
Impact of Stress on Epilepsy: Focus on Neuroinflammation—A Mini Review


Abstract
1. Introduction
2. Acute Stress in Epilepsy
2.1. Acute Stress Effects on Epilepsy in Humans
2.2. Acute Stress in Animal Models of Epilepsy
2.2.1. Acute Stress in Epileptogenesis
2.2.2. Acute Repetitive Stress in Epileptogenesis
2.2.3. Acute Stress in Chronic Phase of Epilepsy
2.2.4. Acute Repetitive Stress in Chronic Phase of Epilepsy
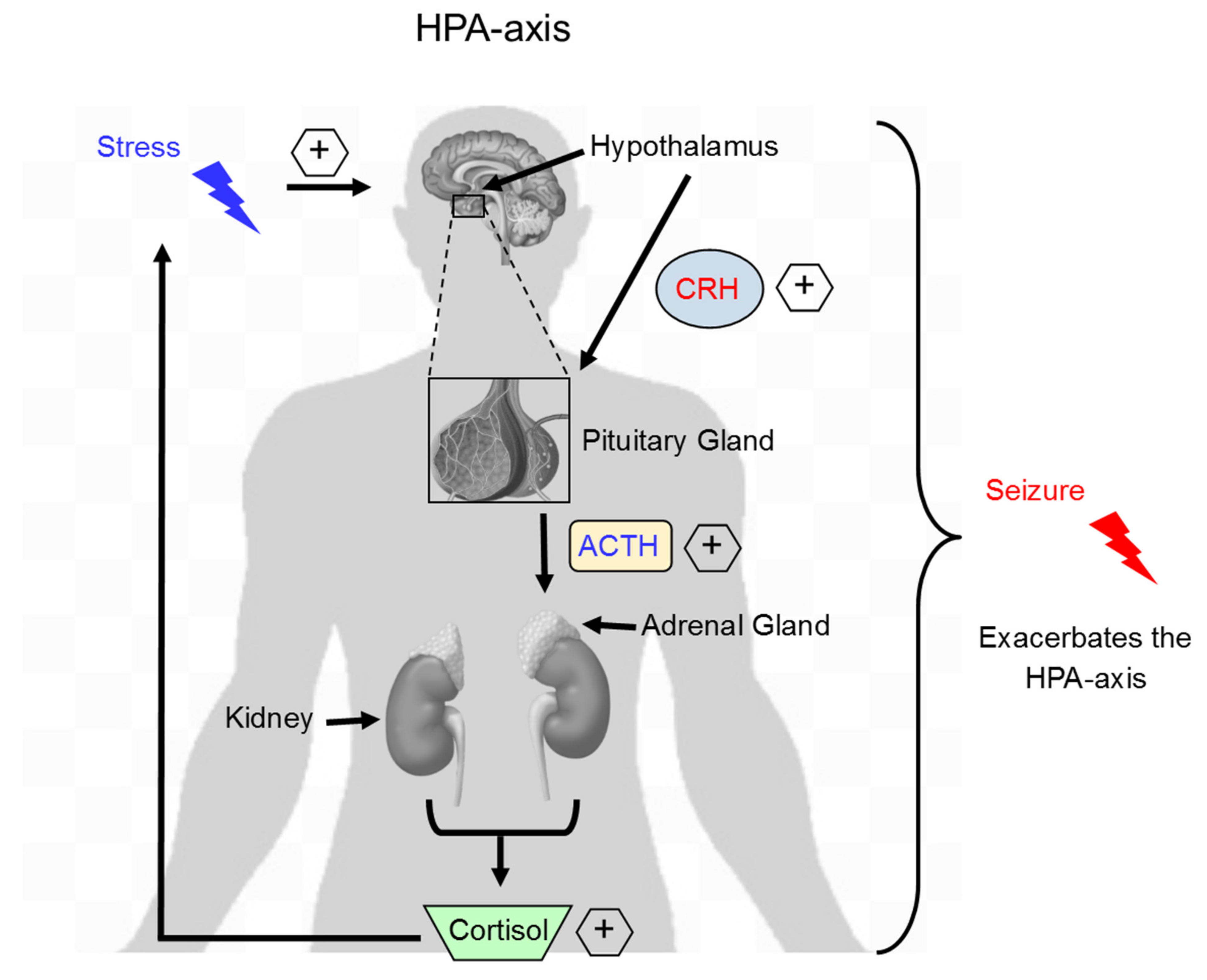
2.3. Acute Stress and Hormonal Changes
2.4. Neuroinflammation and Acute Stress in Epilepsy
3. Chronic Stress in Epilepsy
3.1. Chronic Stress and Seizure Occurrence in Humans
3.2. Chronic Stress in Animal Models of Epilepsy
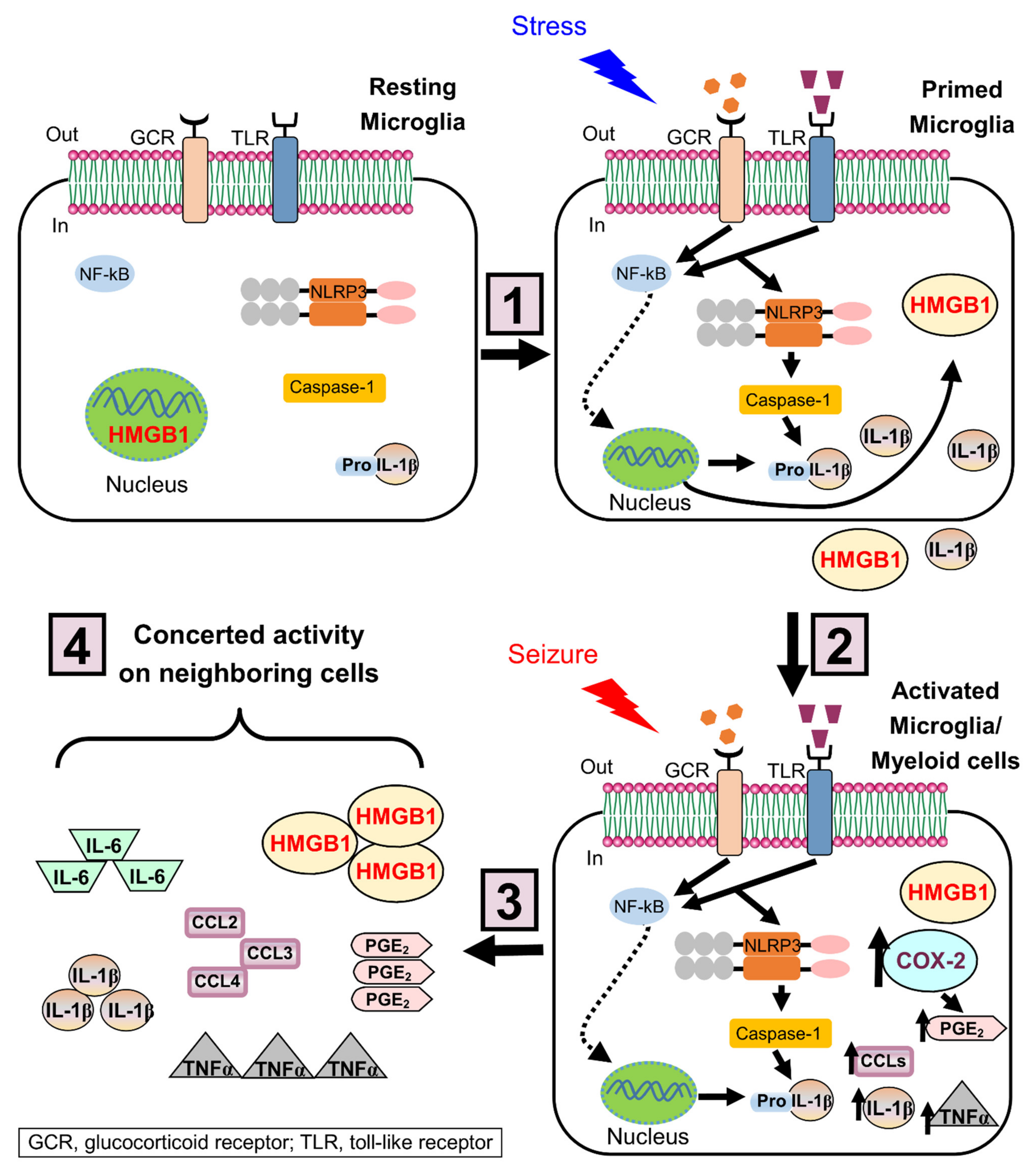
3.3. Neuroinflammation and Chronic Stress in Epilepsy
4. Epilepsy and Depression
4.1. Animal Studies of Epilepsy and Comorbid Depression
4.2. Sex Differences in Comorbid Depression among People with Epilepsy
4.3. Neuroinflammation-Related Depression in Epilepsy
4.4. Neuroinflammation as a Target to Treat Epilepsy and Comorbid Depression
4.5. Potential Biomarkers in Epilepsy and Comorbid Depression
4.6. Stress Primes the Brain to Depression
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Available online: https://www.who.int/mental_health/neurology/epilepsy/report_2019/en/ (accessed on 14 April 2021).
