
Peroxisomal Disorders and Their Mouse Models Point to Essential Roles of Peroxisomes for Retinal Integrity

Abstract
1. Introduction
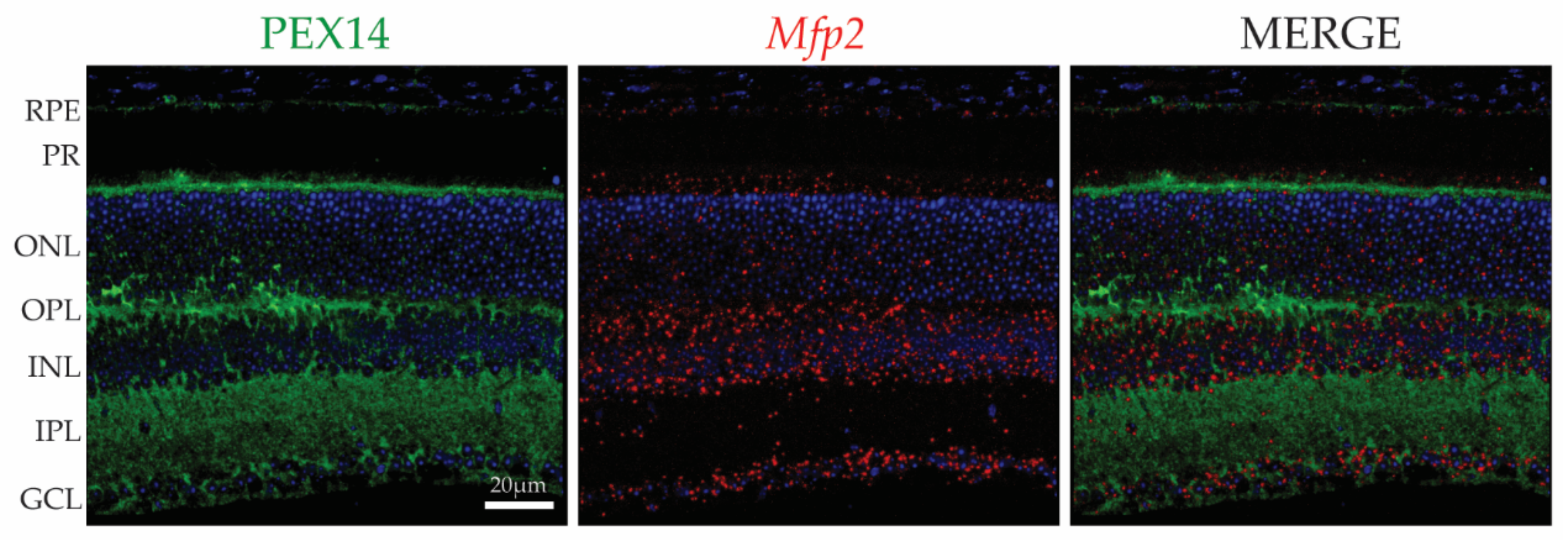
2. The Location of Peroxisomes in the Retina
3. Zellweger Spectrum Disorders
3.1. Retinopathy: A Recurrent Phenomenon
3.2. Histopathology
3.3. Insights form the Pex1 Knock-In Mouse Model
4. Single Enzyme Deficiencies
4.1. Defects in α-Oxidation
4.1.1. Retinitis Pigmentosa in Refsum Disease
4.1.2. Insights from In Vivo and In Vitro Models
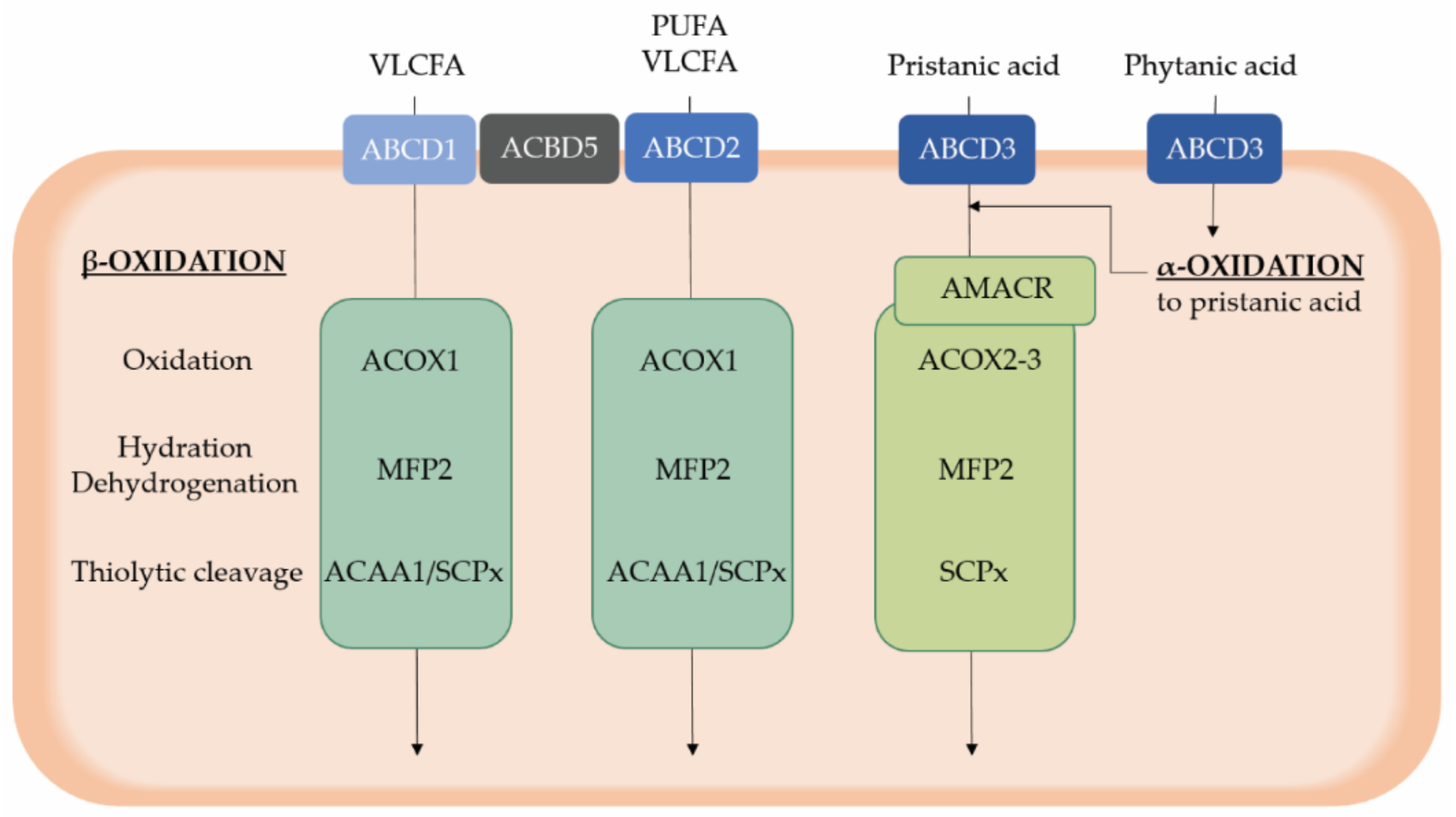
4.2. Peroxisomal β-Oxidation Deficiency
4.2.1. X-Linked Adrenoleukodystrophy
4.2.2. Acyl-CoA Oxidase 1 Deficiency
4.2.3. Multifunctional Protein 2 Deficiency
4.2.4. Deficient Oxidation of Branched Chain Fatty Acids and Bile Acid Synthesis
4.2.5. Acyl-CoA Binding Domain Containing Protein 5 Deficiency
5. Ether Phospholipid Deficiency
5.1. Rhizomelic Chondrodysplasia Punctata
5.2. Insights from the Different Mouse Models
6. Candidate Metabolites Causing Retinopathy
6.1. Saturated Very Long Chain Fatty Acids
6.2. Branched Chain Fatty Acids
6.3. Polyunsaturated Fatty Acids
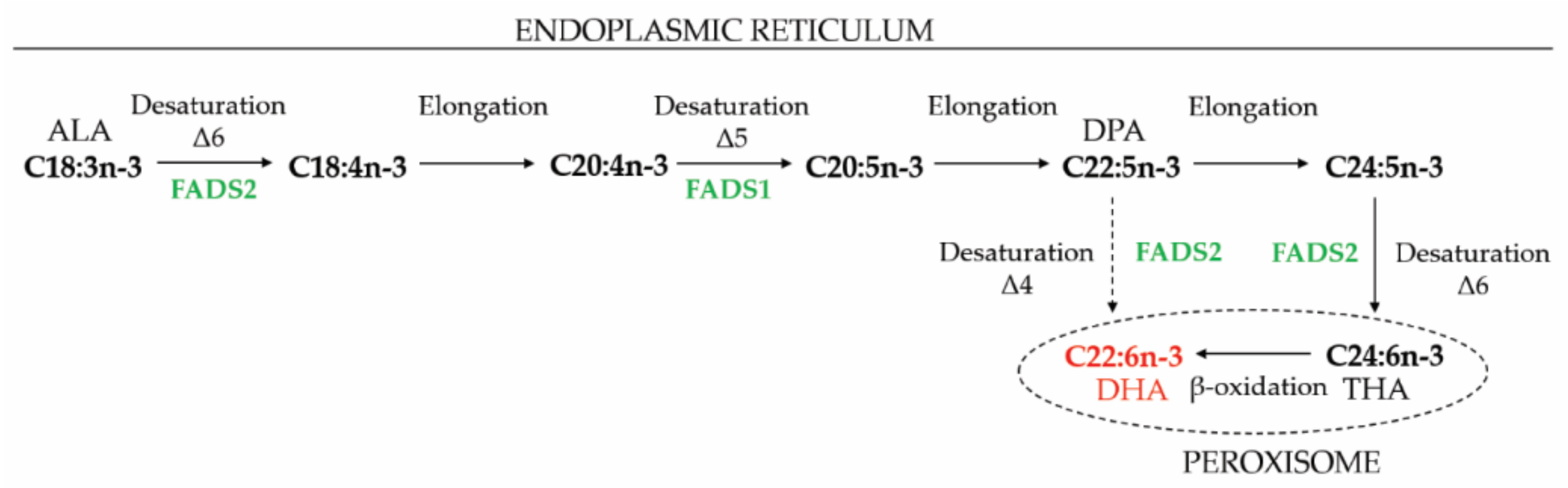
6.3.1. Docosahexaenoic Acid
6.3.2. Very Long Chain Polyunsaturated Fatty Acids
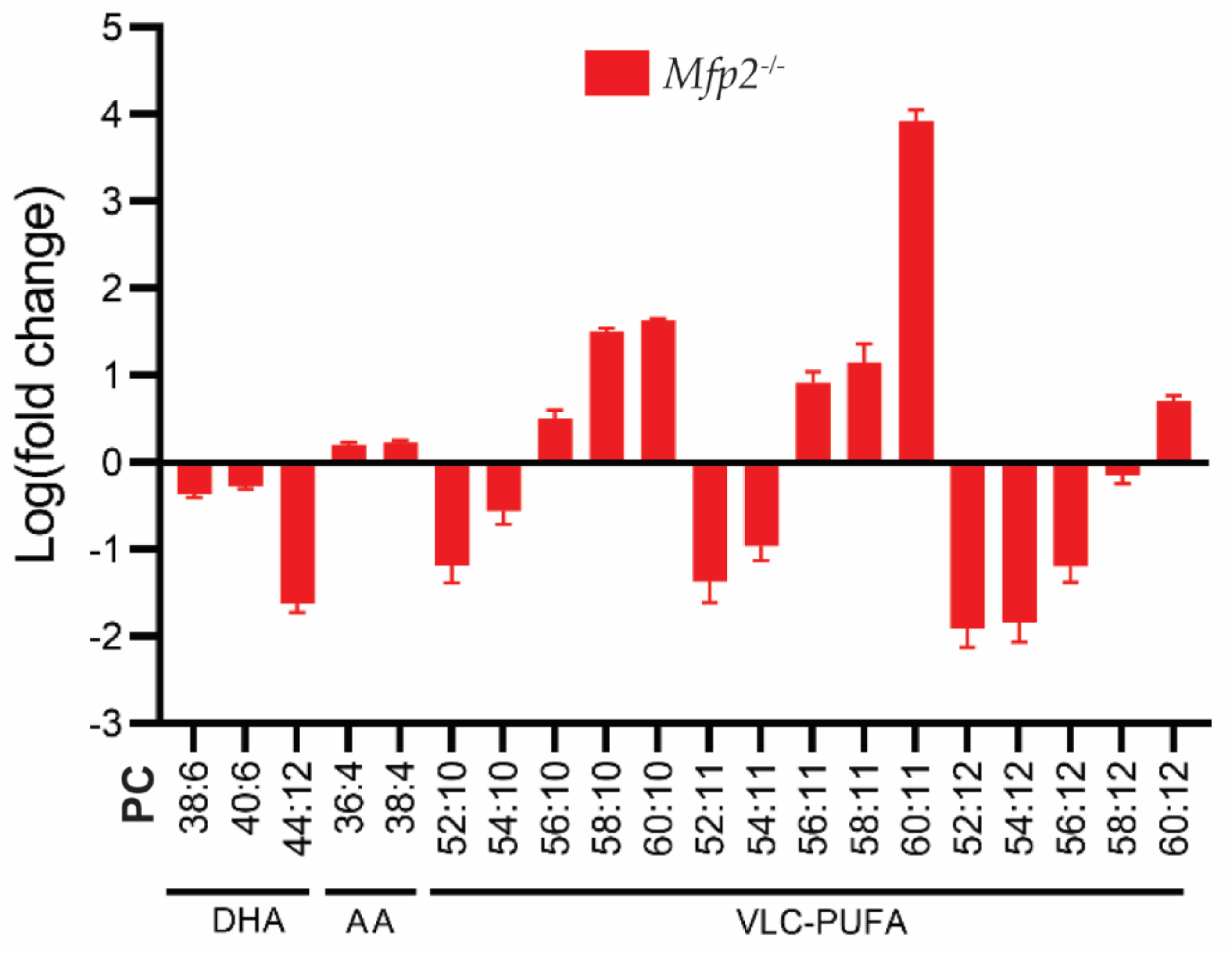
6.3.3. Retinal PUFA Status of Mfp2−/− Mice
6.3.4. The Origin of the Altered PUFA Profile
6.3.5. Link between the Altered PUFA Profile and Retinal Phenotype
7. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Folz, S.J.; Trobe, J.D. The peroxisome and the eye. Surv. Ophthalmol. 1991, 35, 353–368. [Google Scholar] [CrossRef]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef]
- Molday, R.S.; Moritz, O.L. Photoreceptors at a glance. J. Cell Sci. 2015, 128, 4039–4045. [Google Scholar] [CrossRef]
- Baehr, W.; Hanke-Gogokhia, C.; Sharif, A.; Reed, M.; Dahl, T.; Frederick, J.M.; Ying, G. Insights into photoreceptor ciliogenesis revealed by animal models. Prog. Retin. Eye Res. 2019, 71, 26–56. [Google Scholar] [CrossRef]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef]
- Lakkaraju, A.; Umapathy, A.; Tan, L.X.; Daniele, L.; Philp, N.J.; Boesze-Battaglia, K.; Williams, D.S. The cell biology of the retinal pigment epithelium. Prog. Retin. Eye Res. 2020, 100846. [Google Scholar] [CrossRef]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife 2017, 6. [Google Scholar] [CrossRef]
- Islinger, M.; Cardoso, M.J.; Schrader, M. Be different—The diversity of peroxisomes in the animal kingdom. Biochim. Biophys. Acta 2010, 1803, 881–897. [Google Scholar] [CrossRef]
- Robison, W.G., Jr.; Kuwabara, T. Microperoxisomes in retinal pigment epithelium. Investig. Ophthalmol. 1975, 14, 866–872. [Google Scholar]
- Leuenberger, P.M.; Novikoff, A.B. Studies on microperoxisomes. VII. Pigment epithelial cells and other cell types in the retina of rodents. J. Cell Biol. 1975, 65, 324–334. [Google Scholar] [CrossRef]
- Beard, M.E.; Davies, T.; Holloway, M.; Holtzman, E. Peroxisomes in pigment epithelium and Muller cells of amphibian retina possess D-amino acid oxidase as well as catalase. Exp. Eye Res. 1988, 47, 795–806. [Google Scholar] [CrossRef]
- Deguchi, J.; Yamamoto, A.; Fujiki, Y.; Uyama, M.; Tsukahara, I.; Tashiro, Y. Localization of nonspecific lipid transfer protein (nsLTP = sterol carrier protein 2) and acyl-CoA oxidase in peroxisomes of pigment epithelial cells of rat retina. J. Histochem. Cytochem. 1992, 40, 403–410. [Google Scholar] [CrossRef]
- Hazlett, L.D.; Hazlett, J.C.; Ireland, M.; Bradley, R.H. Microperoxisomes in retinal epithelium and tapetum lucidum of the American opossum. Exp. Eye Res. 1978, 27, 343–348. [Google Scholar] [CrossRef]
- Atalla, L.; Fernandez, M.A.; Rao, N.A. Immunohistochemical localization of catalase in ocular tissue. Curr. Eye Res. 1987, 6, 1181–1187. [Google Scholar] [CrossRef]
- Das, Y.; Roose, N.; De Groef, L.; Fransen, M.; Moons, L.; Van Veldhoven, P.P.; Baes, M. Differential distribution of peroxisomal proteins points to specific roles of peroxisomes in the murine retina. Mol. Cell. Biochem. 2019, 456, 53–62. [Google Scholar] [CrossRef]
- Argyriou, C.; Polosa, A.; Cecyre, B.; Hsieh, M.; Di Pietro, E.; Cui, W.; Bouchard, J.F.; Lachapelle, P.; Braverman, N. A longitudinal study of retinopathy in the PEX1-Gly844Asp mouse model for mild Zellweger Spectrum Disorder. Exp. Eye Res. 2019, 186, 107713. [Google Scholar] [CrossRef]
- Zaki, M.S.; Heller, R.; Thoenes, M.; Nurnberg, G.; Stern-Schneider, G.; Nurnberg, P.; Karnati, S.; Swan, D.; Fateen, E.; Nagel-Wolfrum, K.; et al. PEX6 is Expressed in Photoreceptor Cilia and Mutated in Deafblindness with Enamel Dysplasia and Microcephaly. Hum. Mutat. 2016, 37, 170–174. [Google Scholar] [CrossRef]
- Smith, C.E.; Poulter, J.A.; Levin, A.V.; Capasso, J.E.; Price, S.; Ben-Yosef, T.; Sharony, R.; Newman, W.G.; Shore, R.C.; Brookes, S.J.; et al. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur. J. Hum. Genet. 2016, 24, 1565–1571. [Google Scholar] [CrossRef]
- Daniele, L.L.; Caughey, J.; Volland, S.; Sharp, R.C.; Dhingra, A.; Williams, D.S.; Philp, N.J.; Boesze-Battaglia, K. Peroxisome turnover and diurnal modulation of antioxidant activity in retinal pigment epithelia utilizes microtubule-associated protein 1 light chain 3B (LC3B). Am. J. Physiol. Cell Physiol. 2019, 317, C1194–C1204. [Google Scholar] [CrossRef]
