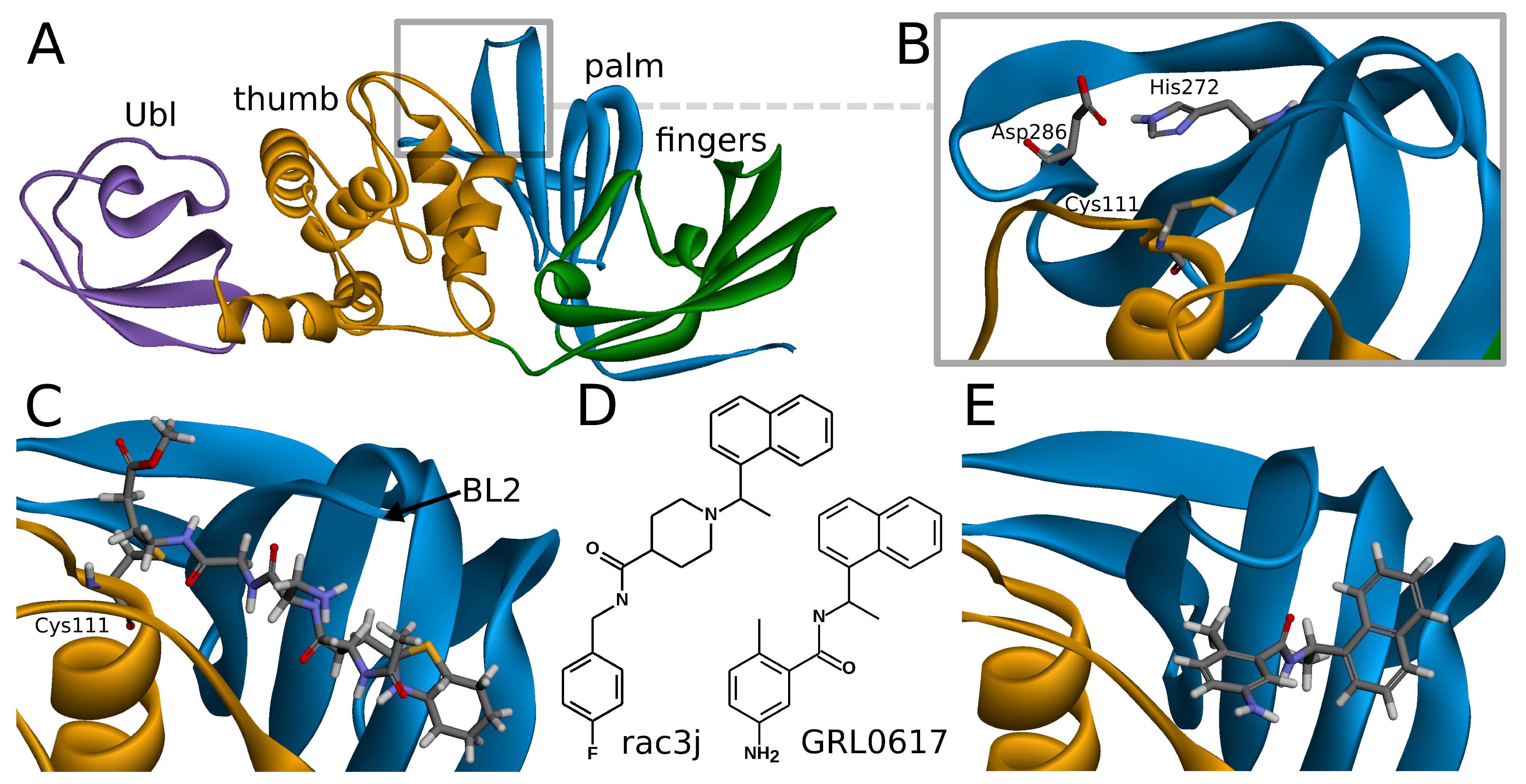
2.1. Overview of SARS-CoV-2 PLpro Structure and Its Known Inhibitors
SARS-CoV-2 PLpro is a monomeric enzyme that may be divided into two main domains—the catalytic domain and the ubiquitin-like (Ubl) domain. The first one is the interesting part in terms of enzymatic functions, as well as inhibiting the protein. It may be further divided into three subdomains: thumb, palm, and fingers (
Figure 1A). The active site is located between palm and thumb, utilizing three main residues, called the catalytic triad: Cys111, His272, and Asp286 (
Figure 1B) [
24].