- Temkin, N.R.; Davis, G.R. Stress as a risk factor for seizures among adults with epilepsy. Epilepsia 1984, 25, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Novakova, B.; Harris, P.R.; Ponnusamy, A.; Reuber, M. The role of stress as a trigger for epileptic seizures: A narrative review of evidence from human and animal studies. Epilepsia 2013, 54, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- McKee, H.R.; Privitera, M.D. Stress as a seizure precipitant: Identification, associated factors, and treatment options. Seizure 2017, 44, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.D.; Taylor, D.C.; Kasper, B.S. Stress, seizures, and epilepsy: Patient narratives. Epilepsy Behav. 2018, 80, 163–172. [Google Scholar] [CrossRef]
- Koolhaas, J.M.; Bartolomucci, A.; Buwalda, B.; de Boer, S.F.; Flugge, G.; Korte, S.M.; Meerlo, P.; Murison, R.; Olivier, B.; Palanza, P.; et al. Stress revisited: A critical evaluation of the stress concept. Neurosci. Biobehav. Rev. 2011, 35, 1291–1301. [Google Scholar] [CrossRef]
- Crosswell, A.D.; Lockwood, K.G. Best practices for stress measurement: How to measure psychological stress in health research. Health Psychol. Open 2020, 7. [Google Scholar] [CrossRef]
- Sawyer, N.T.; Escayg, A. Stress and epilepsy: Multiple models, multiple outcomes. J. Clin. Neurophysiol. 2010, 27, 445–452. [Google Scholar] [CrossRef]
- Galtrey, C.M.; Mula, M.; Cock, H.R. Stress and epilepsy: Fact or fiction, and what can we do about it? Pract. Neurol. 2016, 16, 270–278. [Google Scholar] [CrossRef]
- Kotwas, I.; McGonigal, A.; Bastien-Toniazzo, M.; Bartolomei, F.; Micoulaud-Franchi, J.A. Stress regulation in drug-resistant epilepsy. Epilepsy Behav. 2017, 71 (Pt A), 39–50. [Google Scholar] [CrossRef]
- Gargiulo, Á.J.M.; Scévola, L.; Sarudiansky, M.; Kochen, S.; D’Alessio, L. Epilepsy and Psychiatric Comorbidities: New Approaches and Perspectives. In Psychiatry and Neuroscience Update: From Epistemology to Clinical Psychiatry—Vol. IV; Gargiulo, P.Á., Mesones Arroyo, H.L., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 537–553. [Google Scholar]
- Kanner, A.M.; Balabanov, A. Depression and epilepsy: How closely related are they? Neurology 2002, 58, 27–39. [Google Scholar] [CrossRef]
- Blaszczyk, B.; Czuczwar, S.J. Epilepsy coexisting with depression. Pharmacol Rep. 2016, 68, 1084–1092. [Google Scholar] [CrossRef]
- Elger, C.E.; Johnston, S.A.; Hoppe, C. Diagnosing and treating depression in epilepsy. Seizure 2017, 44, 184–193. [Google Scholar] [CrossRef]
- Bermeo-Ovalle, A. Psychiatric comorbidities go untreated in patients with epilepsy: Ignorance or denial? Epilepsy Behav. 2019, 98 (Pt B), 306–308. [Google Scholar] [CrossRef]
- Chang, B.S.; Krishnan, V.; Dulla, C.G.; Jette, N.; Marsh, E.D.; Dacks, P.A.; Whittemore, V.; Poduri, A.; NINDS/AES Epilepsy Research Benchmark Stewards Committee. Epilepsy Benchmarks Area I: Understanding the Causes of the Epilepsies and Epilepsy-Related Neurologic, Psychiatric, and Somatic Conditions. Epilepsy Curr. 2020, 20, 5–13. [Google Scholar] [CrossRef]
- Gandy, M.; Modi, A.C.; Wagner, J.L.; LaFrance, J.W.C.; Reuber, M.; Tang, V.; Valente, K.D.; Goldstein, L.H.; Donald, K.A.; Rayner, G.; et al. Managing Depression and Anxiety in People with Epilepsy: A Survey of Epilepsy Health Professionals by The ILAE Psychology Task Force. Epilepsia Open 2020, 6, 127–139. [Google Scholar] [CrossRef]
- Mazarati, A.M.; Lewis, M.L.; Pittman, Q.J. Neurobehavioral comorbidities of epilepsy: Role of inflammation. Epilepsia 2017, 58, 48–56. [Google Scholar] [CrossRef]
- Slavich, G.M.; Irwin, M.R. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol. Bull. 2014, 140, 774–815. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef]
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Russo, S.J. Immune and Neuroendocrine Mechanisms of Stress Vulnerability and Resilience. Neuropsychopharmacology 2017, 42, 62–80. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Shaikh, M.F.; Shah, S.; Kumari, Y.; Othman, I. Role of inflammation in epilepsy and neurobehavioral comorbidities: Implication for therapy. Eur. J. Pharmacol. 2018, 837, 145–155. [Google Scholar] [CrossRef]
- Hayley, S.; Hakim, A.M.; Albert, P.R. Depression, dementia and immune dysregulation. Brain 2020. [Google Scholar] [CrossRef]
- Becker, C.; Bouvier, E.; Ghestem, A.; Siyoucef, S.; Claverie, D.; Camus, F.; Bartolomei, F.; Benoliel, J.J.; Bernard, C. Predicting and treating stress-induced vulnerability to epilepsy and depression. Ann. Neurol. 2015, 78, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders. Neurobiol. Stress 2016, 4, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J. Primed for Problems: Stress Confers Vulnerability to Epilepsy and Associated Comorbidities. Epilepsy Curr. 2015, 15, 344–346. [Google Scholar] [CrossRef]
- Kohler, O.; Krogh, J.; Mors, O.; Benros, M.E. Inflammation in Depression and the Potential for Anti-Inflammatory Treatment. Curr. Neuropharmacol. 2016, 14, 732–742. [Google Scholar] [CrossRef]
- Husain, M.I.; Chaudhry, I.B.; Khoso, A.B.; Husain, M.O.; Hodsoll, J.; Ansari, M.A.; Naqvi, H.A.; Minhas, F.A.; Carvalho, A.F.; Meyer, J.H.; et al. Minocycline and celecoxib as adjunctive treatments for bipolar depression: A multicentre, factorial design randomised controlled trial. Lancet Psychiatry 2020, 7, 515–527. [Google Scholar] [CrossRef]
- Miller, A.H.; Pariante, C.M. Trial failures of anti-inflammatory drugs in depression. Lancet Psychiatry 2020, 7, 837. [Google Scholar] [CrossRef]
- Privitera, M.; Walters, M.; Lee, I.; Polak, E.; Fleck, A.; Schwieterman, D.; Haut, S.R. Characteristics of people with self-reported stress-precipitated seizures. Epilepsy Behav. 2014, 41, 74–77. [Google Scholar] [CrossRef]
- Stevens, J.R. Emotional activation of the electroencephalogram in patients with convulsive disorders. J. Nerv. Ment. Dis. 1959, 128, 339–351. [Google Scholar] [CrossRef]
- Feldman, R.G.; Paul, N.L. Identity of emotional triggers in epilepsy. J. Nerv. Ment. Dis. 1976, 162, 345–353. [Google Scholar] [CrossRef]
- Van der Kop, M.L.; Ekstrom, A.M.; Arida, R.M. Reduction in seizure frequency with a high-intensity fitness program (CrossFit): A case report. Epilepsy Behav. Rep. 2020, 13, 100354. [Google Scholar] [CrossRef]
- Arida, R.M.; Scorza, F.A.; Terra, V.C.; Scorza, C.A.; de Almeida, A.C.; Cavalheiro, E.A. Physical exercise in epilepsy: What kind of stressor is it? Epilepsy Behav. 2009, 16, 381–387. [Google Scholar] [CrossRef]
- Arida, R.M.; Teixeira-Machado, L. The Contribution of Physical Exercise to Brain Resilience. Front. Behav. Neurosci. 2020, 14, 626–769. [Google Scholar]
- Canzian, J.; Franscescon, F.; Muller, T.E.; Stefanello, F.V.; Souza, T.P.; Rosa, L.V.; Rosemberg, D.B. Stress increases susceptibility to pentylenetetrazole-induced seizures in adult zebrafish. Epilepsy Behav. 2021, 114 (Pt A), 107557. [Google Scholar] [CrossRef]
- Pericic, D.; Jazvinscak, M.; Svob, D.; Mirkovic, K. Swim stress alters the behavioural response of mice to GABA-related and some GABA-unrelated convulsants. Epilepsy Res. 2001, 43, 145–152. [Google Scholar] [CrossRef]
- Homayoun, H.; Dehpour, A.R. Differential contribution of cholecystokinin receptors to stress-induced modulation of seizure and nociception thresholds in mice. Pharmacol. Biochem. Behav. 2004, 78, 209–215. [Google Scholar] [CrossRef]