- Das, Y.; Swinkels, D.; Kocherlakota, S.; Vinckier, S.; Vaz, F.M.; Wever, E.; van Kampen, A.H.C.; Jun, B.; Do, K.V.; Moons, L.; et al. Peroxisomal Multifunctional Protein 2 Deficiency Perturbs Lipid Homeostasis in the Retina and Causes Visual Dysfunction in Mice. Front. Cell Dev. Biol. 2021, 9, 632930. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Morita, M. ABC Transporter Subfamily D: Distinct Differences in Behavior between ABCD1-3 and ABCD4 in Subcellular Localization, Function, and Human Disease. Biomed. Res. Int. 2016, 2016, 6786245. [Google Scholar] [CrossRef]
- Lo, W.K.; Bernstein, M.H. Daily patterns of the retinal pigment epithelium. Microperoxisomes and phagosomes. Exp. Eye Res. 1981, 32, 1–10. [Google Scholar] [CrossRef]
- Argyriou, C.; D’Agostino, M.D.; Braverman, N. Peroxisome biogenesis disorders. Transl. Sci. Rare Dis. 2016, 1, 111–144. [Google Scholar] [CrossRef]
- Ebberink, M.S.; Mooijer, P.A.; Gootjes, J.; Koster, J.; Wanders, R.J.; Waterham, H.R. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum. Mutat. 2011, 32, 59–69. [Google Scholar] [CrossRef]
- Berendse, K.; Engelen, M.; Ferdinandusse, S.; Majoie, C.B.; Waterham, H.R.; Vaz, F.M.; Koelman, J.H.; Barth, P.G.; Wanders, R.J.; Poll-The, B.T. Zellweger spectrum disorders: Clinical manifestations in patients surviving into adulthood. J. Inherit. Metab. Dis. 2016, 39, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Courtney, R.J.; Pennesi, M.E. Interval spectral-domain optical coherence tomography and electrophysiology findings in neonatal adrenoleukodystrophy. JAMA Ophthalmol. 2013, 131, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Noguer, M.T.; Martinez, M. Visual follow-up in peroxisomal-disorder patients treated with docosahexaenoic Acid ethyl ester. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2277–2285. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.R.; Kriss, A.; Taylor, D.; Coffey, R.; Pembrey, M. Follow-up and diagnostic reappraisal of 75 patients with Leber’s congenital amaurosis. Am. J. Ophthalmol. 1989, 107, 624–631. [Google Scholar] [CrossRef]
- Majewski, J.; Wang, Z.; Lopez, I.; Al Humaid, S.; Ren, H.; Racine, J.; Bazinet, A.; Mitchel, G.; Braverman, N.; Koenekoop, R.K. A new ocular phenotype associated with an unexpected but known systemic disorder and mutation: Novel use of genomic diagnostics and exome sequencing. J. Med. Genet. 2011, 48, 593–596. [Google Scholar] [CrossRef]
- Michelakakis, H.M.; Zafeiriou, D.I.; Moraitou, M.S.; Gootjes, J.; Wanders, R.J. PEX1 deficiency presenting as Leber congenital amaurosis. Pediatr. Neurol. 2004, 31, 146–149. [Google Scholar] [CrossRef]
- Raas-Rothschild, A.; Wanders, R.J.; Mooijer, P.A.; Gootjes, J.; Waterham, H.R.; Gutman, A.; Suzuki, Y.; Shimozawa, N.; Kondo, N.; Eshel, G.; et al. A PEX6-defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents. Am. J. Hum. Genet. 2002, 70, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, G.; Sanchez-Navarro, I.; Aller, E.; Jaijo, T.; Fuster-Garcia, C.; Rodriguez-Munoz, A.; Vallejo, E.; Telleria, J.J.; Vazquez, S.; Beltran, S.; et al. Exome sequencing identifies PEX6 mutations in three cases diagnosed with Retinitis Pigmentosa and hearing impairment. Mol. Vis. 2020, 26, 216–225. [Google Scholar]
- Barillari, M.R.; Karali, M.; Di Iorio, V.; Contaldo, M.; Piccolo, V.; Esposito, M.; Costa, G.; Argenziano, G.; Serpico, R.; Carotenuto, M.; et al. Mild form of Zellweger Spectrum Disorders (ZSD) due to variants in PEX1: Detailed clinical investigation in a 9-years-old female. Mol. Genet. Metab. Rep. 2020, 24, 100615. [Google Scholar] [CrossRef]
- Daich Varela, M.; Jani, P.; Zein, W.M.; D’Souza, P.; Wolfe, L.; Chisholm, J.; Zalewski, C.; Adams, D.; Warner, B.M.; Huryn, L.A.; et al. The peroxisomal disorder spectrum and Heimler syndrome: Deep phenotyping and review of the literature. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 618–630. [Google Scholar] [CrossRef]
- De Munter, S.; Verheijden, S.; Regal, L.; Baes, M. Peroxisomal Disorders: A Review on Cerebellar Pathologies. Brain Pathol. 2015, 25, 663–678. [Google Scholar] [CrossRef]
- Cohen, S.M.; Brown, F.R., 3rd; Martyn, L.; Moser, H.W.; Chen, W.; Kistenmacher, M.; Punnett, H.; de la Cruz, Z.C.; Chan, N.R.; Green, W.R. Ocular histopathologic and biochemical studies of the cerebrohepatorenal syndrome (Zellweger’s syndrome) and its relationship to neonatal adrenoleukodystrophy. Am. J. Ophthalmol. 1983, 96, 488–501. [Google Scholar] [CrossRef]
- Glasgow, B.J.; Brown, H.H.; Hannah, J.B.; Foos, R.Y. Ocular pathologic findings in neonatal adrenoleukodystrophy. Ophthalmology 1987, 94, 1054–1060. [Google Scholar] [CrossRef]
- Tao, L.W.; Wu, Z.; Guymer, R.H.; Luu, C.D. Ellipsoid zone on optical coherence tomography: A review. Clin. Exp. Ophthalmol. 2016, 44, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Ratbi, I.; Jaouad, I.C.; Elorch, H.; Al-Sheqaih, N.; Elalloussi, M.; Lyahyai, J.; Berraho, A.; Newman, W.G.; Sefiani, A. Severe early onset retinitis pigmentosa in a Moroccan patient with Heimler syndrome due to novel homozygous mutation of PEX1 gene. Eur. J. Med. Genet. 2016, 59, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.J.; Hu, F.Y.; Xu, P.; Qi, Y.H.; Li, J.K.; Zhang, Y.J.; Chen, F.; Chang, Q.; Song, F.; Shen, S.M.; et al. Expanding the clinical and genetic spectrum of Heimler syndrome. Orphanet J. Rare Dis. 2019, 14, 290. [Google Scholar] [CrossRef] [PubMed]