Although SARS-CoV-2 PLpro is a relatively short known protein, its great significance for the virus, as well as being an immensely valid molecular target for novel potential drugs, led to great attention on it in the scientific world. Thus, multiple crystal structures of this enzyme are already available, including apo-protein [
24,
25] as well as structures of PLpro with ubiquitin, ISG15 [
35] or inhibitors [
24,
25,
30]. There are two main classes of SARS-CoV-2 PLpro inhibitors, with both solved tertiary structures of protein–inhibitor complexes and results of in vitro studies regarding their binding affinities.
Covalent inhibitors represent one of the most important types of compounds studied so far. In this group, the main direction seems to be focused on the peptide scaffolds [
30]. There are also other proposals for nonpeptide, covalent inhibitors, such as ebselen [
37], its derivatives [
31], and disulfiram [
38]. In the case of the peptide inhibitors, the covalent bond is formed with one of the amino acids of the catalytic triad—Cys111. As the structures of those compounds form quite long chains, the large part of the molecule is placed out of the active site and lies under the blocking loop 2 (BL2) in the palm subdomain (
Figure 1C) [
30].
Noncovalent PLpro inhibitors are the second main type of potential anti-SARS-CoV-2 drugs. In this group, the most significant and the most extensively studied compounds include inhibitors known to act on SARS-CoV PLpro and their derivatives. Such a direction is reasonable due to the high sequential similarity (90%) and identity (83%) of PLpro between SARS-CoV and SARS-CoV-2 [
24]. The SARS-CoV PLpro noncovalent inhibitors are in a big part members of one structural group of compounds, namely, the derivatives of N-[1-(naphthalen-1-yl)ethyl]benzamide (e.g., GRL0617) [
7]. Other proposals include, most notably, derivatives of N-benzyl-1-[1-(naphthalen-1-yl)ethyl]piperidine-4-carboxamide (e.g., rac3j), with a different arrangement of the scaffold in the center of the molecule [
35] (
Figure 1D). A recently developed class of GRL0617 derivatives retains its central N-ethylbenzamide part, but replaces the naphthyl group with a 2-phenylthiophene moiety [
36]. Importantly, the noncovalent inhibitors do not bind at the active site, but instead nearby, below the BL2, similarly to the part of the structure of the peptide, covalent inhibitors (
Figure 1E). This binding site is placed at the interface between palm and thumb. Residues from both of these subdomains take part in forming protein–ligand interactions. While most of the site is quite rigid, the crucial BL2 is a flexible loop, exhibiting considerable induced fit effects. Our analysis of available structures as well as conformations from explicit solvent molecular dynamics simulations implies that the conformation of the BL2 varies significantly, depending on the presence and the type of the inhibitor. Thus, not every tertiary structure of PLpro is equally suitable for computer-aided drug design and there is a need to rationally select applicable ones.
At present, new SARS-CoV-2 PLpro crystal structures are constantly determined, and to date, there are over 30 available at the Protein Data Bank (PDB) (
Supplementary Table S1). From the structure-based drug design perspective, the most crucial factor is the presence and the type of ligand at the binding site of interest. Thus, the PLpro structures may be divided into a few main groups. The first one includes crystals with the apo-enzyme, namely PDB IDs: 6wrh, 6wzu, 6xg3 [
24], 7cjd [
25], 6w9c, 7d47, 7d6h, and 7nfv. Because of the induced fit effect at the BL2, the apo conformations are in most cases the least useful ones for an application in structure-based computational methods. The second type of PLpro structures contains natural ligands, namely ubiquitin (PDB ID: 6xaa [
35]) and ISG15 (PDB IDs: 6yva [
11] and 6xa9 [
35]). Those structures should intuitively be more suitable than the apo ones. However, a more detailed analysis of the BL2 conformation and our validation show that the structures with ubiquitin or ISG15 are also not sufficient for the in silico screening. The third group contains PLpro with covalent, peptide inhibitors (PDB IDs: 6wuu and 6wx4 [
30]). Covalent inhibitors, apart from an obvious covalent bond, present a distinct binding mode compared to the noncovalent compounds. Once again, this is especially visible in the BL2 conformation, therefore limiting these crystal structures utility for noncovalent inhibitors’ design. The fourth group includes PLpro with cocrystallized GRL0617 or its close derivatives, namely PDB IDs: 7jir, 7jit, 7jiv, 7jiw [
24], 7cmd [
25], 7jn2, 7koj, 7kok, 7kol, 7krx, 7jrn, and 7cjm. These structures are well suited for the in silico screening. All the ligands in this group retain the N-[1-(naphthalen-1-yl)ethyl]benzamide scaffold of the GRL0617, varying only in the substituents attached to the phenyl group. Thus, the conformations of the BL2 remain almost exactly the same, and the aforementioned PDB entries exhibit only slight differences. Selection of one representative PLpro structure from this set is therefore a sufficient strategy. The next group contains GRL0617 derivatives with N-ethylbenzamide scaffold retained but with the naphthyl group replaced with a 2-phenylthiophene moiety (PDB IDs: 7lbr, 7lbs, 7llf, 7llz, and 7los [
36]). However, the altered fragment of the inhibitors is placed between the BL2 and the rest of the palm subdomain and in turn does not cause any significant BL2 conformational rearrangements compared to GRL0617-bound structures. The next type of the PLpro structure is the PDB ID: 7e35 with the derivative of rac3j. As the inhibitor cocrystallized in this entry possesses a distinct scaffold than GRL0617, the conformation of BL2 varies slightly in this structure. Thus, it may be utilized alternatively to or together with the aforementioned noncovalent inhibitor-bound PLpro structures in order to design potentially more diverse compounds. The last PLpro structure type is the PDB ID: 7m1y with ebselen. However, this compound is bound at a distinct site, and thus this PLpro conformation is not suitable for design of noncovalent inhibitors, similarly to apo-structures.