- Shirzadian, A.; Ostadhadi, S.; Hassanipour, M.; Shafaroodi, H.; Khoshnoodi, M.; Haj-Mirzaian, A.; Sharifzadeh, M.; Amiri, S.; Ghasemi, M.; Dehpour, A.R. Acute foot-shock stress decreased seizure susceptibility against pentylenetetrazole-induced seizures in mice: Interaction between endogenous opioids and cannabinoids. Epilepsy Behav. 2018, 87, 25–31. [Google Scholar] [CrossRef]
- Maggio, N.; Shavit Stein, E.; Segal, M. Complex modulation by stress of the effect of seizures on long term potentiation in mouse hippocampal slices. Hippocampus 2017, 27, 860–870. [Google Scholar] [CrossRef]
- Weizman, R.; Weizman, A.; Kook, K.A.; Vocci, F.; Deutsch, S.I.; Paul, S.M. Repeated swim stress alters brain benzodiazepine receptors measured in vivo. J. Pharmacol. Exp. Ther. 1989, 249, 701–707. [Google Scholar]
- Becker, C.; Mancic, A.; Ghestem, A.; Poillerat, V.; Claverie, D.; Bartolomei, F.; Brouillard, F.; Benoliel, J.J.; Bernard, C. Antioxidant treatment after epileptogenesis onset prevents comorbidities in rats sensitized by a past stressful event. Epilepsia 2019, 60, 648–655. [Google Scholar] [CrossRef]
- Cain, D.P.; Corcoran, M.E. Intracerebral beta-endorphin, met-enkephalin and morphine: Kindling of seizures and handling-induced potentiation of epileptiform effects. Life Sci. 1984, 34, 2535–2542. [Google Scholar] [CrossRef]
- Cain, D.P.; Corcoran, M.E. Epileptiform effects of met-enkephalin, beta-endorphin and morphine: Kindling of generalized seizures and potentiation of epileptiform effects by handling. Brain Res. 1985, 338, 327–336. [Google Scholar] [CrossRef]
- Takeshita, H.; Matsuda, K.; Komatsu, K.; Ito, K.; Kawahara, R.; Hazama, H. Effects of immobilization stress on hippocampal interictal discharges in hippocampal kindled rats. Jpn. J. Psychiatry Neurol. 1991, 45, 388–390. [Google Scholar] [CrossRef]
- Beldhuis, H.J.; Koolhaas, J.M.; Bohus, B. Effect of different agonistic experiences on behavioural seizures in fully amygdala kindled rats. Neurosci. Lett. 1992, 141, 1–4. [Google Scholar] [CrossRef][Green Version]
- Schridde, U.; van Luijtelaar, G. Corticosterone increases spike-wave discharges in a dose- and time-dependent manner in WAG/Rij rats. Pharmacol. Biochem. Behav. 2004, 78, 369–375. [Google Scholar] [CrossRef]
- Forcelli, P.A.; Orefice, L.L.; Heinrichs, S.C. Neural, endocrine and electroencephalographic hyperreactivity to human contact: A diathesis-stress model of seizure susceptibility in El mice. Brain Res. 2007, 1144, 248–256. [Google Scholar] [CrossRef]
- Sawyer, N.T.; Papale, L.A.; Eliason, J.; Neigh, G.N.; Escayg, A. Scn8a voltage-gated sodium channel mutation alters seizure and anxiety responses to acute stress. Psychoneuroendocrinology 2014, 39, 225–236. [Google Scholar] [CrossRef]
- Tolmacheva, E.A.; Oitzl, M.S.; van Luijtelaar, G. Stress, glucocorticoids and absences in a genetic epilepsy model. Horm. Behav. 2012, 61, 706–710. [Google Scholar] [CrossRef]
- Szafarczyk, A.; Caracchini, M.; Rondouin, G.; Ixart, G.; Malaval, F.; Assenmacher, I. Plasma ACTH and corticosterone responses to limbic kindling in the rat. Exp. Neurol. 1986, 92, 583–590. [Google Scholar] [CrossRef]
- Karst, H.; Bosma, A.; Hendriksen, E.; Kamphuis, W.; de Kloet, E.R.; Joels, M. Effect of adrenalectomy in kindled rats. Neuroendocrinology 1997, 66, 348–359. [Google Scholar] [CrossRef]
- Joels, M. Stress, the hippocampus, and epilepsy. Epilepsia 2009, 50, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, M.; Beghi, E.; RESt-1 Group. Accidents in patients with epilepsy: Types, circumstances, and complications: A European cohort study. Epilepsia 2004, 45, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E. Accidents and injuries in patients with epilepsy. Expert Rev. Neurother. 2009, 9, 291–298. [Google Scholar] [CrossRef]
- Zhu, Y.; Klomparens, E.A.; Guo, S.; Geng, X. Neuroinflammation caused by mental stress: The effect of chronic restraint stress and acute repeated social defeat stress in mice. Neurol. Res. 2019, 41, 762–769. [Google Scholar] [CrossRef]
- Weber, M.D.; Godbout, J.P.; Sheridan, J.F. Repeated Social Defeat, Neuroinflammation, and Behavior: Monocytes Carry the Signal. Neuropsychopharmacology 2017, 42, 46–61. [Google Scholar] [CrossRef]
- Ramirez, K.; Fornaguera-Trias, J.; Sheridan, J.F. Stress-Induced Microglia Activation and Monocyte Trafficking to the Brain Underlie the Development of Anxiety and Depression. Curr. Top. Behav. Neurosci. 2017, 31, 155–172. [Google Scholar]
- Welcome, M.O.; Mastorakis, N.E. Stress-induced blood brain barrier disruption: Molecular mechanisms and signaling pathways. Pharmacol. Res. 2020, 157. [Google Scholar] [CrossRef]
- Xu, B.; Lian, S.; Li, S.Z.; Guo, J.R.; Wang, J.F.; Wang, D.; Zhang, L.P.; Yang, H.M. GABAB receptor mediate hippocampal neuroinflammation in adolescent male and female mice after cold expose. Brain Res. Bull. 2018, 142, 163–175. [Google Scholar] [CrossRef]
- Iwata, M.; Ota, K.T.; Duman, R.S. The inflammasome: Pathways linking psychological stress, depression, and systemic illnesses. Brain Behav. Immun. 2013, 31, 105–114. [Google Scholar] [CrossRef]
- Espinosa-Garcia, C.; Sayeed, I.; Yousuf, S.; Atif, F.; Sergeeva, E.G.; Neigh, G.N.; Stein, D.G. Stress primes microglial polarization after global ischemia: Therapeutic potential of progesterone. Brain Behav. Immun. 2017, 66, 177–192. [Google Scholar] [CrossRef]
- Fisher, R.S.; Vickrey, B.G.; Gibson, P.; Hermann, B.; Penovich, P.; Scherer, A.; Walker, S.G. The impact of epilepsy from the patient′s perspective II: Views about therapy and health care. Epilepsy Res. 2000, 41, 53–61. [Google Scholar] [CrossRef]
- Frucht, M.M.; Quigg, M.; Schwaner, C.; Fountain, N.B. Distribution of seizure precipitants among epilepsy syndromes. Epilepsia 2000, 41, 1534–1539. [Google Scholar]
- Nakken, K.O.; Solaas, M.H.; Kjeldsen, M.J.; Friis, M.L.; Pellock, J.M.; Corey, L.A. Which seizure-precipitating factors do patients with epilepsy most frequently report? Epilepsy Behav. 2005, 6, 85–89. [Google Scholar] [CrossRef]
- Sperling, M.R.; Schilling, C.A.; Glosser, D.; Tracy, J.I.; Asadi-Pooya, A.A. Self-perception of seizure precipitants and their relation to anxiety level, depression, and health locus of control in epilepsy. Seizure 2008, 17, 302–307. [Google Scholar] [CrossRef]
- Klein, P.; van Passel, L. Effect of stress related to the 9/11/2001 terror attack on seizures in patients with epilepsy. Neurology 2005, 64, 1815–1816. [Google Scholar] [CrossRef]
- Neufeld, M.Y.; Sadeh, M.; Cohn, D.F.; Korczyn, A.D. Stress and epilepsy: The Gulf war experience. Seizure 1994, 3, 135–139. [Google Scholar] [CrossRef]
- Swinkels, W.A.; Engelsman, M.; Kasteleijn-Nolst Trenite, D.G.; Baal, M.G.; de Haan, G.J.; Oosting, J. Influence of an evacuation in February 1995 in The Netherlands on the seizure frequency in patients with epilepsy: A controlled study. Epilepsia 1998, 39, 1203–1207. [Google Scholar] [CrossRef]
- Thapar, A.; Kerr, M.; Harold, G. Stress, anxiety, depression, and epilepsy: Investigating the relationship between psychological factors and seizures. Epilepsy Behav. 2009, 14, 134–140. [Google Scholar] [CrossRef]