- Wangtiraumnuay, N.; Alnabi, W.A.; Tsukikawa, M.; Thau, A.; Capasso, J.; Sharony, R.; Inglehearn, C.F.; Levin, A.V. Ophthalmic manifestations of Heimler syndrome due to PEX6 mutations. Ophthalmic Genet. 2018, 39, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.H.; Barbazetto, I.A.; Chen, R.; Yannuzzi, L.A.; Tsang, S.H.; Spaide, R.F. Macular dystrophy in Heimler syndrome. Ophthalmic Genet. 2011, 32, 97–100. [Google Scholar] [CrossRef]
- Hiebler, S.; Masuda, T.; Hacia, J.G.; Moser, A.B.; Faust, P.L.; Liu, A.; Chowdhury, N.; Huang, N.; Lauer, A.; Bennett, J.; et al. The Pex1-G844D mouse: A model for mild human Zellweger spectrum disorder. Mol. Genet. Metab. 2014, 111, 522–532. [Google Scholar] [CrossRef]
- Jones, B.W.; Watt, C.B.; Marc, R.E. Retinal remodelling. Clin. Exp. Optom. 2005, 88, 282–291. [Google Scholar] [CrossRef]
- Ruether, K.; Baldwin, E.; Casteels, M.; Feher, M.D.; Horn, M.; Kuranoff, S.; Leroy, B.P.; Wanders, R.J.; Wierzbicki, A.S. Adult Refsum disease: A form of tapetoretinal dystrophy accessible to therapy. Surv. Ophthalmol. 2010, 55, 531–538. [Google Scholar] [CrossRef]
- Kemp, S.; Berger, J.; Aubourg, P. X-linked adrenoleukodystrophy: Clinical, metabolic, genetic and pathophysiological aspects. Biochim. Biophys. Acta 2012, 1822, 1465–1474. [Google Scholar] [CrossRef]
- Kaplan, P.W.; Kruse, B.; Tusa, R.J.; Shankroff, J.; Rignani, J.; Moser, H.W. Visual system abnormalities in adrenomyeloneuropathy. Ann. Neurol. 1995, 37, 550–552. [Google Scholar] [CrossRef]
- Ohkuma, Y.; Hayashi, T.; Yoshimine, S.; Tsuneoka, H.; Terao, Y.; Akiyama, M.; Ida, H.; Ohashi, T.; Okumura, A.; Ebihara, N.; et al. Retinal Ganglion Cell Loss in X-linked Adrenoleukodystrophy with an ABCD1 Mutation (Gly266Arg). Neuroophthalmology 2014, 38, 331–335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Traboulsi, E.I.; Maumenee, I.H. Ophthalmologic manifestations of X-linked childhood adrenoleukodystrophy. Ophthalmology 1987, 94, 47–52. [Google Scholar] [CrossRef]
- Grainger, B.T.; Papchenko, T.L.; Danesh-Meyer, H.V. Optic nerve atrophy in adrenoleukodystrophy detectable by optic coherence tomography. J. Clin. Neurosci. 2010, 17, 122–124. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Jimenez-Sanchez, G.; Koster, J.; Denis, S.; Van Roermund, C.W.; Silva-Zolezzi, I.; Moser, A.B.; Visser, W.F.; Gulluoglu, M.; Durmaz, O.; et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum. Mol. Genet. 2015, 24, 361–370. [Google Scholar] [CrossRef]
- Haugarvoll, K.; Johansson, S.; Tzoulis, C.; Haukanes, B.I.; Bredrup, C.; Neckelmann, G.; Boman, H.; Knappskog, P.M.; Bindoff, L.A. MRI characterisation of adult onset alpha-methylacyl-coA racemase deficiency diagnosed by exome sequencing. Orphanet J. Rare Dis. 2013, 8, 1. [Google Scholar] [CrossRef]
- Stewart, M.W.; Vavra, M.W.; Whaley, N.R. Fundus Findings in a Patient with alpha-methlyacyl-coa racemase deficiency. Retin. Cases Brief Rep. 2011, 5, 262–266. [Google Scholar] [CrossRef]
- Kapina, V.; Sedel, F.; Truffert, A.; Horvath, J.; Wanders, R.J.; Waterham, H.R.; Picard, F. Relapsing rhabdomyolysis due to peroxisomal alpha-methylacyl-coa racemase deficiency. Neurology 2010, 75, 1300–1302. [Google Scholar] [CrossRef]
- Smith, E.H.; Gavrilov, D.K.; Oglesbee, D.; Freeman, W.D.; Vavra, M.W.; Matern, D.; Tortorelli, S. An adult onset case of alpha-methyl-acyl-CoA racemase deficiency. J. Inherit. Metab. Dis. 2010, 33 (Suppl. S3), S349–S353. [Google Scholar] [CrossRef]
- Thompson, S.A.; Calvin, J.; Hogg, S.; Ferdinandusse, S.; Wanders, R.J.; Barker, R.A. Relapsing encephalopathy in a patient with alpha-methylacyl-CoA racemase deficiency. BMJ Case Rep. 2009, 2009, bcr08.2008.0814. [Google Scholar] [CrossRef]
- Clarke, C.E.; Alger, S.; Preece, M.A.; Burdon, M.A.; Chavda, S.; Denis, S.; Ferdinandusse, S.; Wanders, R.J. Tremor and deep white matter changes in alpha-methylacyl-CoA racemase deficiency. Neurology 2004, 63, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Dick, D.; Horvath, R.; Chinnery, P.F. AMACR mutations cause late-onset autosomal recessive cerebellar ataxia. Neurology 2011, 76, 1768–1770. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Denis, S.; Clayton, P.T.; Graham, A.; Rees, J.E.; Allen, J.T.; McLean, B.N.; Brown, A.Y.; Vreken, P.; Waterham, H.R.; et al. Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Nat. Genet. 2000, 24, 188–191. [Google Scholar] [CrossRef]
- McLean, B.N.; Allen, J.; Ferdinandusse, S.; Wanders, R.J. A new defect of peroxisomal function involving pristanic acid: A case report. J. Neurol. Neurosurg. Psychiatry 2002, 72, 396–399. [Google Scholar] [CrossRef]
- Carrozzo, R.; Bellini, C.; Lucioli, S.; Deodato, F.; Cassandrini, D.; Cassanello, M.; Caruso, U.; Rizzo, C.; Rizza, T.; Napolitano, M.L.; et al. Peroxisomal acyl-CoA-oxidase deficiency: Two new cases. Am. J. Med. Genet. A 2008, 146A, 1676–1681. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Hogenhout, E.M.; Koster, J.; van Roermund, C.W.T.; IJlst, L.; Moser, A.B.; Wanders, R.J.; Waterham, H.R. Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency. Hum. Mutat. 2007, 28, 904–912. [Google Scholar] [CrossRef]
- Suzuki, Y.; Iai, M.; Kamei, A.; Tanabe, Y.; Chida, S.; Yamaguchi, S.; Zhang, Z.; Takemoto, Y.; Shimozawa, N.; Kondo, N. Peroxisomal acyl CoA oxidase deficiency. J. Pediatr. 2002, 140, 128–130. [Google Scholar] [CrossRef]
