [Google Scholar] [CrossRef]
- Wardell, S.E.; Boonyaratanakornkit, V.; Adelman, J.S.; Aronheim, A.; Edwards, D.P. Jun dimerization protein 2 functions as a progesterone receptor N-terminal domain coactivator. Mol. Cell. Biol. 2002, 22, 5451–5466. [Google Scholar] [CrossRef]
- Wardell, S.E.; Kwok, S.C.; Sherman, L.; Hodges, R.S.; Edwards, D.P. Regulation of the amino-terminal transcription activation domain of progesterone receptor by a cofactor-induced protein folding mechanism. Mol. Cell. Biol. 2005, 25, 8792–8808. [Google Scholar] [CrossRef]
- Hill, K.K.; Roemer, S.C.; Jones, D.N.; Churchill, M.E.; Edwards, D.P. A progesterone receptor coactivator (JDP2) mediates activity through interaction with residues in the carboxyl-terminal extension of the DNA binding domain. J. Biol. Chem. 2009, 284, 24415–24424. [Google Scholar] [CrossRef]
- Chiou, S.S.; Wang, S.S.; Wu, D.C.; Lin, Y.C.; Kao, L.P.; Kuo, K.K.; Wu, C.C.; Chai, C.Y.; Lin, C.L.; Lee, C.Y.; et al. Control of oxidative stress and generation of induced pluripotent stem cell-like cells by Jun dimerization protein 2. Cancers 2013, 5, 959–984. [Google Scholar] [CrossRef]
- Tanigawa, S.; Lee, C.H.; Lin, C.S.; Ku, C.C.; Hasegawa, H.; Qin, S.; Kawahara, A.; Korenori, Y.; Miyamori, K.; Noguchi, M.; et al. Jun dimerization protein 2 is a critical component of the Nrf2/MafK complex regulating the response to ROS homeostasis. Cell. Death. Dis. 2013, 4, e921. [Google Scholar] [CrossRef] [PubMed]
- Maciejak, A.; Kiliszek, M.; Michalak, M.; Tulacz, D.; Opolski, G.; Matlak, K.; Dobrzycki, S.; Segiet, A.; Gora, M.; Burzynska, B. Gene expression profiling reveals potential prognostic biomarkers associated with the progression of heart failure. Genome Med. 2015, 7, 26. [Google Scholar] [CrossRef]
- Qiu, L.; Liu, X. Identification of key genes involved in myocardial infarction. Eur. J. Med. Res. 2019, 24, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, N. Uncovering potential differentially expressed miRNAs and targeted mRNAs in myocardial infarction based on integrating analysis. Mol. Med. Rep. 2020, 22, 4383–4395. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, J.; Huang, X. Rosuvastatin attenuates myocardial ischemia-reperfusion injury via upregulating mir-17-3p-mediated autophagy. Cell Reprogram. 2019, 21, 323–330. [Google Scholar] [CrossRef]
- Huang, S.; Tao, W.; Guo, Z.; Cao, J.; Huang, X. Suppression of long noncoding RNA TTTY15 attenuates hypoxia-induced cardiomyocytes injury by targeting miR-455-5p. Gene 2019, 701, 1–8. [Google Scholar] [CrossRef]
- Kehat, I.; Heinrich, R.; Ben-Izhak, O.; Miyazaki, H.; Gutkind, J.S.; Aronheim, A. Inhibition of basic leucine zipper transcription is a major mediator of atrial dilatation. Cardiovasc. Res. 2006, 70, 543–554. [Google Scholar] [CrossRef]
- Heger, J.; Bornbaum, J.; Würfel, A.; Hill, C.; Brockmann, N.; Gáspár, R.; Pálóczi, J.; Varga, Z.V.; Sárközy, M.; Bencsik, P.; et al. JDP2 overexpression provokes cardiac dysfunction in mice. Sci. Rep. 2018, 8, 7647. [Google Scholar] [CrossRef]
- Parahuleva, M.S.; Kockskämper, J.; Heger, J.; Grimm, W.; Scherer, A.; Bühler, S.; Kreutz, J.; Schulz, R.; Euler, G. Structural, Pro-Inflammatory and Calcium Handling Remodeling Underlies Spontaneous Onset of Paroxysmal Atrial Fibrillation in JDP2-Overexpressing Mice. Int. J. Mol. Sci. 2020, 21, 9095. [Google Scholar] [CrossRef]
- Hill, C.; Würfel, A.; Heger, J.; Meyering, B.; Schlüter, K.; Weber, M.; Ferdinandy, P.; Aronheim, A.; Schulz, R.; Euler, G. Inhibition of AP-1 signaling by JDP2 overexpression protects cardiomyocytes against hypertrophy and apoptosis induction. Cardiovasc. Res. 2013, 9, 121–128. [Google Scholar] [CrossRef]
- Kalfon, R.; Haas, T.; Shofti, R.; Moskovitz, J.D.; Schwartz, O.; Suss-Toby, E.; Aronheim, A. c-Jun dimerization protein 2 (JDP2) deficiency promotes cardiac hypertrophy and dysfunction in response to pressure overload. Int. J. Cardiol. 2017, 249, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Kalfon, R.; Friedman, T.; Eliachar, S.; Shofti, R.; Haas, T.; Koren, L.; Moskovitz, J.D.; Hai, T.; Aronheim, A. JDP2 and ATF3 deficiencies dampen maladaptive cardiac remodeling and preserve cardiac function. PLoS ONE 2019, 14, e0213081. [Google Scholar] [CrossRef]
- Maack, C.; Eschenhagen, T.; Hamdani, N.; Heinzel, F.R.; Lyon, A.R.; Manstein, D.J.; Metzger, J.; Papp, Z.; Tocchetti, C.G.; Yilmaz, M.B.; et al. Treatments targeting inotropy. Eur. Heart J. 2019, 40, 3626–3644. [Google Scholar] [CrossRef]