- Eggers, A.E. Temporal lobe epilepsy is a disease of faulty neuronal resonators rather than oscillators, and all seizures are provoked, usually by stress. Med. Hypotheses 2007, 69, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Eggers, A.E. Redrawing Papez′ circuit: A theory about how acute stress becomes chronic and causes disease. Med. Hypotheses 2007, 69, 852–857. [Google Scholar] [CrossRef]
- Weiss, I.C.; Pryce, C.R.; Jongen-Relo, A.L.; Nanz-Bahr, N.I.; Feldon, J. Effect of social isolation on stress-related behavioural and neuroendocrine state in the rat. Behav. Brain Res. 2004, 152, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Manouze, H.; Ghestem, A.; Poillerat, V.; Bennis, M.; Ba-M′hamed, S.; Benoliel, J.J.; Becker, C.; Bernard, C. Effects of Single Cage Housing on Stress, Cognitive, and Seizure Parameters in the Rat and Mouse Pilocarpine Models of Epilepsy. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Chadda, R.; Devaud, L.L. Sex differences in effects of mild chronic stress on seizure risk and GABAA receptors in rats. Pharmacol. Biochem. Behav. 2004, 78, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Nomura, H.; Murakami, Y.; Taki, K.; Takahata, H.; Watanabe, H. Long-term social isolation enhances picrotoxin seizure susceptibility in mice: Up-regulatory role of endogenous brain allopregnanolone in GABAergic systems. Pharmacol. Biochem. Behav. 2003, 75, 831–835. [Google Scholar] [CrossRef]
- Serra, M.; Pisu, M.G.; Littera, M.; Papi, G.; Sanna, E.; Tuveri, F.; Usala, L.; Purdy, R.H.; Biggio, G. Social isolation-induced decreases in both the abundance of neuroactive steroids and GABA(A) receptor function in rat brain. J. Neurochem. 2000, 75, 732–740. [Google Scholar] [CrossRef]
- Wei, K.; Bao, W.; Zhao, Z.; Zhou, W.; Liu, J.; Wei, Y.; Li, M.; Wu, X.; Liu, B.; Du, Y.; et al. Changes of the brain activities after chronic restraint stress in rats: A study based onF-FDG PET. Neurosci. Lett. 2018, 665, 104–109. [Google Scholar] [CrossRef]
- Jones, N.C.; Lee, H.E.; Yang, M.; Rees, S.M.; Morris, M.J.; O′Brien, T.J.; Salzberg, M.R. Repeatedly stressed rats have enhanced vulnerability to amygdala kindling epileptogenesis. Psychoneuroendocrinology 2013, 38, 263–270. [Google Scholar] [CrossRef]
- MacKenzie, G.; Maguire, J. Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Res. 2015, 109, 13–27. [Google Scholar] [CrossRef]
- Tian, R.H.; Li, J.Y. Induction of Epileptic Seizures in Mouse Models of Chronic Restraint Stress. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2018, 40, 656–659. [Google Scholar]
- Zhu, X.; Dong, J.; Xia, Z.; Zhang, A.; Chao, J.; Yao, H. Repeated restraint stress increases seizure susceptibility by activation of hippocampal endoplasmic reticulum stress. Neurochem. Int. 2017, 110, 25–37. [Google Scholar] [CrossRef]
- Pechlivanova, D.M.; Stoynev, A.G.; Tchekalarova, J.D. The effects of chronic losartan pretreatment on restraint stress-induced changes in motor activity, nociception and pentylenetetrazol generalized seizures in rats. Folia Med. 2011, 53, 69–73. [Google Scholar] [CrossRef]
- Skwara, A.J.; Karwoski, T.E.; Czambel, R.K.; Rubin, R.T.; Rhodes, M.E. Influence of environmental enrichment on hypothalamic-pituitary-adrenal (HPA) responses to single-dose nicotine, continuous nicotine by osmotic mini-pumps, and nicotine withdrawal by mecamylamine in male and female rats. Behav. Brain Res. 2012, 234, 1–10. [Google Scholar] [CrossRef]
- Dezsi, G.; Ozturk, E.; Salzberg, M.R.; Morris, M.; O′Brien, T.J.; Jones, N.C. Environmental enrichment imparts disease-modifying and transgenerational effects on genetically-determined epilepsy and anxiety. Neurobiol. Dis. 2016, 93, 129–136. [Google Scholar] [CrossRef]
- Gorantla, V.R.; Thomas, S.E.; Millis, R.M. Environmental Enrichment and Brain Neuroplasticity in the Kainate Rat Model of Temporal Lobe Epilepsy. J. Epilepsy Res. 2019, 9, 51–64. [Google Scholar] [CrossRef]
- Korbey, S.M.; Heinrichs, S.C.; Leussis, M.P. Seizure susceptibility and locus ceruleus activation are reduced following environmental enrichment in an animal model of epilepsy. Epilepsy Behav. 2008, 12, 30–38. [Google Scholar] [CrossRef]
- Manno, I.; Macchi, F.; Caleo, M.; Bozzi, Y. Environmental enrichment reduces spontaneous seizures in the Q54 transgenic mouse model of temporal lobe epilepsy. Epilepsia 2011, 52, 113–117. [Google Scholar] [CrossRef]
- Vrinda, M.; Sasidharan, A.; Aparna, S.; Srikumar, B.N.; Kutty, B.M.; Shankaranarayana Rao, B.S. Enriched environment attenuates behavioral seizures and depression in chronic temporal lobe epilepsy. Epilepsia 2017, 58, 1148–1158. [Google Scholar] [CrossRef]
- Young, D.; Lawlor, P.A.; Leone, P.; Dragunow, M.; During, M.J. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat. Med. 1999, 5, 448–453. [Google Scholar] [CrossRef]
- Koh, S.; Magid, R.; Chung, H.; Stine, C.D.; Wilson, D.N. Depressive behavior and selective down-regulation of serotonin receptor expression after early-life seizures: Reversal by environmental enrichment. Epilepsy Behav. 2007, 10, 26–31. [Google Scholar] [CrossRef]
- Akyuz, E.; Eroglu, E. Envisioning the crosstalk between environmental enrichment and epilepsy: A novel perspective. Epilepsy Behav. 2021, 115, 107660. [Google Scholar] [CrossRef]
- Choi, J.; Min, H.J.; Shin, J.S. Increased levels of HMGB1 and pro-inflammatory cytokines in children with febrile seizures. J. Neuroinflammation 2011, 8, 135. [Google Scholar] [CrossRef]
- Sakuma, H.; Tanuma, N.; Kuki, I.; Takahashi, Y.; Shiomi, M.; Hayashi, M. Intrathecal overproduction of proinflammatory cytokines and chemokines in febrile infection-related refractory status epilepticus. J. Neurol. Neurosurg. Psychiatry 2015, 86, 820–822. [Google Scholar] [CrossRef]
- Rakhade, S.N.; Jensen, F.E. Epileptogenesis in the immature brain: Emerging mechanisms. Nat. Rev. Neurol. 2009, 5, 380–391. [Google Scholar] [CrossRef]
- Beach, T.G.; Woodhurst, W.B.; MacDonald, D.B.; Jones, M.W. Reactive microglia in hippocampal sclerosis associated with human temporal lobe epilepsy. Neurosci. Lett. 1995, 191, 27–30. [Google Scholar] [CrossRef]
- Crespel, A.; Coubes, P.; Rousset, M.C.; Brana, C.; Rougier, A.; Rondouin, G.; Bockaert, J.; Baldy-Moulinier, M.; Lerner-Natoli, M. Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res. 2002, 952, 159–169. [Google Scholar] [CrossRef]
- Aronica, E.; Bauer, S.; Bozzi, Y.; Caleo, M.; Dingledine, R.; Gorter, J.A.; Henshall, D.C.; Kaufer, D.; Koh, S.; Loscher, W.; et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017, 58, 27–38. [Google Scholar] [CrossRef]
- Klein, P.; Dingledine, R.; Aronica, E.; Bernard, C.; Blumcke, I.; Boison, D.; Brodie, M.J.; Brooks-Kayal, A.R.; Engel, J., Jr.; Forcelli, P.A.; et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 2018, 59, 37–66. [Google Scholar] [CrossRef]
- Van Vliet, E.A.; Aronica, E.; Vezzani, A.; Ravizza, T. Review: Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: Emerging evidence from preclinical and clinical studies. Neuropathol. Appl. Neurobiol. 2018, 1, 91–111. [Google Scholar] [CrossRef]
- Varvel, N.H.; Jiang, J.; Dingledine, R. Candidate drug targets for prevention or modification of epilepsy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 229–247. [Google Scholar] [CrossRef]