- Suzuki, Y.; Shimozawa, N.; Yajima, S.; Tomatsu, S.; Kondo, N.; Nakada, Y.; Akaboshi, S.; Lai, M.; Tanabe, Y.; Hashimoto, T.; et al. Novel subtype of peroxisomal acyl-CoA oxidase deficiency and bifunctional enzyme deficiency with detectable enzyme protein: Identification by means of complementation analysis. Am. J. Hum. Genet. 1994, 54, 36–43. [Google Scholar]
- Wang, R.Y.; Monuki, E.S.; Powers, J.; Schwartz, P.H.; Watkins, P.A.; Shi, Y.; Moser, A.; Shrier, D.A.; Waterham, H.R.; Nugent, D.J.; et al. Effects of hematopoietic stem cell transplantation on acyl-CoA oxidase deficiency: A sibling comparison study. J. Inherit. Metab. Dis. 2014, 37, 791–799. [Google Scholar] [CrossRef]
- Kurian, M.A.; Ryan, S.; Besley, G.T.; Wanders, R.J.; King, M.D. Straight-chain acyl-CoA oxidase deficiency presenting with dysmorphia, neurodevelopmental autistic-type regression and a selective pattern of leukodystrophy. J. Inherit. Metab. Dis. 2004, 27, 105–108. [Google Scholar] [CrossRef]
- Poll-The, B.T.; Roels, F.; Ogier, H.; Scotto, J.; Vamecq, J.; Schutgens, R.B.; Wanders, R.J.; van Roermund, C.W.; van Wijland, M.J.; Schram, A.W.; et al. A new peroxisomal disorder with enlarged peroxisomes and a specific deficiency of acyl-CoA oxidase (pseudo-neonatal adrenoleukodystrophy). Am. J. Hum. Genet. 1988, 42, 422–434. [Google Scholar]
- Ferdinandusse, S.; Barker, S.; Lachlan, K.; Duran, M.; Waterham, H.R.; Wanders, R.J.; Hammans, S. Adult peroxisomal acyl-coenzyme A oxidase deficiency with cerebellar and brainstem atrophy. J. Neurol. Neurosurg. Psychiatry 2010, 81, 310–312. [Google Scholar] [CrossRef]
- Vilarinho, S.; Sari, S.; Mazzacuva, F.; Bilguvar, K.; Esendagli-Yilmaz, G.; Jain, D.; Akyol, G.; Dalgic, B.; Gunel, M.; Clayton, P.T.; et al. ACOX2 deficiency: A disorder of bile acid synthesis with transaminase elevation, liver fibrosis, ataxia, and cognitive impairment. Proc. Natl. Acad. Sci. USA 2016, 113, 11289–11293. [Google Scholar] [CrossRef]
- Monte, M.J.; Alonso-Pena, M.; Briz, O.; Herraez, E.; Berasain, C.; Argemi, J.; Prieto, J.; Marin, J.J.G. ACOX2 deficiency: An inborn error of bile acid synthesis identified in an adolescent with persistent hypertransaminasemia. J. Hepatol. 2017, 66, 581–588. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; van Roermund, C.W.T.; Preece, M.A.; Koster, J.; Ebberink, M.S.; Waterham, H.R.; Wanders, R.J.A. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta 2018, 1864, 952–958. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Mooyer, P.A.; Dekker, C.; Duran, M.; Soorani-Lunsing, R.J.; Boltshauser, E.; Macaya, A.; Gartner, J.; Majoie, C.B.; et al. Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann. Neurol. 2006, 59, 92–104. [Google Scholar] [CrossRef]
- Amor, D.J.; Marsh, A.P.; Storey, E.; Tankard, R.; Gillies, G.; Delatycki, M.B.; Pope, K.; Bromhead, C.; Leventer, R.J.; Bahlo, M.; et al. Heterozygous mutations in HSD17B4 cause juvenile peroxisomal D-bifunctional protein deficiency. Neurol. Genet. 2016, 2, e114. [Google Scholar] [CrossRef] [PubMed]
- Lines, M.A.; Jobling, R.; Brady, L.; Marshall, C.R.; Scherer, S.W.; Rodriguez, A.R.; Lee, L.; Lang, A.E.; Mestre, T.A.; Wanders, R.J.; et al. Peroxisomal D-bifunctional protein deficiency: Three adults diagnosed by whole-exome sequencing. Neurology 2014, 82, 963–968. [Google Scholar] [CrossRef] [PubMed]
- McMillan, H.J.; Worthylake, T.; Schwartzentruber, J.; Gottlieb, C.C.; Lawrence, S.E.; Mackenzie, A.; Beaulieu, C.L.; Mooyer, P.A.; Wanders, R.J.; Majewski, J.; et al. Specific combination of compound heterozygous mutations in 17beta-hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D-bifunctional protein deficiency. Orphanet J. Rare Dis. 2012, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Kostopoulos, P.; Denis, S.; Rusch, H.; Overmars, H.; Dillmann, U.; Reith, W.; Haas, D.; Wanders, R.J.; Duran, M.; et al. Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Am. J. Hum. Genet. 2006, 78, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Falkenberg, K.D.; Koster, J.; Mooyer, P.A.; Jones, R.; van Roermund, C.W.T.; Pizzino, A.; Schrader, M.; Wanders, R.J.A.; Vanderver, A.; et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J. Med. Genet. 2017, 54, 330–337. [Google Scholar] [CrossRef]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef]
- Bartlett, M.; Nasiri, N.; Pressman, R.; Bademci, G.; Forghani, I. First reported adult patient with retinal dystrophy and leukodystrophy caused by a novel ACBD5 variant: A case report and review of literature. Am. J. Med. Genet. A 2021, 4, 1236–1241. [Google Scholar] [CrossRef]
- Malheiro, A.R.; da Silva, T.F.; Brites, P. Plasmalogens and fatty alcohols in rhizomelic chondrodysplasia punctata and Sjogren-Larsson syndrome. J. Inherit. Metab. Dis. 2015, 38, 111–121. [Google Scholar] [CrossRef]
- Saab, S.; Mazzocco, J.; Creuzot-Garcher, C.P.; Bron, A.M.; Bretillon, L.; Acar, N. Plasmalogens in the retina: From occurrence in retinal cell membranes to potential involvement in pathophysiology of retinal diseases. Biochimie 2014, 107 Pt A, 58–65. [Google Scholar] [CrossRef]
- Braverman, N.; Chen, L.; Lin, P.; Obie, C.; Steel, G.; Douglas, P.; Chakraborty, P.K.; Clarke, J.T.; Boneh, A.; Moser, A.; et al. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum. Mutat. 2002, 20, 284–297. [Google Scholar] [CrossRef]