- Koren, L.; Alishekevitz, D.; Elhanani, O.; Nevelsky, A.; Hai, T.; Kehat, I.; Shaked, Y.; Aronheim, A. ATF3-dependent cross-talk between cardiomyocytes and macrophages promotes cardiac maladaptive remodelling. Int. J. Cardiol. 2015, 198, 232–240. [Google Scholar] [CrossRef]
- Weidenfeld-Baranboim, K.; Hasin, T.; Darlyuk, I.; Heinrich, R.; Elhanani, O.; Pan, J.; Yokoyama, K.K.; Aronheim, A. The ubiquitously expressed bZIP inhibitor, JDP2, suppresses the transcription of its homologue immediate early gene counterpart, ATF3. Nucleic Acids Res. 2009, 37, 2194–2203. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Chaves, A.; Chen, J.; Kelley, R.; Jones, K.; Weed, H.G.; Gardner, K.L.; Gangi, L.; Yamaguchi, M.; Klomkleaw, W.; et al. Transgenic mice with cardiac-specific expression of activating transcription factor 3, a stress-inducible gene, have conduction abnormalities and contractile dysfunction. Am. J. Pathol. 2001, 159, 639–650. [Google Scholar] [CrossRef]
- Koren, L.; Elhanani, O.; Kehat, I.; Hai, T.; Aronheim, A. Adult Cardiac Expression of the Activating Transcription Factor 3, ATF3, Promotes Ventricular Hypertrophy. PLoS ONE 2013, 8, e68396. [Google Scholar] [CrossRef] [PubMed]
- VanderBrink, B.A.; Sellitto, C.; Saba, S.; Link, M.S.; Zhu, W.; Homoud, M.K.; Estes, N.A., III; Paul, D.L.; Wang, P.J. Connexin40-deficient mice exhibit atrioventricular nodal and infra-Hisian conduction abnormalities. J. Cardiovasc. Electrophysiol. 2000, 11, 1270–1276. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, D.; Xie, X.; Wang, S.; Wang, Z.; Zhao, W.; Xu, H.; Zheng, L. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic. Res. Cardiol. 2016, 111, 63. [Google Scholar] [CrossRef]
- Leuschner, F.; Nahrendorf, M. Novel functions of macrophages in the heart: Insights into electrical conduction, stress, and diastolic dysfunction. Eur. Heart J. 2020, 41, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef]
- Boos, C.J. Infection and atrial fibrillation: Inflammation begets AF. Eur. Heart J. 2020, 41, 1120–1122. [Google Scholar] [CrossRef]
- Müller, F.U.; Lewin, G.; Baba, H.A.; Bokník, P.; Fabritz, L.; Kirchhefer, U.; Kirchhof, P.; Loser, K.; Matus, M.; Neumann, J.; et al. Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J. Biol. Chem. 2005, 280, 6906–6914. [Google Scholar] [CrossRef]
- Stümpel, F.T.; Stein, J.; Himmler, K.; Scholz, B.; Seidl, M.D.; Skryabin, B.V.; Müller, F.U. Homozygous CREM-IbΔC-X Overexpressing Mice Are a Reliable and Effective Disease Model for Atrial Fibrillation. Front. Pharmacol. 2018, 9, 706. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| JDP2 Expression | Disease/Provocation | Cell Type | Species | Ventricular Effects | Atrial Effects | Reference |
|---|---|---|---|---|---|---|
 | MI | PBMCs | human | predictor for HF development | [18] | |
| MI | PBMCs | human | central part in protein-protein network | [19] | |
| MI | PBMCs | human | target of hsa-mir-17-3p | [20] | |
| hypoxia | CMs | human | [22] | ||
| overexpression | CMs/heart | juvenile mice | atrial dilatation, AF | [23] | |
| overexpression | CMs/heart | adult mice | ventricular dysfunction | [24] | |
| overexpression | CMs/heart | adult mice | atrial dilatation, AF | [25] | |
| overexpression | isolated CMs | adult mice | contractile dysfunction, protection vs. hypertrophy and apoptosis | [26] | |
 | KO + pressure overload | global | mice | ventricular dysfunction | [27] | |
| KO + ATF3 KO + pressure overload | global | mice | preserved ventricular function | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Euler, G.; Kockskämper, J.; Schulz, R.; Parahuleva, M.S. JDP2, a Novel Molecular Key in Heart Failure and Atrial Fibrillation? Int. J. Mol. Sci. 2021, 22, 4110. https://doi.org/10.3390/ijms22084110
Euler G, Kockskämper J, Schulz R, Parahuleva MS. JDP2, a Novel Molecular Key in Heart Failure and Atrial Fibrillation? International Journal of Molecular Sciences. 2021; 22(8):4110. https://doi.org/10.3390/ijms22084110
Chicago/Turabian StyleEuler, Gerhild, Jens Kockskämper, Rainer Schulz, and Mariana S. Parahuleva. 2021. "JDP2, a Novel Molecular Key in Heart Failure and Atrial Fibrillation?" International Journal of Molecular Sciences 22, no. 8: 4110. https://doi.org/10.3390/ijms22084110
APA StyleEuler, G., Kockskämper, J., Schulz, R., & Parahuleva, M. S. (2021). JDP2, a Novel Molecular Key in Heart Failure and Atrial Fibrillation? International Journal of Molecular Sciences, 22(8), 4110. https://doi.org/10.3390/ijms22084110