- Balosso, S.; Maroso, M.; Sanchez-Alavez, M.; Ravizza, T.; Frasca, A.; Bartfai, T.; Vezzani, A. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain 2008, 131 (Pt 12), 3256–3265. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Iori, V.; Wright, C.I.; French, J.; Vezzani, A. Interleukin-1beta biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 2011, 8, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Bianchi, M.E.; Vezzani, A. Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: The importance of IL-1beta and high-mobility group box 1. J. Intern. Med. 2011, 270, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav. Immun. 2008, 22, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Bartfai, T.; Bianchi, M.; Rossetti, C.; French, J. Therapeutic potential of new antiinflammatory drugs. Epilepsia 2011, 52, 67–69. [Google Scholar] [CrossRef]
- Vezzani, A.; Maroso, M.; Balosso, S.; Sanchez, M.A.; Bartfai, T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav. Immun. 2011, 25, 1281–1289. [Google Scholar] [CrossRef]
- Tynan, R.J.; Naicker, S.; Hinwood, M.; Nalivaiko, E.; Buller, K.M.; Pow, D.V.; Day, T.A.; Walker, F.R. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain Behav. Immun. 2010, 24, 1058–1068. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; Stiner, L.M.; Bailey, M.T.; Nelson, R.J.; Godbout, J.P.; Sheridan, J.F. beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci 2011, 31, 6277–6288. [Google Scholar] [CrossRef]
- Wang, M.; Chen, Y. Inflammation: A Network in the Pathogenesis of Status Epilepticus. Front. Mol. Neurosci. 2018, 11, 341. [Google Scholar] [CrossRef]
- Meng, F.; Yao, L. The role of inflammation in epileptogenesis. Acta Epileptol. 2020, 2. [Google Scholar] [CrossRef]
- Benson, M.J.; Manzanero, S.; Borges, K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia 2015, 56, 895–905. [Google Scholar] [CrossRef]
- Wu, W.; Li, Y.; Wei, Y.; Bosco, D.B.; Xie, M.; Zhao, M.G.; Richardson, J.R.; Wu, L.J. Microglial depletion aggravates the severity of acute and chronic seizures in mice. Brain Behav. Immun. 2020, 89, 245–255. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef]
- Sa-Pereira, I.; Brites, D.; Brito, M.A. Neurovascular unit: A focus on pericytes. Mol. Neurobiol. 2012, 45, 327–347. [Google Scholar] [CrossRef]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, 5665–5674. [Google Scholar] [CrossRef]
- Ozdemir, H.H.; Akil, E.; Acar, A.; Tamam, Y.; Varol, S.; Cevik, M.U.; Arikanoglu, A. Changes in serum albumin levels and neutrophil-lymphocyte ratio in patients with convulsive status epilepticus. Int. J. Neurosci. 2017, 127, 417–420. [Google Scholar] [CrossRef]
- Scholz, M.; Cinatl, J.; Schadel-Hopfner, M.; Windolf, J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med. Res. Rev. 2007, 27, 401–416. [Google Scholar] [CrossRef]
- Stowe, A.M.; Adair-Kirk, T.L.; Gonzales, E.R.; Perez, R.S.; Shah, A.R.; Park, T.S.; Gidday, J.M. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol. Dis. 2009, 35, 82–90. [Google Scholar] [CrossRef]
- Chang, K.H.; Hsu, Y.C.; Chang, M.Y.; Lin, C.L.; Wu, T.N.; Hwang, B.F.; Chen, C.Y.; Liu, H.C.; Kao, C.H. A Large-Scale Study Indicates Increase in the Risk of Epilepsy in Patients With Different Risk Factors, Including Rheumatoid Arthritis. Medicine 2015, 94, 1485. [Google Scholar] [CrossRef]
- Lance, E.I.; Sreenivasan, A.K.; Zabel, T.A.; Kossoff, E.H.; Comi, A.M. Aspirin use in Sturge-Weber syndrome: Side effects and clinical outcomes. J. Child Neurol. 2013, 28, 213–218. [Google Scholar] [CrossRef]
- Semple, B.D.; O′Brien, T.J.; Gimlin, K.; Wright, D.K.; Kim, S.E.; Casillas-Espinosa, P.M.; Webster, K.M.; Petrou, S.; Noble-Haeusslein, L.J. Interleukin-1 Receptor in Seizure Susceptibility after Traumatic Injury to the Pediatric Brain. J. Neurosci. 2017, 37, 7864–7877. [Google Scholar] [CrossRef]
- Taraschenko, O.; Fox, H.S.; Zekeridou, A.; Pittock, S.J.; Eldridge, E.; Farukhuddin, F.; Al-Saleem, F.; Devi Kattala, C.; Dessain, S.K.; Casale, G.; et al. Seizures and memory impairment induced by patient-derived anti-N-methyl-D-aspartate receptor antibodies in mice are attenuated by anakinra, an interleukin-1 receptor antagonist. Epilepsia 2021, 62, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, S.; Villeneuve, N.; Trebuchon, A.; Kaphan, E.; Lepine, A.; McGonigal, A.; Roubertie, A.; Barthez, M.A.; Trommsdorff, V.; Lefranc, J.; et al. Anti-tumor necrosis factor alpha therapy (adalimumab) in Rasmussen′s encephalitis: An open pilot study. Epilepsia 2016, 57, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Quan, Y.; Ganesh, T.; Pouliot, W.A.; Dudek, F.E.; Dingledine, R. Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proc. Natl. Acad. Sci. USA 2013, 110, 3591–3596. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yang, M.S.; Quan, Y.; Gueorguieva, P.; Ganesh, T.; Dingledine, R. Therapeutic window for cyclooxygenase-2 related anti-inflammatory therapy after status epilepticus. Neurobiol. Dis. 2015, 76, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Ganesh, T.; Lelutiu, N.; Gueorguieva, P.; Dingledine, R. Inhibition of the prostaglandin EP2 receptor is neuroprotective and accelerates functional recovery in a rat model of organophosphorus induced status epilepticus. Neuropharmacology 2015, 93, 15–27. [Google Scholar] [CrossRef]
- Rojas, A.; Ganesh, T.; Wang, W.; Wang, J.; Dingledine, R. A rat model of organophosphate-induced status epilepticus and the beneficial effects of EP2 receptor inhibition. Neurobiol. Dis. 2020, 133, 104399. [Google Scholar] [CrossRef]
- Dudek, K.A.; Dion-Albert, L.; Lebel, M.; LeClair, K.; Labrecque, S.; Tuck, E.; Ferrer Perez, C.; Golden, S.A.; Tamminga, C.; Turecki, G.; et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc. Natl. Acad. Sci. USA 2020, 117, 3326–3336. [Google Scholar] [CrossRef]
- Xu, G.; Li, Y.; Ma, C.; Wang, C.; Sun, Z.; Shen, Y.; Liu, L.; Li, S.; Zhang, X.; Cong, B. Restraint Stress Induced Hyperpermeability and Damage of the Blood-Brain Barrier in the Amygdala of Adult Rats. Front. Mol. Neurosci. 2019, 12, 32. [Google Scholar] [CrossRef]
- Friedman, D.E.; Kung, D.H.; Laowattana, S.; Kass, J.S.; Hrachovy, R.A.; Levin, H.S. Identifying depression in epilepsy in a busy clinical setting is enhanced with systematic screening. Seizure 2009, 18, 429–433. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, Y.S.; Yang, T.W.; Kwon, O.Y. Optimal cutoff score of the Neurological Disorders Depression Inventory for Epilepsy (NDDI-E) for detecting major depressive disorder: A meta-analysis. Epilepsy Behav. 2019, 92, 61–70. [Google Scholar] [CrossRef]
- LaFrance, W.C., Jr.; Kanner, A.M.; Hermann, B. Psychiatric comorbidities in epilepsy. Int. Rev. Neurobiol. 2008, 83, 347–383. [Google Scholar] [PubMed]
- Mula, M.; Sander, J.W. Negative effects of antiepileptic drugs on mood in patients with epilepsy. Drug Saf. 2007, 30, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Kanner, A.M. Most antidepressant drugs are safe for patients with epilepsy at therapeutic doses: A review of the evidence. Epilepsy Behav. 2016, 61, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, K. What can animal models tell us about depressive disorders. In Modeling Neuropsychiatric Disorders in Laboratory Animals; Woodhead Publishing: Cambridge, UK, 2016. [Google Scholar]
- Porsolt, R.D.; Le Pichon, M.; Jalfre, M. Depression: A new animal model sensitive to antidepressant treatments. Nature 1977, 266, 730–732. [Google Scholar] [CrossRef] [PubMed]
- NIMH. Available online: https://grants.nih.gov/grants/guide/notice-files/NOT-MH-19-053.html (accessed on 14 April 2021).