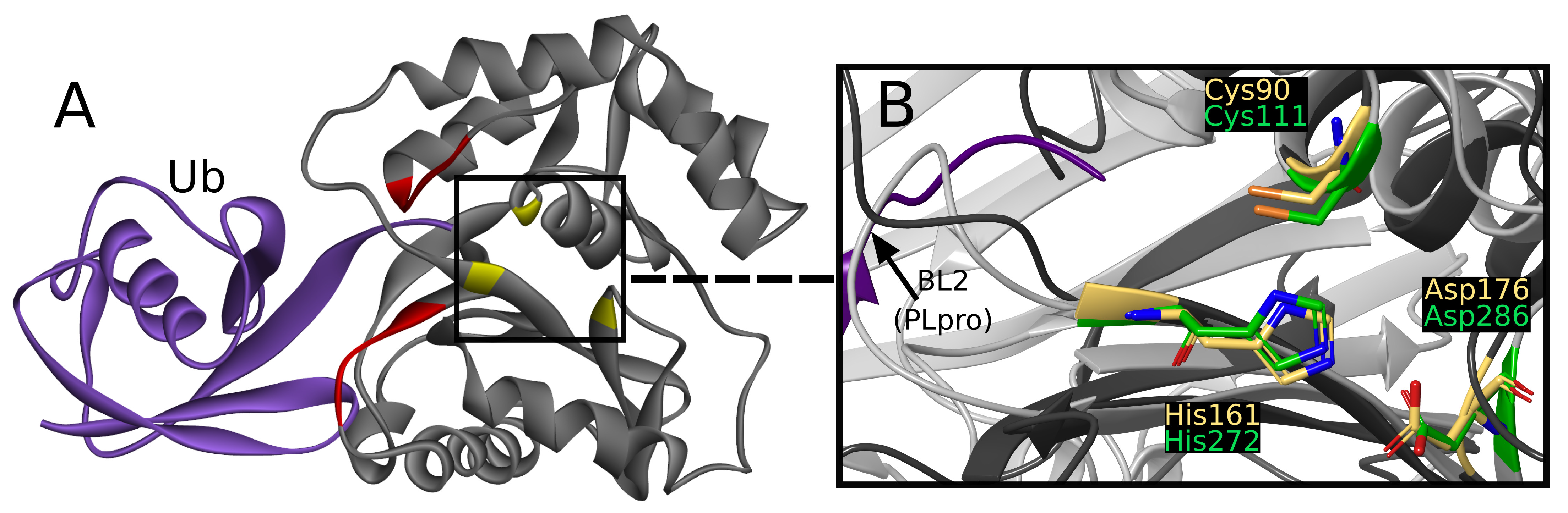
2.2. Overview of Human UCH-L1 and Its Similarity to SARS-CoV-2 PLpro
UCH-L1 is a proteolytic enzyme that hydrolyzes the peptide bond with glycine at ubiquitin’s C-terminus. Thus far, eight crystallographic structures have been solved, one of which was cocrystallized with a covalent, peptide inhibitor [
39] and other three with ubiquitin, which is considered to interact with two sites created with residues 5–10 and 211–216 (
Figure 2A) [
40].
In native state, UCH-L1 exists as a monomer. However, it was shown that this protein, unlike other UCH family members, can create an asymmetric homodimer while exhibiting additional ubiquitin ligase activity [
41,
42]. Its secondary structure comprises, similarly to UCH-L3, a helix-
-helix sandwich fold consisting of a right lobe—five
-helices, and left lobe—two
-helices and six
-strands [
41]. The highly folded protein backbone creates the 5
2 knot with a core length of 215 amino acids (residues 5-219), and a slipknot 3
1 containing 159 amino acids (residues 6-164) [
43,
44].