- Van den Brink, D.M.; Brites, P.; Haasjes, J.; Wierzbicki, A.S.; Mitchell, J.; Lambert-Hamill, M.; de Belleroche, J.; Jansen, G.A.; Waterham, H.R.; Wanders, R.J. Identification of PEX7 as the second gene involved in Refsum disease. Am. J. Hum. Genet. 2003, 72, 471–477. [Google Scholar] [CrossRef]
- Bernstein, P.S.; Lloyd, M.B.; O’Day, W.T.; Bok, D. Effect of phytanic acid on cultured retinal pigment epithelium: An in vitro model for Refsum’s disease. Exp. Eye Res. 1992, 55, 869–878. [Google Scholar] [CrossRef]
- Young, S.P.; Johnson, A.W.; Muller, D.P. Effects of phytanic acid on the vitamin E status, lipid composition and physical properties of retinal cell membranes: Implications for adult Refsum disease. Clin. Sci. 2001, 101, 697–705. [Google Scholar] [CrossRef]
- Steinberg, D.; Avigan, J.; Mize, C.E.; Baxter, J.H.; Cammermeyer, J.; Fales, H.M.; Highet, P.F. Effects of dietary phytol and phytanic acid in animals. J. Lipid Res. 1966, 7, 684–691. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Zomer, A.W.; Komen, J.C.; van den Brink, C.E.; Thanos, M.; Hamers, F.P.; Wanders, R.J.; van der Saag, P.T.; Poll-The, B.T.; Brites, P. Ataxia with loss of Purkinje cells in a mouse model for Refsum disease. Proc. Natl. Acad. Sci. USA 2008, 105, 17712–17717. [Google Scholar] [CrossRef] [PubMed]
- Violante, S.; Achetib, N.; van Roermund, C.W.T.; Hagen, J.; Dodatko, T.; Vaz, F.M.; Waterham, H.R.; Chen, H.; Baes, M.; Yu, C.; et al. Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 2019, 33, 4355–4364. [Google Scholar] [CrossRef] [PubMed]
- Voss, A.; Reinhart, M.; Sankarappa, S.; Sprecher, H. The metabolism of 7,10,13,16,19-docosapentaenoic acid to 4,7,10,13,16,19-docosahexaenoic acid in rat liver is independent of a 4-desaturase. J. Biol. Chem. 1991, 266, 19995–20000. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Mooijer, P.A.; Zhang, Z.; Reddy, J.K.; Spector, A.A.; Wanders, R.J. Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J. Lipid Res. 2001, 42, 1987–1995. [Google Scholar] [CrossRef]
- Wang, N.; Anderson, R.E. Synthesis of docosahexaenoic acid by retina and retinal pigment epithelium. Biochemistry 1993, 32, 13703–13709. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, N.P.; Pennacchiotti, G.L.; Sprecher, H.; Aveldano, M.I. Active synthesis of C24:5, n-3 fatty acid in retina. Biochem. J. 1996, 316 Pt 3, 859–864. [Google Scholar] [CrossRef][Green Version]
- Simon, M.V.; Agnolazza, D.L.; German, O.L.; Garelli, A.; Politi, L.E.; Agbaga, M.P.; Anderson, R.E.; Rotstein, N.P. Synthesis of docosahexaenoic acid from eicosapentaenoic acid in retina neurons protects photoreceptors from oxidative stress. J. Neurochem. 2016, 136, 931–946. [Google Scholar] [CrossRef]
- Bazan, H.E.; Careaga, M.M.; Sprecher, H.; Bazan, N.G. Chain elongation and desaturation of eicosapentaenoate to docosahexaenoate and phospholipid labeling in the rat retina in vivo. Biochim. Biophys. Acta 1982, 712, 123–128. [Google Scholar] [CrossRef]
- Park, H.G.; Park, W.J.; Kothapalli, K.S.; Brenna, J.T. The fatty acid desaturase 2 (FADS2) gene product catalyzes Delta4 desaturation to yield n-3 docosahexaenoic acid and n-6 docosapentaenoic acid in human cells. FASEB J. 2015, 29, 3911–3919. [Google Scholar] [CrossRef]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef]
- Fan, C.Y.; Pan, J.; Chu, R.; Lee, D.; Kluckman, K.D.; Usuda, N.; Singh, I.; Yeldandi, A.V.; Rao, M.S.; Maeda, N.; et al. Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J. Biol. Chem. 1996, 271, 24698–24710. [Google Scholar] [CrossRef]
- Baes, M.; Huyghe, S.; Carmeliet, P.; Declercq, P.E.; Collen, D.; Mannaerts, G.P.; Van Veldhoven, P.P. Inactivation of the peroxisomal multifunctional protein-2 in mice impedes the degradation of not only 2-methyl-branched fatty acids and bile acid intermediates but also of very long chain fatty acids. J. Biol. Chem. 2000, 275, 16329–16336. [Google Scholar] [CrossRef]
- Setchell, K.D.; Heubi, J.E.; Bove, K.E.; O’Connell, N.C.; Brewsaugh, T.; Steinberg, S.J.; Moser, A.; Squires, R.H., Jr. Liver disease caused by failure to racemize trihydroxycholestanoic acid: Gene mutation and effect of bile acid therapy. Gastroenterology 2003, 124, 217–232. [Google Scholar] [CrossRef]
- Savolainen, K.; Kotti, T.J.; Schmitz, W.; Savolainen, T.I.; Sormunen, R.T.; Ilves, M.; Vainio, S.J.; Conzelmann, E.; Hiltunen, J.K. A mouse model for alpha-methylacyl-CoA racemase deficiency: Adjustment of bile acid synthesis and intolerance to dietary methyl-branched lipids. Hum. Mol. Genet. 2004, 13, 955–965. [Google Scholar] [CrossRef]
- Costello, J.L.; Castro, I.G.; Hacker, C.; Schrader, T.A.; Metz, J.; Zeuschner, D.; Azadi, A.S.; Godinho, L.F.; Costina, V.; Findeisen, P.; et al. ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 2017, 216, 331–342. [Google Scholar] [CrossRef]
- Hua, R.; Cheng, D.; Coyaud, E.; Freeman, S.; Di Pietro, E.; Wang, Y.; Vissa, A.; Yip, C.M.; Fairn, G.D.; Braverman, N.; et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 2017, 216, 367–377. [Google Scholar] [CrossRef]
- Nazarko, T.Y.; Ozeki, K.; Till, A.; Ramakrishnan, G.; Lotfi, P.; Yan, M.; Subramani, S. Peroxisomal Atg37 binds Atg30 or palmitoyl-CoA to regulate phagophore formation during pexophagy. J. Cell Biol. 2014, 204, 541–557. [Google Scholar] [CrossRef]