- Molendijk, M.L.; de Kloet, E.R. FORCED SWIM STRESSOR: Trends in usage and mechanistic consideration. Eur. J. Neurosci. 2021. [Google Scholar] [CrossRef]
- Scheggi, S.; De Montis, M.G.; Gambarana, C. Making Sense of Rodent Models of Anhedonia. Int. J. Neuropsychopharmacol. 2018, 21, 1049–1065. [Google Scholar] [CrossRef]
- Hammen, C. Stress and depression. Annu. Rev. Clin. Psychol. 2005, 1, 293–319. [Google Scholar] [CrossRef]
- Duman, C.H. Models of depression. Vitam. Horm. 2010, 82, 1–21. [Google Scholar]
- Wood, S.K.; Bhatnagar, S. Resilience to the effects of social stress: Evidence from clinical and preclinical studies on the role of coping strategies. Neurobiol. Stress 2015, 1, 164–173. [Google Scholar] [CrossRef]
- Wood, S.K.; Walker, H.E.; Valentino, R.J.; Bhatnagar, S. Individual differences in reactivity to social stress predict susceptibility and resilience to a depressive phenotype: Role of corticotropin-releasing factor. Endocrinology 2010, 151, 1795–1805. [Google Scholar] [CrossRef]
- Schetter, C.D.; Dolbier, C. Resilience in the Context of Chronic Stress and Health in Adults. Soc. Personal. Psychol. Compass 2011, 5, 634–652. [Google Scholar] [CrossRef]
- Planchez, B.; Surget, A.; Belzung, C. Animal models of major depression: Drawbacks and challenges. J. Neural Transm. (Vienna) 2019, 126, 1383–1408. [Google Scholar] [CrossRef]
- Epps, S.A.; Weinshenker, D. Rhythm and blues: Animal models of epilepsy and depression comorbidity. Biochem. Pharmacol. 2013, 85, 135–146. [Google Scholar] [CrossRef]
- Mazarati, A.; Siddarth, P.; Baldwin, R.A.; Shin, D.; Caplan, R.; Sankar, R. Depression after status epilepticus: Behavioural and biochemical deficits and effects of fluoxetine. Brain 2008, 131 (Pt 8), 2071–2083. [Google Scholar] [CrossRef]
- Klein, S.; Bankstahl, J.P.; Loscher, W.; Bankstahl, M. Sucrose consumption test reveals pharmacoresistant depression-associated behavior in two mouse models of temporal lobe epilepsy. Exp. Neurol. 2015, 263, 263–271. [Google Scholar] [CrossRef]
- Phillips, K.F.; Deshpande, L.S. Repeated low-dose organophosphate DFP exposure leads to the development of depression and cognitive impairment in a rat model of Gulf War Illness. Neurotoxicology 2016, 52, 127–133. [Google Scholar] [CrossRef]
- Zanirati, G.; Azevedo, P.N.; Venturin, G.T.; Greggio, S.; Alcara, A.M.; Zimmer, E.R.; Feltes, P.K.; DaCosta, J.C. Depression comorbidity in epileptic rats is related to brain glucose hypometabolism and hypersynchronicity in the metabolic network architecture. Epilepsia 2018, 59, 923–934. [Google Scholar] [CrossRef]
- Upadhya, D.; Kodali, M.; Gitai, D.; Castro, O.W.; Zanirati, G.; Upadhya, R.; Attaluri, S.; Mitra, E.; Shuai, B.; Hattiangady, B.; et al. A Model of Chronic Temporal Lobe Epilepsy Presenting Constantly Rhythmic and Robust Spontaneous Seizures, Co-morbidities and Hippocampal Neuropathology. Aging Dis. 2019, 10, 915–936. [Google Scholar] [CrossRef]
- Mazarati, A.M.; Shin, D.; Kwon, Y.S.; Bragin, A.; Pineda, E.; Tio, D.; Taylor, A.N.; Sankar, R. Elevated plasma corticosterone level and depressive behavior in experimental temporal lobe epilepsy. Neurobiol. Dis. 2009, 34, 457–461. [Google Scholar] [CrossRef]
- Hooper, A.; Paracha, R.; Maguire, J. Seizure-induced activation of the HPA axis increases seizure frequency and comorbid depression-like behaviors. Epilepsy Behav. 2018, 78, 124–133. [Google Scholar] [CrossRef]
- Wulsin, A.C.; Franco-Villanueva, A.; Romancheck, C.; Morano, R.L.; Smith, B.L.; Packard, B.A.; Danzer, S.C.; Herman, J.P. Functional disruption of stress modulatory circuits in a model of temporal lobe epilepsy. PLoS ONE 2018, 13, 0197955. [Google Scholar] [CrossRef]
- Lenck-Santini, P.P. Cognitive and behavioral comorbidities in epilepsy: The treacherous nature of animal models. Epilepsy Curr. 2013, 13, 182–183. [Google Scholar] [CrossRef]
- Christian, C.A.; Reddy, D.S.; Maguire, J.; Forcelli, P.A. Sex Differences in the Epilepsies and Associated Comorbidities: Implications for Use and Development of Pharmacotherapies. Pharmacol. Rev. 2020, 72, 767–800. [Google Scholar] [CrossRef]
- Gaus, V.; Kiep, H.; Holtkamp, M.; Burkert, S.; Kendel, F. Gender differences in depression, but not in anxiety in people with epilepsy. Seizure 2015, 32, 37–42. [Google Scholar] [CrossRef]
- Liu, Z.; Yin, R.; Fan, Z.; Fan, H.; Wu, H.; Shen, B.; Wu, S.; Kuang, F. Gender Differences in Associated and Predictive Factors of Anxiety and Depression in People With Epilepsy. Front. Psychiatry 2020, 11, 670. [Google Scholar] [CrossRef]
- Oliveira, C.V.; Grigoletto, J.; Funck, V.R.; Ribeiro, L.R.; Royes, L.F.; Fighera, M.R.; Furian, A.F.; Oliveira, M.S. Evaluation of potential gender-related differences in behavioral and cognitive alterations following pilocarpine-induced status epilepticus in C57BL/6 mice. Physiol. Behav. 2015, 143, 142–150. [Google Scholar] [CrossRef]
- Bekhbat, M.; Neigh, G.N. Stress-induced neuroimmune priming in males and females: Comparable but not identical. Brain Behav. Immun. 2018, 73, 149–150. [Google Scholar] [CrossRef]
- Fonken, L.K.; Frank, M.G.; Gaudet, A.D.; D′Angelo, H.M.; Daut, R.A.; Hampson, E.C.; Ayala, M.T.; Watkins, L.R.; Maier, S.F. Neuroinflammatory priming to stress is differentially regulated in male and female rats. Brain Behav. Immun. 2018, 70, 257–267. [Google Scholar] [CrossRef]
- Fonken, L.K.; Frank, M.G.; Gaudet, A.D.; Maier, S.F. Stress and aging act through common mechanisms to elicit neuroinflammatory priming. Brain Behav. Immun. 2018, 73, 133–148. [Google Scholar] [CrossRef]
- Frank, M.G.; Fonken, L.K.; Annis, J.L.; Watkins, L.R.; Maier, S.F. Stress disinhibits microglia via down-regulation of CD200R: A mechanism of neuroinflammatory priming. Brain Behav. Immun. 2018, 69, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.C.; Wohleb, E.S.; Zhang, Y.; Fogaca, M.; Hare, B.; Duman, R.S. Persistent Increase in Microglial RAGE Contributes to Chronic Stress-Induced Priming of Depressive-like Behavior. Biol. Psychiatry 2018, 83, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Garcia, C.; Atif, F.; Yousuf, S.; Sayeed, I.; Neigh, G.N.; Stein, D.G. Progesterone Attenuates Stress-Induced NLRP3 Inflammasome Activation and Enhances Autophagy following Ischemic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3740. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.C.; O′Brien, T.J. Stress, epilepsy, and psychiatric comorbidity: How can animal models inform the clinic? Epilepsy Behav. 2013, 26, 363–369. [Google Scholar] [CrossRef]
- Peng, W.F.; Fan, F.; Li, X.; Zhang, Q.Q.; Ding, J.; Wang, X. Different behavioral and pathological changes between epilepsy-associated depression and primary depression models. Epilepsy Behav. 2018, 83, 212–218. [Google Scholar] [CrossRef]
- Cardamone, L.; Salzberg, M.R.; O′Brien, T.J.; Jones, N.C. Antidepressant therapy in epilepsy: Can treating the comorbidities affect the underlying disorder? Br. J. Pharmacol. 2013, 168, 1531–1554. [Google Scholar] [CrossRef]
- Maguire, M.J.; Weston, J.; Singh, J.; Marson, A.G. Antidepressants for people with epilepsy and depression. Cochrane Database Syst. Rev. 2014, CD010682. [Google Scholar] [CrossRef]