The active site is created by Cys90, His161, and Asp176, together called the catalytic triad [
45]. The catalytic triad of UCHs shows close resemblance to PLpro active site (
Figure 2B) [
46]. To the best of our knowledge, a comparative study of UCH-L1 and SARS-CoV-2 PLpro has not yet been conducted. Middle East respiratory syndrome coronavirus (MERS-CoV) PLpro and human UCH-L3 have been previously used together in experimental work in inhibitor research [
47].
While superimposing PLpro and UCH-L1 structures by catalytic triad, the resemblance of
-strands beneath PLpro BL2,
-helices that include Cys90, and loops above active site (
Figure 2B) can be seen. The similarity is sufficient to consider the risk of nonselectivity, although existing differences create possibilities of designing selective PLpro inhibitors.
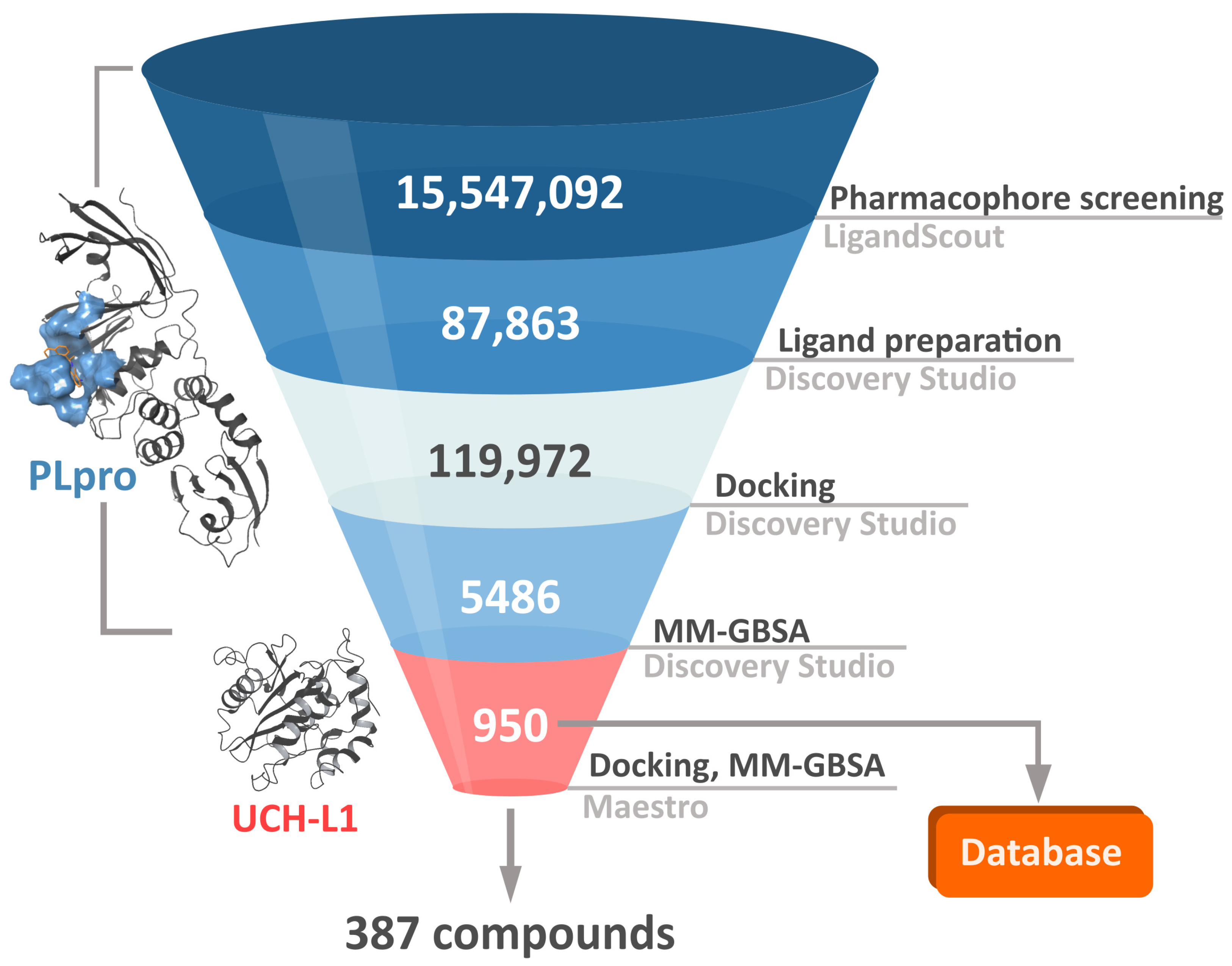
2.3. Virtual Screening Workflow
In order to both efficiently and accurately select potential PLpro inhibitors, we established a multistep workflow, including a diverse range of computational techniques. We started with relatively fast methods and, moving to the next steps, employed more accurate and time-costly ones (
Figure 3). As a chemical space to screen, we picked a library of over 15 million drug-like, diverse compounds from the ENAMINE REAL database.