- Yagita, Y.; Shinohara, K.; Abe, Y.; Nakagawa, K.; Al-Owain, M.; Alkuraya, F.S.; Fujiki, Y. Deficiency of a Retinal Dystrophy Protein, Acyl-CoA Binding Domain-containing 5 (ACBD5), Impairs Peroxisomal beta-Oxidation of Very-long-chain Fatty Acids. J. Biol. Chem. 2017, 292, 691–705. [Google Scholar] [CrossRef]
- Darwisch, W.; von Spangenberg, M.; Lehmann, J.; Singin, Ö.; Deubert, G.; Kühl, S.; Roos, J.; Horstmann, H.; Körber, C.; Hoppe, S.; et al. Cerebellar and hepatic alterations in ACBD5-deficient mice are associated with unexpected, distinct alterations in cellular lipid homeostasis. Commun. Biol. 2020, 3, 713. [Google Scholar] [CrossRef]
- Dorninger, F.; Forss-Petter, S.; Berger, J. From peroxisomal disorders to common neurodegenerative diseases—The role of ether phospholipids in the nervous system. FEBS Lett. 2017, 591, 2761–2788. [Google Scholar] [CrossRef]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [CrossRef]
- Brites, P.; Motley, A.M.; Gressens, P.; Mooyer, P.A.; Ploegaert, I.; Everts, V.; Evrard, P.; Carmeliet, P.; Dewerchin, M.; Schoonjans, L.; et al. Impaired neuronal migration and endochondral ossification in Pex7 knockout mice: A model for rhizomelic chondrodysplasia punctata. Hum. Mol. Genet. 2003, 12, 2255–2267. [Google Scholar] [CrossRef]
- Braverman, N.; Zhang, R.; Chen, L.; Nimmo, G.; Scheper, S.; Tran, T.; Chaudhury, R.; Moser, A.; Steinberg, S. A Pex7 hypomorphic mouse model for plasmalogen deficiency affecting the lens and skeleton. Mol. Genet. Metab. 2010, 99, 408–416. [Google Scholar] [CrossRef]
- Rodemer, C.; Thai, T.P.; Brugger, B.; Kaercher, T.; Werner, H.; Nave, K.A.; Wieland, F.; Gorgas, K.; Just, W.W. Inactivation of ether lipid biosynthesis causes male infertility, defects in eye development and optic nerve hypoplasia in mice. Hum. Mol. Genet. 2003, 12, 1881–1895. [Google Scholar] [CrossRef]
- Liegel, R.; Chang, B.; Dubielzig, R.; Sidjanin, D.J. Blind sterile 2 (bs2), a hypomorphic mutation in Agps, results in cataracts and male sterility in mice. Mol. Genet. Metab. 2011, 103, 51–59. [Google Scholar] [CrossRef]
- Liegel, R.P.; Ronchetti, A.; Sidjanin, D.J. Alkylglycerone phosphate synthase (AGPS) deficient mice: Models for rhizomelic chondrodysplasia punctate type 3 (RCDP3) malformation syndrome. Mol. Genet. Metab. Rep. 2014, 1, 299–311. [Google Scholar] [CrossRef]
- Saab, S.; Buteau, B.; Leclere, L.; Bron, A.M.; Creuzot-Garcher, C.P.; Bretillon, L.; Acar, N. Involvement of plasmalogens in post-natal retinal vascular development. PLoS ONE 2014, 9, e101076. [Google Scholar] [CrossRef]
- Jaspers, Y.R.J.; Ferdinandusse, S.; Dijkstra, I.M.E.; Barendsen, R.W.; van Lenthe, H.; Kulik, W.; Engelen, M.; Goorden, S.M.I.; Vaz, F.M.; Kemp, S. Comparison of the Diagnostic Performance of C26:0-Lysophosphatidylcholine and Very Long-Chain Fatty Acids Analysis for Peroxisomal Disorders. Front. Cell Dev. Biol. 2020, 8, 690. [Google Scholar] [CrossRef]
- Huyghe, S.; Schmalbruch, H.; Hulshagen, L.; Veldhoven, P.V.; Baes, M.; Hartmann, D. Peroxisomal multifunctional protein-2 deficiency causes motor deficits and glial lesions in the adult central nervous system. Am. J. Pathol. 2006, 168, 1321–1334. [Google Scholar] [CrossRef][Green Version]
- Fliesler, S.J.; Anderson, R.E. Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid Res. 1983, 22, 79–131. [Google Scholar] [CrossRef]
- Anderson, R.E.; Maude, M.B. Lipids of ocular tissues. 8. The effects of essential fatty acid deficiency on the phospholipids of the photoreceptor membranes of rat retina. Arch. Biochem. Biophys. 1972, 151, 270–276. [Google Scholar] [CrossRef]
- Tinoco, J. Dietary requirements and functions of alpha-linolenic acid in animals. Prog. Lipid Res. 1982, 21, 1–45. [Google Scholar] [CrossRef]
- Anderson, D.M.; Ablonczy, Z.; Koutalos, Y.; Spraggins, J.; Crouch, R.K.; Caprioli, R.M.; Schey, K.L. High resolution MALDI imaging mass spectrometry of retinal tissue lipids. J. Am. Soc. Mass Spectrom. 2014, 25, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Aveldano, M.I. A novel group of very long chain polyenoic fatty acids in dipolyunsaturated phosphatidylcholines from vertebrate retina. J. Biol. Chem. 1987, 262, 1172–1179. [Google Scholar] [CrossRef]
- Aveldano, M.I.; Sprecher, H. Very long chain (C24 to C36) polyenoic fatty acids of the n-3 and n-6 series in dipolyunsaturated phosphatidylcholines from bovine retina. J. Biol. Chem. 1987, 262, 1180–1186. [Google Scholar] [CrossRef]
- Agbaga, M.P.; Mandal, M.N.; Anderson, R.E. Retinal very long-chain PUFAs: New insights from studies on ELOVL4 protein. J. Lipid Res. 2010, 51, 1624–1642. [Google Scholar] [CrossRef]
- Agbaga, M.P.; Merriman, D.K.; Brush, R.S.; Lydic, T.A.; Conley, S.M.; Naash, M.I.; Jackson, S.; Woods, A.S.; Reid, G.E.; Busik, J.V.; et al. Differential composition of DHA and very-long-chain PUFAs in rod and cone photoreceptors. J. Lipid Res. 2018, 59, 1586–1596. [Google Scholar] [CrossRef]
- Bazan, N.G. Overview of how N32 and N34 elovanoids sustain sight by protecting retinal pigment epithelial cells and photoreceptors. J. Lipid Res. 2021, 100058. [Google Scholar] [CrossRef]
- Benolken, R.M.; Anderson, R.E.; Wheeler, T.G. Membrane fatty acids associated with the electrical response in visual excitation. Science 1973, 182, 1253–1254. [Google Scholar] [CrossRef]
- Wheeler, T.G.; Benolken, R.M.; Anderson, R.E. Visual membranes: Specificity of fatty acid precursors for the electrical response to illumination. Science 1975, 188, 1312–1314. [Google Scholar] [CrossRef]