- Singh, T.; Goel, R.K. Epilepsy Associated Depression: An Update on Current Scenario, Suggested Mechanisms, and Opportunities. Neurochem. Res. 2021. [Google Scholar] [CrossRef]
- Kenis, G.; Maes, M. Effects of antidepressants on the production of cytokines. Int. J. Neuropsychopharmacol. 2002, 5, 401–412. [Google Scholar] [CrossRef]
- Sutcigil, L.; Oktenli, C.; Musabak, U.; Bozkurt, A.; Cansever, A.; Uzun, O.; Sanisoglu, S.Y.; Yesilova, Z.; Ozmenler, N.; Ozsahin, A.; et al. Pro- and anti-inflammatory cytokine balance in major depression: Effect of sertraline therapy. Clin. Dev. Immunol. 2007. [Google Scholar] [CrossRef]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef]
- Piletz, J.E.; Halaris, A.; Iqbal, O.; Hoppensteadt, D.; Fareed, J.; Zhu, H.; Sinacore, J.; Devane, C.L. Pro-inflammatory biomakers in depression: Treatment with venlafaxine. World J. Biol. Psychiatry 2009, 10, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Fornaro, M.; Martino, M.; Battaglia, F.; Colicchio, S.; Perugi, G. Increase in IL-6 levels among major depressive disorder patients after a 6-week treatment with duloxetine 60 mg/day: A preliminary observation. Neuropsychiatr. Dis. Treat. 2011, 7, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Pineda, E.A.; Hensler, J.G.; Sankar, R.; Shin, D.; Burke, T.F.; Mazarati, A.M. Interleukin-1beta causes fluoxetine resistance in an animal model of epilepsy-associated depression. Neurotherapeutics 2012, 9, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; Santoro, M.; D′Agostino, G.; Constanti, A.; Russo, E. Role of Histone Deacetylases (HDACs) in Epilepsy and Epileptogenesis. Curr. Pharm. Des. 2017, 23, 5546–5562. [Google Scholar] [CrossRef]
- Citraro, R.; Leo, A.; De Caro, C.; Nesci, V.; Gallo Cantafio, M.E.; Amodio, N.; Mattace Raso, G.; Lama, A.; Russo, R.; Calignano, A.; et al. Effects of Histone Deacetylase Inhibitors on the Development of Epilepsy and Psychiatric Comorbidity in WAG/Rij Rats. Mol. Neurobiol. 2020, 57, 408–421. [Google Scholar] [CrossRef]
- Reddy, S.D.; Clossen, B.L.; Reddy, D.S. Epigenetic Histone Deacetylation Inhibition Prevents the Development and Persistence of Temporal Lobe Epilepsy. J. Pharmacol. Exp. Ther. 2018, 364, 97–109. [Google Scholar] [CrossRef]
- Hu, Q.P.; Mao, D.A. Histone deacetylase inhibitor SAHA attenuates post-seizure hippocampal microglia TLR4/MYD88 signaling and inhibits TLR4 gene expression via histone acetylation. BMC Neurosci. 2016, 17, 22. [Google Scholar] [CrossRef]
- Singh, V.; Bhatia, H.S.; Kumar, A.; de Oliveira, A.C.; Fiebich, B.L. Histone deacetylase inhibitors valproic acid and sodium butyrate enhance prostaglandins release in lipopolysaccharide-activated primary microglia. Neuroscience 2014, 265, 147–157. [Google Scholar] [CrossRef]
- Bialer, M.; Johannessen, S.I.; Koepp, M.J.; Levy, R.H.; Perucca, E.; Perucca, P.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the Fifteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XV). I. Drugs in preclinical and early clinical development. Epilepsia 2020, 61, 2340–2364. [Google Scholar] [CrossRef]
- Jyonouchi, H.; Geng, L. Intractable Epilepsy (IE) and Responses to Anakinra, a Human Recombinant IL-1 Receptor Agonist (IL-1ra): Case Reports. J. Clin. Cell. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Godfred, R.M.; Parikh, M.S.; Haltiner, A.M.; Caylor, L.M.; Sepkuty, J.P.; Doherty, M.J. Does aspirin use make it harder to collect seizures during elective video-EEG telemetry? Epilepsy Behav. 2013, 27, 115–117. [Google Scholar] [CrossRef]
- Jun, J.S.; Lee, S.T.; Kim, R.; Chu, K.; Lee, S.K. Tocilizumab treatment for new onset refractory status epilepticus. Ann. Neurol. 2018, 84, 940–945. [Google Scholar] [CrossRef]
- Nowak, M.; Strzelczyk, A.; Reif, P.S.; Schorlemmer, K.; Bauer, S.; Norwood, B.A.; Oertel, W.H.; Rosenow, F.; Strik, H.; Hamer, H.M. Minocycline as potent anticonvulsant in a patient with astrocytoma and drug resistant epilepsy. Seizure 2012, 21, 227–228. [Google Scholar] [CrossRef]
- Rojas, A.; Amaradhi, R.; Banik, A.; Jiang, C.; Abreu-Melon, J.; Wang, S.; Dingledine, R.; Ganesh, T. A Novel Second-Generation EP2 Receptor Antagonist Reduces Neuroinflammation and Gliosis After Status Epilepticus in Rats. Neurotherapeutics 2021. [Google Scholar] [CrossRef]
- Zhu, K.; Hu, M.; Yuan, B.; Liu, J.X.; Liu, Y. Aspirin attenuates spontaneous recurrent seizures in the chronically epileptic mice. Neurol. Res. 2017, 39, 744–757. [Google Scholar] [CrossRef]
- Wang, N.; Mi, X.; Gao, B.; Gu, J.; Wang, W.; Zhang, Y.; Wang, X. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 2015, 287, 144–156. [Google Scholar] [CrossRef]
- Iori, V.; Iyer, A.M.; Ravizza, T.; Beltrame, L.; Paracchini, L.; Marchini, S.; Cerovic, M.; Hill, C.; Ferrari, M.; Zucchetti, M.; et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol. Dis. 2017, 99, 12–23. [Google Scholar] [CrossRef]
- Walker, L.E.; Janigro, D.; Heinemann, U.; Riikonen, R.; Bernard, C.; Patel, M. WONOEP appraisal: Molecular and cellular biomarkers for epilepsy. Epilepsia 2016, 57, 1354–1362. [Google Scholar] [CrossRef]
- Ravizza, T.; Onat, F.Y.; Brooks-Kayal, A.R.; Depaulis, A.; Galanopoulou, A.S.; Mazarati, A.; Numis, A.L.; Sankar, R.; Friedman, A. WONOEP appraisal: Biomarkers of epilepsy-associated comorbidities. Epilepsia 2017, 58, 331–342. [Google Scholar] [CrossRef]
- Rojas, A.; Jiang, J.; Ganesh, T.; Yang, M.S.; Lelutiu, N.; Gueorguieva, P.; Dingledine, R. Cyclooxygenase-2 in epilepsy. Epilepsia 2014, 55, 17–25. [Google Scholar] [CrossRef]
- Weber, M.D.; Frank, M.G.; Tracey, K.J.; Watkins, L.R.; Maier, S.F. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: A priming stimulus of microglia and the NLRP3 inflammasome. J. Neurosci. 2015, 35, 316–324. [Google Scholar] [CrossRef]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress sounds the alarmin: The role of the danger-associated molecular pattern HMGB1 in stress-induced neuroinflammatory priming. Brain Behav. Immun. 2015, 48, 1–7. [Google Scholar] [CrossRef]
- Walker, F.R.; Nilsson, M.; Jones, K. Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr. Drug Targets 2013, 14, 1262–1276. [Google Scholar] [CrossRef]
- Calcia, M.A.; Bonsall, D.R.; Bloomfield, P.S.; Selvaraj, S.; Barichello, T.; Howes, O.D. Stress and neuroinflammation: A systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology 2016, 233, 1637–1650. [Google Scholar] [CrossRef]
- Neher, J.J.; Cunningham, C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019, 40, 358–374. [Google Scholar] [CrossRef]
- Koo, J.W.; Wohleb, E.S. How Stress Shapes Neuroimmune Function: Implications for the Neurobiology of Psychiatric Disorders. Biol. Psychiatry 2020, 20, 32067–32069. [Google Scholar]
- Alcocer-Gomez, E.; de Miguel, M.; Casas-Barquero, N.; Nunez-Vasco, J.; Sanchez-Alcazar, J.A.; Fernandez-Rodriguez, A.; Cordero, M.D. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav. Immun. 2014, 36, 111–117. [Google Scholar] [CrossRef]