The first step of our workflow was to efficiently screen the above-mentioned large ligand library. For this purpose, we selected pharmacophore screening, using LigandScout [
48]. As traditional ligand-based methods may have difficulties in finding novel structure groups, we utilized a mixed pharmacophore based on protein–ligand complexes obtained from the PDB. It contains descriptors derived both from a ligand’s chemical structure and from ligand–protein interactions. To further enhance our model’s ability to properly filter a broader spectrum of potential PLpro inhibitors, we merged a few such pharmacophores into a complex one. As components we used structures with SARS-CoV-2 PLpro cocrystallized with noncovalent inhibitors similar to GRL0617 [
24] or covalent inhibitor [
30], as well as SARS-CoV PLpro with a noncovalent inhibitor of another type than may be encountered in SARS-CoV-2 PLpro crystals present at the time, the derivative of N-benzyl-1-[1-(naphthalen-1-yl)ethyl]piperidine-4-carboxamide [
49]. In this case, elements of ligand-based drug design have an advantage of being biased toward compounds similar to those exhibiting moderate successes in both SARS-CoV and SARS-CoV-2 in vitro studies. However, as we aim to find potentially superior drug candidates, the procedure able to seek also slightly different structural groups is preferable. The decision to utilize a merged model consisting of various protein–ligand complexes allowed us to obtain a less restrictive pharmacophore, capable of spotting compounds with more diverse chemical structures compared to those from crystals.
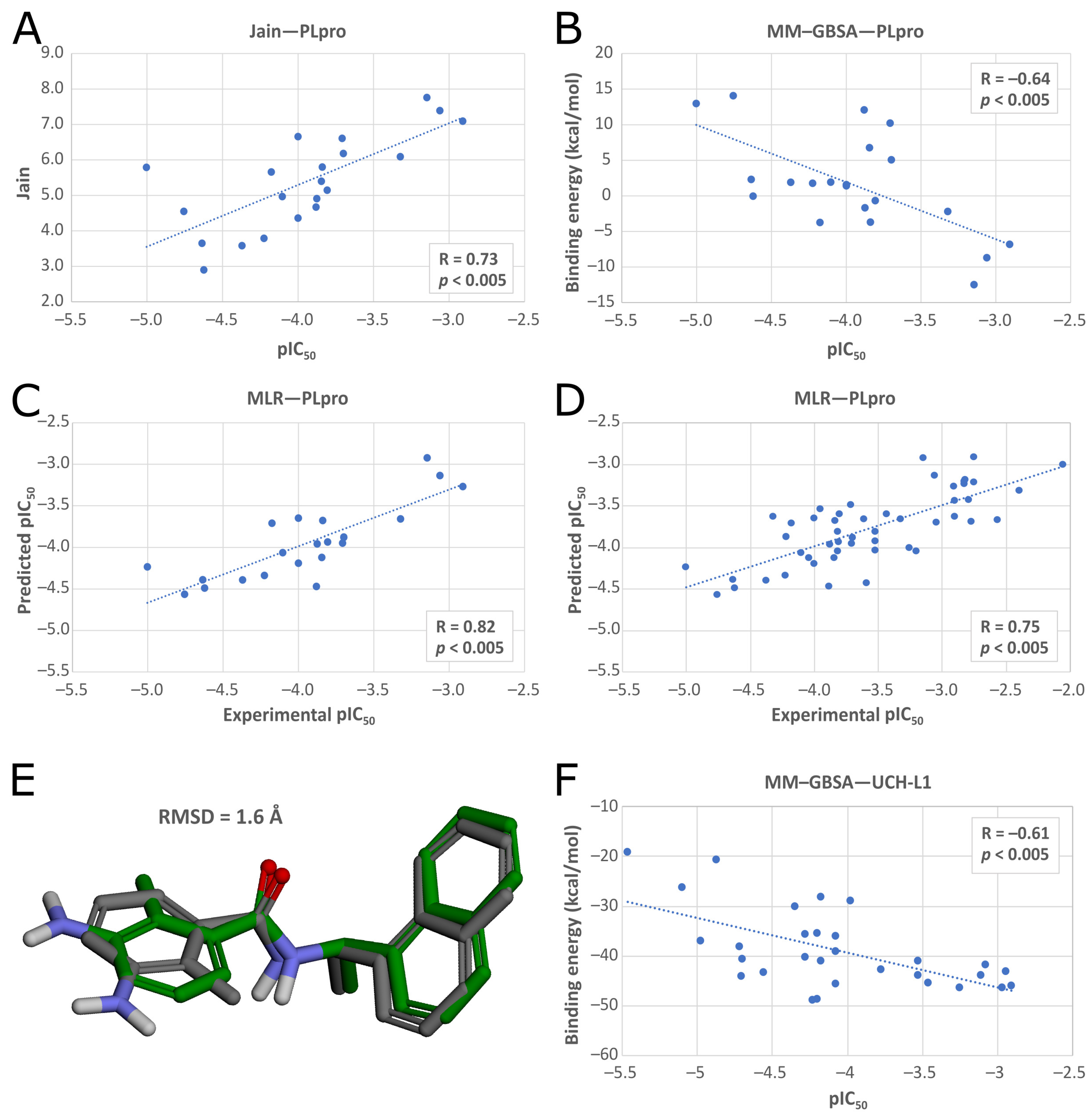
In the second phase of the screening campaign, we picked around 88,000 compounds with the best scoring function values from the previous step. In this part, we estimated binding affinities of selected compounds toward the PLpro binding site. We selected molecular docking as a semiaccurate and effective method to achieve this task. Additionally, this is a structure-based technique which allowed us to put more emphasis on the molecular target and consequentially allowed us to potentially find compounds more distinct from the PLpro inhibitors known so far. We utilized the Discovery Studio CDOCKER protocol—an accurate, rigid-protein docking program [
50]. We used PDB ID: 7jn2 structure as a model of the PLpro, as it performed best during our validation. Docked compounds were scored with Jain function. Then, as a more accurate measure of binding affinity prediction, we calculated binding energies of the 5486 best-scored compounds using the molecular mechanics–generalized Born and surface area solvation (MM–GBSA) method. After this step, we left 950 potential PLpro inhibitors. As our validation showed, in this case, both Jain and MM–GBSA binding energy correlated well with experimental pIC