- Senapati, S.; Gragg, M.; Samuels, I.S.; Parmar, V.M.; Maeda, A.; Park, P.S. Effect of dietary docosahexaenoic acid on rhodopsin content and packing in photoreceptor cell membranes. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1403–1413. [Google Scholar] [CrossRef]
- Weisinger, H.S.; Vingrys, A.J.; Sinclair, A.J. Effect of dietary n-3 deficiency on the electroretinogram in the guinea pig. Ann. Nutr. Metab. 1996, 40, 91–98. [Google Scholar] [CrossRef]
- Weisinger, H.S.; Vingrys, A.J.; Bui, B.V.; Sinclair, A.J. Effects of dietary n-3 fatty acid deficiency and repletion in the guinea pig retina. Investig. Ophthalmol. Vis. Sci. 1999, 40, 327–338. [Google Scholar]
- Neuringer, M.; Connor, W.E.; Van Petten, C.; Barstad, L. Dietary omega-3 fatty acid deficiency and visual loss in infant rhesus monkeys. J. Clin. Investig. 1984, 73, 272–276. [Google Scholar] [CrossRef]
- Neuringer, M.; Connor, W.E.; Lin, D.S.; Barstad, L.; Luck, S. Biochemical and functional effects of prenatal and postnatal omega 3 fatty acid deficiency on retina and brain in rhesus monkeys. Proc. Natl. Acad. Sci. USA 1986, 83, 4021–4025. [Google Scholar] [CrossRef]
- Jeffrey, B.G.; Mitchell, D.C.; Gibson, R.A.; Neuringer, M. n-3 fatty acid deficiency alters recovery of the rod photoresponse in rhesus monkeys. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2806–2814. [Google Scholar]
- Uauy, R.D.; Birch, D.G.; Birch, E.E.; Tyson, J.E.; Hoffman, D.R. Effect of dietary omega-3 fatty acids on retinal function of very-low-birth-weight neonates. Pediatr. Res. 1990, 28, 485–492. [Google Scholar] [CrossRef]
- Birch, E.E.; Birch, D.G.; Hoffman, D.R.; Uauy, R. Dietary essential fatty acid supply and visual acuity development. Investig. Ophthalmol. Vis. Sci. 1992, 33, 3242–3253. [Google Scholar]
- Birch, D.G.; Birch, E.E.; Hoffman, D.R.; Uauy, R.D. Retinal development in very-low-birth-weight infants fed diets differing in omega-3 fatty acids. Investig. Ophthalmol. Vis. Sci. 1992, 33, 2365–2376. [Google Scholar]
- O’Connor, D.L.; Hall, R.; Adamkin, D.; Auestad, N.; Castillo, M.; Connor, W.E.; Connor, S.L.; Fitzgerald, K.; Groh-Wargo, S.; Hartmann, E.E.; et al. Growth and development in preterm infants fed long-chain polyunsaturated fatty acids: A prospective, randomized controlled trial. Pediatrics 2001, 108, 359–371. [Google Scholar] [CrossRef]
- Martinez, M. Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney and retina of patients with peroxisomal disorders. Brain Res. 1992, 583, 171–182. [Google Scholar] [CrossRef]
- Paker, A.M.; Sunness, J.S.; Brereton, N.H.; Speedie, L.J.; Albanna, L.; Dharmaraj, S.; Moser, A.B.; Jones, R.O.; Raymond, G.V. Docosahexaenoic acid therapy in peroxisomal diseases: Results of a double-blind, randomized trial. Neurology 2010, 75, 826–830. [Google Scholar] [CrossRef]
- Deak, F.; Anderson, R.E.; Fessler, J.L.; Sherry, D.M. Novel Cellular Functions of Very Long Chain-Fatty Acids: Insight from ELOVL4 Mutations. Front. Cell. Neurosci. 2019, 13, 428. [Google Scholar] [CrossRef]
- Suh, M.; Clandinin, M.T. 20:5n-3 but not 22:6n-3 is a preferred substrate for synthesis of n-3 very-long- chain fatty acids (C24-C36) in retina. Curr. Eye Res. 2005, 30, 959–968. [Google Scholar] [CrossRef]
- Yu, M.; Benham, A.; Logan, S.; Brush, R.S.; Mandal, M.N.; Anderson, R.E.; Agbaga, M.P. ELOVL4 protein preferentially elongates 20:5n3 to very long chain PUFAs over 20:4n6 and 22:6n3. J. Lipid Res. 2012, 53, 494–504. [Google Scholar] [CrossRef]
- Bennett, L.D.; Hopiavuori, B.R.; Brush, R.S.; Chan, M.; Van Hook, M.J.; Thoreson, W.B.; Anderson, R.E. Examination of VLC-PUFA-deficient photoreceptor terminals. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4063–4072. [Google Scholar] [CrossRef]
- Logan, S.; Anderson, R.E. Dominant Stargardt Macular Dystrophy (STGD3) and ELOVL4. Adv. Exp. Med. Biol. 2014, 801, 447–453. [Google Scholar] [CrossRef]
- Poulos, A.; Sharp, P.; Johnson, D.; Easton, C. The occurrence of polyenoic very long chain fatty acids with greater than 32 carbon atoms in molecular species of phosphatidylcholine in normal and peroxisome-deficient (Zellweger’s syndrome) brain. Biochem. J. 1988, 253, 645–650. [Google Scholar] [CrossRef]
- Poulos, A.; Sharp, P.; Singh, H.; Johnson, D.; Fellenberg, A.; Pollard, A. Detection of a homologous series of C26-C38 polyenoic fatty acids in the brain of patients without peroxisomes (Zellweger’s syndrome). Biochem. J. 1986, 235, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.; Poulos, A.; Fellenberg, A.; Johnson, D. Structure and lipid distribution of polyenoic very-long-chain fatty acids in the brain of peroxisome-deficient patients (Zellweger syndrome). Biochem. J. 1987, 248, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.S.; Calandria, J.M.; Gordon, W.C.; Jun, B.; Zhou, Y.; Gelfman, C.M.; Li, S.; Jin, M.; Knott, E.J.; Chang, B.; et al. Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell survival. Nat. Commun. 2015, 6, 6228. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.H.; Chan, J.P.; Cazenave-Gassiot, A.; Poh, R.W.; Foo, J.C.; Galam, D.L.; Ghosh, S.; Nguyen, L.N.; Barathi, V.A.; Yeo, S.W.; et al. Mfsd2a Is a Transporter for the Essential omega-3 Fatty Acid Docosahexaenoic Acid (DHA) in Eye and Is Important for Photoreceptor Cell Development. J. Biol. Chem. 2016, 291, 10501–10514. [Google Scholar] [CrossRef] [PubMed]