- Cristina de Brito Toscano, E.; Leandro Marciano Vieira, E.; Boni Rocha Dias, B.; Vidigal Caliari, M.; Paula Goncalves, A.; Varela Giannetti, A.; Mauricio Siqueira, J.; Kimie Suemoto, C.; Elaine Paraizo Leite, R.; Nitrini, R.; et al. NLRP3 and NLRP1 inflammasomes are up-regulated in patients with mesial temporal lobe epilepsy and may contribute to overexpression of caspase-1 and IL-beta in sclerotic hippocampi. Brain Res. 2021, 1752, 147230. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Liu, Y.Z.; Shen, X.L.; Wu, T.Y.; Zhang, T.; Wang, W.; Wang, Y.X.; Jiang, C.L. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, 6. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, Y.; Yang, T.; Song, M.; Wang, C.; Yao, Y.; Fan, H. Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors. Front. Mol. Neurosci. 2019, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, F.N.; Costa, A.P.; Ghisleni, G.; Diaz, A.P.; Rodrigues, A.L.S.; Peluffo, H.; Kaster, M.P. NLRP3 inflammasome-driven pathways in depression: Clinical and preclinical findings. Brain Behav. Immun. 2017, 64, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Fonken, L.K.; Watkins, L.R.; Maier, S.F. Acute stress induces chronic neuroinflammatory, microglial and behavioral priming: A role for potentiated NLRP3 inflammasome activation. Brain Behav. Immun. 2020, 89, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Huesmann, G.R.; Bove, R. Treating Stress to Improve Neurologic Outcomes. Pract. Neurol. 2018, 40–48. [Google Scholar]
- Takahashi, A.; Flanigan, M.E.; McEwen, B.S.; Russo, S.J. Aggression, Social Stress, and the Immune System in Humans and Animal Models. Front. Behav. Neurosci. 2018, 12, 56. [Google Scholar] [CrossRef]
- Kim, J.E.; Cho, K.O. Functional Nutrients for Epilepsy. Nutrients 2019, 11, 1309. [Google Scholar] [CrossRef]
- Arida, R.M.; de Almeida, A.C.; Cavalheiro, E.A.; Scorza, F.A. Experimental and clinical findings from physical exercise as complementary therapy for epilepsy. Epilepsy Behav. 2013, 26, 273–278. [Google Scholar] [CrossRef]
- Thompson, N.J.; Patel, A.H.; Selwa, L.M.; Stoll, S.C.; Begley, C.E.; Johnson, E.K.; Fraser, R.T. Expanding the efficacy of Project UPLIFT: Distance delivery of mindfulness-based depression prevention to people with epilepsy. J. Consult. Clin. Psychol. 2015, 83, 304–313. [Google Scholar] [CrossRef]
- Haut, S.R.; Privitera, M. Author response: Behavioral interventions as a treatment for epilepsy: A multicenter randomized controlled trial. Neurology 2018, 91, 722. [Google Scholar] [CrossRef]
- Novakova, B.; Harris, P.R.; Rawlings, G.H.; Reuber, M. Coping with stress: A pilot study of a self-help stress management intervention for patients with epileptic or psychogenic nonepileptic seizures. Epilepsy Behav. 2019, 94, 169–177. [Google Scholar] [CrossRef]
- Privitera, M.; Haut, S.R.; Lipton, R.B.; McGinley, J.S.; Cornes, S. Seizure self-prediction in a randomized controlled trial of stress management. Neurology 2019, 93, 2021–2031. [Google Scholar] [CrossRef]
- Gandy, M.; Karin, E.; McDonald, S.; Meares, S.; Scott, A.J.; Titov, N.; Dear, B.F. A feasibility trial of an internet-delivered psychological intervention to manage mental health and functional outcomes in neurological disorders. J. Psychosom. Res. 2020, 136, 110173. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| (A) Clinical studies | |||
| Drug | Mechanism of drug/target | Outcome | Ref |
| Anakinra | IL-1ra | Reduced number of seizures | [183] |
| Reduced peripheral blood monocytes cytokine production: IL-1β/IL-10 ratio | |||
| Aspirin | COX-2 inhibitor | Reduced seizure frequency | [184] |
| This study lacks of inflammation analysis | |||
| Tocilizumab | IL-6 receptor | SE was terminated in most patients | [185] |
| IL-6 levels were normalized | |||
| 2 out of 7 patients experienced severe adverse events related to infection during therapy | |||
| Minocycline | Microglial activation inhibitor | Reduced seizure frequency | [186] |
| This study lacks of inflammation analysis | |||
| (B) Preclinical studies | |||
| Fluoxetine | Pilocarpine-induced SE | Failed to exert antiepileptogenic effects | [148] |
| No improvement in depressive-like behaviors | |||
| SSRI | This study lacks of inflammation analysis | ||
| Fluoxetine + IL-1ra | Pilocarpine-induced SE | Failed to exert antiepileptogenic effects | [174] |
| Improved depressive-like behaviors | |||
| SSRI + IL-1ra | This study lacks of inflammation analysis | ||
| VPA and NaB | Genetic model of absence epilepsy (WAG/Rij Rats) | Reduced absence seizures | [178,179] |
| Improved depressive-like behaviors | |||
| Hippocampus kindling model | Inhibited development of kindling epileptogenesis | ||
| HDACs | These studies lack of inflammation analysis | ||
| SAHA | Kainic acid-induced SE | Attenuated kainic acid-induced seizures | [180] |
| Suppressed microglial activation in the hippocampus | |||
| HDAC | Inhibited TLR4, MYD88, NF-κB and IL-1β expression | ||
| TG6-10-1 | Organophosphorus-induced SE | Reduced hippocampal neuroinflammation and gliosis, mitigate neuronal injury or BBB breakdown | [127] |
| EP2 antagonist | |||
| TG8-260 | Pilocarpine-induced SE | Reduced hippocampal neuroinflammation and gliosis but, in distinction to the earlier generation EP2 antagonist, did not mitigate neuronal injury or BBB breakdown | [187] |
| EP2 antagonist | |||
| Aspirin | Pilocarpine-induced SE | Reduced seizure frequency and duration | [188] |
| COX-2 inhibitor | Reduced pro-inflammatory cytokine production: PGE2, IL-6 and TNF-α in the hippocampus | ||
| Minocycline | Pilocarpine-induced SE | Inhibited microglial activation | [189] |
| Reduced hippocampal neuroinflammation | |||
| Microglial activation inhibitor | Prevented neuronal loss | ||
| Reduced seizure frequency, duration, and severity | |||
| GAO-3-02 | Amygdala-kindled model | Reduced seizure severity | [182] |
| Pilocarpine-induced SE | Reduced cognitive and memory deficits | ||
| Synaptamide | This study lacks of inflammation analysis | ||
| miR-146a | Intra-amygdaloid injection of kainic acid | Reduced seizure progression and frequency | [190] |
| IL-1R1/TLR4 activation | This study lacks of inflammation analysis | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinosa-Garcia, C.; Zeleke, H.; Rojas, A. Impact of Stress on Epilepsy: Focus on Neuroinflammation—A Mini Review. Int. J. Mol. Sci. 2021, 22, 4061. https://doi.org/10.3390/ijms22084061
Espinosa-Garcia C, Zeleke H, Rojas A. Impact of Stress on Epilepsy: Focus on Neuroinflammation—A Mini Review. International Journal of Molecular Sciences. 2021; 22(8):4061. https://doi.org/10.3390/ijms22084061
Chicago/Turabian StyleEspinosa-Garcia, Claudia, Helena Zeleke, and Asheebo Rojas. 2021. "Impact of Stress on Epilepsy: Focus on Neuroinflammation—A Mini Review" International Journal of Molecular Sciences 22, no. 8: 4061. https://doi.org/10.3390/ijms22084061
APA StyleEspinosa-Garcia, C., Zeleke, H., & Rojas, A. (2021). Impact of Stress on Epilepsy: Focus on Neuroinflammation—A Mini Review. International Journal of Molecular Sciences, 22(8), 4061. https://doi.org/10.3390/ijms22084061