50 values of PLpro ligands known so far. Thus, in order to more accurately estimate the potential inhibitors’ binding affinities, we established our own consensus function based on the multiple linear regression (MLR) for Jain and the binding energy from MM–GBSA. The combined model outperformed both its components alone when it comes to the correlation with the in vitro data. Using this model, we calculated predicted pIC
50 values for the potential PLpro inhibitors.
The third step of the project was aimed to predict the chosen compounds’ selectivity and to filter those that potentially have a weak to no affinity toward human UCH-L1. Here, once again, we employed molecular docking. Based on the comparison of different methods and on the validation results, we decided to use Schrödinger Glide [
51]. We docked 950 potential PLpro inhibitors to the model of UCH-L1 based on the PDB ID: 4jkj. For the obtained protein–ligand complexes, we calculated their binding energies with the MM–GBSA method. The results for the top candidates for PLpro inhibitors, regarding both PLpro and UCH-L1 calculations, are deposited in the database (
https://plpro-inhibitors.cent.uw.edu.pl) to facilitate future work.
Finally, it is important to stress that one of the most significant difficulties in the drug design process is the potential toxicity of the drug candidates. It may be time- and money-saving to assess the potential drug’s toxicity at the early stages of the design. For this purpose, computational techniques are becoming increasingly useful [
52]. In the case of the potential PLpro inhibitors, we focused mainly on the human analogous protein UCH-L1. However, there are many more factors needed to be taken into account. Thus, from the compounds with potentially low affinities toward UCH-L1, we selected 20 with lowest predicted IC
50 values toward PLpro, based on our MLR model. Then, we assessed the potential toxicity of these selected molecules. For this purpose, we utilized the Toxicity Estimation Software Tool (TEST). We estimated the mutagenicity, developmental toxicity, and rat LD
50 of the selected 20 compounds.
2.5. Analysis of the Best Scored Compounds
After all main phases of our screening, we obtained 950 potential PLpro inhibitors. Three hundred eighty-seven of those may be treated as potentially selective, meaning that they should potentially bind well to PLpro and weakly to UCH-L1. We selected the 20 best selective compounds according to their calculated pIC
50 toward PLpro using our MLR model (
Table 1,
Supplementary Figure S2). Their IC
50 values come between 159 and 505 nM. This suggests a potentially higher affinity toward PLpro binding site than for up-to-date synthesized inhibitors. For these 20 compounds, we conducted detailed analysis of their binding poses, the protein–ligand interactions they form, as well as their chemical structures.