- Lobanova, E.S.; Schuhmann, K.; Finkelstein, S.; Lewis, T.R.; Cady, M.A.; Hao, Y.; Keuthan, C.; Ash, J.D.; Burns, M.E.; Shevchenko, A.; et al. Disrupted Blood-Retina Lysophosphatidylcholine Transport Impairs Photoreceptor Health but Not Visual Signal Transduction. J. Neurosci. 2019, 39, 9689–9701. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Reveles, J.; Dhingra, A.; Alexander, D.; Bragin, A.; Philp, N.J.; Boesze-Battaglia, K. Phagocytosis-dependent ketogenesis in retinal pigment epithelium. J. Biol. Chem. 2017, 292, 8038–8047. [Google Scholar] [CrossRef]
- German, O.L.; Agnolazza, D.L.; Politi, L.E.; Rotstein, N.P. Light, lipids and photoreceptor survival: Live or let die? Photochem. Photobiol. Sci. 2015, 14, 1737–1753. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Marcheselli, V.L.; Serhan, C.N.; Bazan, N.G. Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. USA 2004, 101, 8491–8496. [Google Scholar] [CrossRef]
- Jun, B.; Mukherjee, P.K.; Asatryan, A.; Kautzmann, M.A.; Heap, J.; Gordon, W.C.; Bhattacharjee, S.; Yang, R.; Petasis, N.A.; Bazan, N.G. Elovanoids are novel cell-specific lipid mediators necessary for neuroprotective signaling for photoreceptor cell integrity. Sci. Rep. 2017, 7, 5279. [Google Scholar] [CrossRef]
- Kautzmann, M.I.; Gordon, W.C.; Jun, B.; Do, K.V.; Matherne, B.J.; Fang, Z.; Bazan, N.G. Membrane-type frizzled-related protein regulates lipidome and transcription for photoreceptor function. FASEB J. 2020, 34, 912–929. [Google Scholar] [CrossRef]
- Shindou, H.; Koso, H.; Sasaki, J.; Nakanishi, H.; Sagara, H.; Nakagawa, K.M.; Takahashi, Y.; Hishikawa, D.; Iizuka-Hishikawa, Y.; Tokumasu, F.; et al. Docosahexaenoic acid preserves visual function by maintaining correct disc morphology in retinal photoreceptor cells. J. Biol. Chem. 2017, 292, 12054–12064. [Google Scholar] [CrossRef]
- Handa, J.T. How does the macula protect itself from oxidative stress? Mol. Asp. Med. 2012, 33, 418–435. [Google Scholar] [CrossRef]
- Abe, S.; Nagai, T.; Masukawa, M.; Okumoto, K.; Homma, Y.; Fujiki, Y.; Mizuno, K. Localization of Protein Kinase NDR2 to Peroxisomes and Its Role in Ciliogenesis. J. Biol. Chem. 2017, 292, 4089–4098. [Google Scholar] [CrossRef]
- Miyamoto, T.; Hosoba, K.; Itabashi, T.; Iwane, A.H.; Akutsu, S.N.; Ochiai, H.; Saito, Y.; Yamamoto, T.; Matsuura, S. Insufficiency of ciliary cholesterol in hereditary Zellweger syndrome. EMBO J. 2020, 39, e103499. [Google Scholar] [CrossRef]
- Albert, A.; Alexander, D.; Boesze-Battaglia, K. Cholesterol in the rod outer segment: A complex role in a “simple” system. Chem. Phys. Lipids 2016, 199, 94–105. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Peroxisomal Disorder | Biochemical Abnormalities | Ocular Phenotype | |||||
|---|---|---|---|---|---|---|---|---|
| VLCFA | DHA | PRIS | PHYT | D/THCA | PL | |||
| Peroxisome biogenesis disorders | Zellweger spectrum disorders 1 [4,26,28,29,30,31,33,37,40,41] | ↑ | ↓ | ↑ | ↑ | ↑ | ↓ | - varied pigmentary retinopathy, including retinitis pigmentosa - optic atrophy, cataract, glaucoma and nystagmus |
| RCDP type 1 and 5 [3,26] | - | - | - | - | - | ↓ | - retina is spared - cataract | |
| Single enzyme deficiencies | Refsum disease [49] | - | - | - | ↑ | - | - | - retinitis pigmentosa is the cardinal symptom - miosis, attenuated pupillary light responses, iris atrophy and cataract |
| X-ALD [50,51,52,53,54] | ↑ | - | - | - | - | - | - retina is spared - decreased visual acuity due to extensive brain lesions and demyelination of visual tract | |
| ABCD3 deficiency [55] | - | - | - | - | ↑ | - | - no ocular symptoms | |
| AMACR deficiency [56,57,58,59,60,61,62,63,64] | - | - | ↑ | (↑) | ↑ | - | - retinitis pigmentosa - optic atrophy, cataract and visual field defects | |
| ACOX1 deficiency [65,66,67,68,69,70,71,72] | ↑ | NK | - | - | - | - | - pigmentary retinopathy - optic atrophy | |
| ACOX2 deficiency [73,74,75] | - | - | - | - | ↑ | - | - no ocular symptoms | |
| MFP2 deficiency 2 [76,77,78,79] | ↑ | ↓ | ↑ | ↑ | ↑ | - | - pigmentary retinopathy - optic atrophy, cataract | |
| SCPx deficiency [80] | - | - | ↑ | - | (↑) | - | - no ocular symptoms | |
| ACBD5 deficiency [81,82,83] | ↑ | - | - | - | - | - | - cone-rod dystrophy and retinal pigmentary changes - optic atrophy | |
| RCDP type 2, 3 and 4 [84,85] | - | - | - | - | - | ↓ | - retina is spared - cataract | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, Y.; Swinkels, D.; Baes, M. Peroxisomal Disorders and Their Mouse Models Point to Essential Roles of Peroxisomes for Retinal Integrity. Int. J. Mol. Sci. 2021, 22, 4101. https://doi.org/10.3390/ijms22084101
Das Y, Swinkels D, Baes M. Peroxisomal Disorders and Their Mouse Models Point to Essential Roles of Peroxisomes for Retinal Integrity. International Journal of Molecular Sciences. 2021; 22(8):4101. https://doi.org/10.3390/ijms22084101
Chicago/Turabian StyleDas, Yannick, Daniëlle Swinkels, and Myriam Baes. 2021. "Peroxisomal Disorders and Their Mouse Models Point to Essential Roles of Peroxisomes for Retinal Integrity" International Journal of Molecular Sciences 22, no. 8: 4101. https://doi.org/10.3390/ijms22084101
APA StyleDas, Y., Swinkels, D., & Baes, M. (2021). Peroxisomal Disorders and Their Mouse Models Point to Essential Roles of Peroxisomes for Retinal Integrity. International Journal of Molecular Sciences, 22(8), 4101. https://doi.org/10.3390/ijms22084101