2.5.1. Chemical Structures and PLpro Binding Modes
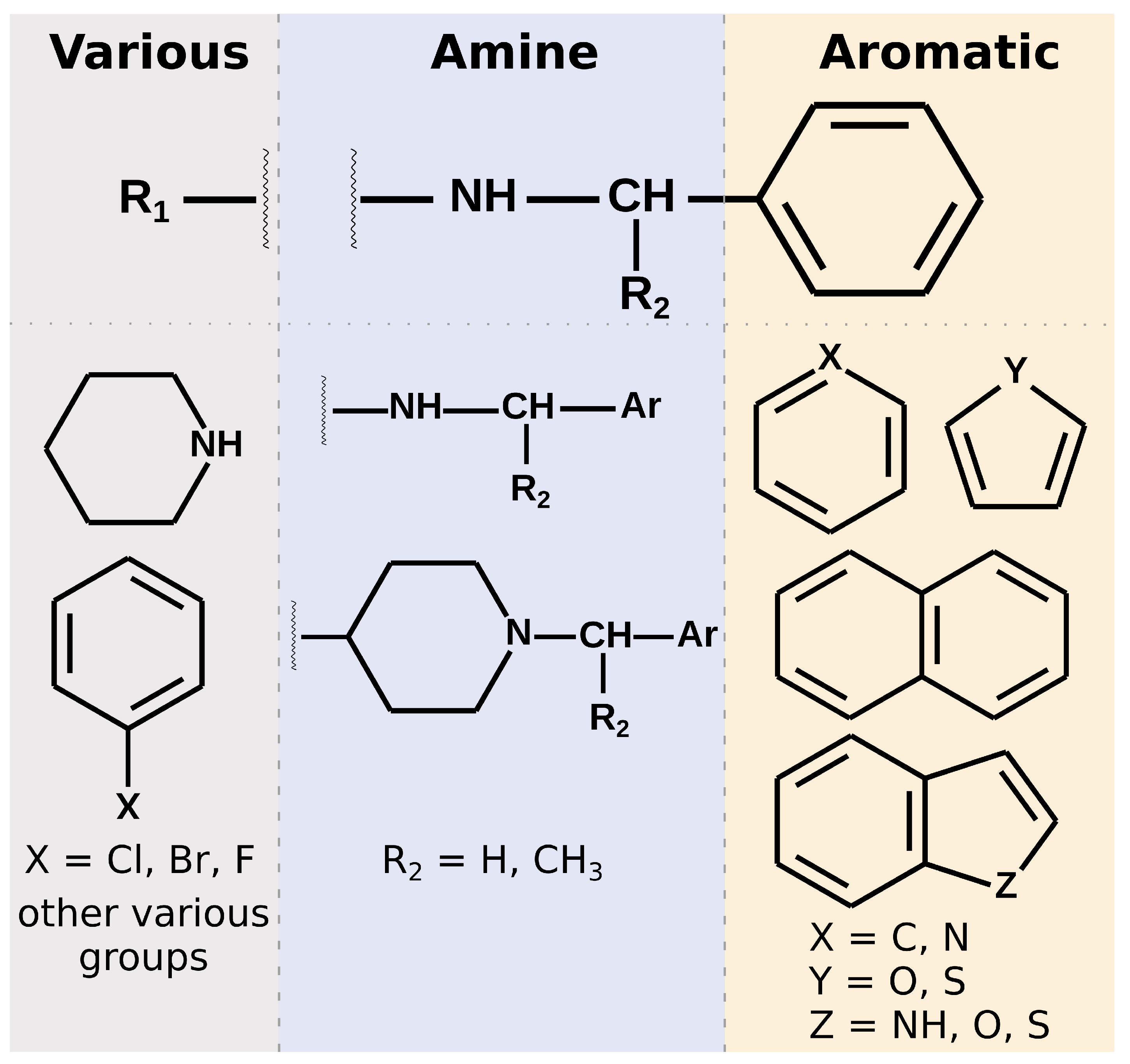
Most of the selected compounds possess similar structural features compared to the SARS-CoV-2 PLpro noncovalent inhibitors known so far. This is partially an expected outcome considering the structure–ligand-based character of the first step of the screening in LigandScout. However, it also shows that the latter phases in Discovery Studio favor similar compounds, even though there are also slightly different ones among the subset obtained after pharmacophore screening. The analysis of the compounds’ binding modes also shows that, in most cases, the 20 selected molecules adopt poses analogical to those from crystals with complexes of PLpro with noncovalent inhibitors. To simplify the analysis, the compounds’ structures may be divided into three parts. When looking from the perspective as in
Figure 1E, BL2 is placed above the inhibitor, the naphthyl group, closer to the fingers subdomain, is situated on the right side, whereas the part near the catalytic triad is located on the left. The right part usually consists of aromatic rings that most often form
–
interactions with Tyr268. The central part comprises crucial hydrophilic groups able to form important hydrogen bonds or salt bridges with nearby residues. Usually, it contains secondary amine groups or amide bonds. The right and central fragments are, in most cases, connected with a methylene group, similarly to the known PLpro inhibitors. The structure of the left part is more diversified and may form not only some hydrogen bonds or salt bridges but also other interactions and, in some cases, only weak ones (
Figure 6).
In detail, the right part of the crystal ligands is built by a naphthalene and forms
–
T-shaped interactions with Tyr268. In our set of the top 20 potential inhibitors, 18 compounds possess an aromatic ring (11) or a polycyclic aromatic scaffold (7) in this part. Among these 18, 12 ligands form interactions with Tyr268, ten of which have a
–
T-shaped character and the latter two are of
-S nature (specific type of the
–lone pair interaction). Interestingly, six of the
–
interactions are formed by polycyclic aromatic structures and only four by single aromatic rings, despite the higher occurrence of the latter. This indicates that in this part of the inhibitor, it is probably preferred to use polycyclic aromatic scaffolds. In the case of the aromatic rings that do not form interactions with Tyr268, they tend to create only other weak interactions instead, whereas the energetically substantially favorable contacts are present in the other parts of those compounds. In those cases, the binding mode of the whole molecule is also slightly different than in crystals (
Figure 7A,D) or in our 12 potential inhibitors described above that strongly interact with Tyr268 (
Figure 7B,E).
The central part of the noncovalent inhibitors from the crystal structures contains amide groups. In some cases, there may be also present a piperidine connected via the nitrogen atom to the right part of the molecule and via the carbon atom in position 4 to the amide linker in the left (e.g., PDB ID: 7e35). This part of the inhibitor is crucial for the proper steric fit to the narrowest pocket of the binding site, just under the BL2. The functional groups present in the center of the molecule are responsible for the most important interactions with nearby amino acids. These include mainly hydrogen bonds or in some cases salt bridges with Asp164, Tyr264, Tyr268, and Gln269. In the case of the latter two, the interactions are formed by the main chains of these amino acids. Thus, the induced fit effect is of great significance in this context, especially the conformation of the backbone of BL2. Hence, it may be difficult to spot such interactions for specific chemical structures of the inhibitors, specific conformations of PLpro or their combinations. Because we conducted docking to only a single, rigid PLpro structure, it is possible to miss some of the potentially important interactions, as our potential inhibitors slightly vary compared to known, crystallized, noncovalent inhibitors. However, the PLpro model based on PDB ID: 7jn2, that we used in this study, comprises a BL2 conformation with Tyr268 and Gln269 placed similarly to the most of the other crystal, inhibitor-bound structures. The utilization of a representative PLpro structure allows us to model the behavior of this crucial fragment of potential inhibitors in a satisfactory manner.
All of our 20 potential inhibitors in the central part of the molecule contain functional groups that create strong interactions with the binding site. Seventeen compounds possess a secondary amine group, while the other three—tertiary amine in a heterocyclic ring. Additionally, two compounds with secondary amine groups also include concurrent tertiary heterocyclic amines. Three other molecules have a second functional group of another type in the main part—amide, hydroxyl, or ester. All 20 compounds form salt bridges with Asp164. Six potential inhibitors create hydrogen bonds with Tyr273. Seven compounds form –cation interactions with Tyr264. There are also three molecules with hydrogen bonds with Gln269. Interestingly, these compounds possess more than one functional group in the central part, suggesting that it may be a valid strategy to include in this fragment multiple groups able to create hydrogen bonds or salt bridges.
Both the chemical structures and consequently the interactions formed by the left part of the PLpro inhibitors exhibit a greater variety compared to the rest of the molecule. This fragment of the crystal ligands consists of an aromatic ring or a polycyclic aromatic scaffold. However, it seems to serve little to no purpose itself when it comes to interacting with binding site residues. In some cases there are hydrogen bonds between substituents attached to the aromatic ring and Gln269, Tyr268 or Glu167. Hence, there is room to work on this part of the new potential inhibitors and achieve a structure more suitable for creating a larger number of important interactions with the binding site, compared to the known chemical compounds. In the case of our potential inhibitors, this part is also diversified. Ten of our compounds possess a heterocyclic scaffold, seven of which being a piperidine. Most of these molecules have binding poses placed in such a way to facilitate creating a salt bridge with Asp164, concurrently to a similar interaction of the same amino acid with the central part of the inhibitor. These are usually the compounds that adopt an overall slightly different binding mode than in crystals. They put a bigger emphasis on the interactions of the left part of the inhibitor and do not always form
–
interactions with Tyr268 with the right fragment, which is a characteristic feature of the crystal complexes. Thus, these compounds have the binding poses directed slightly more toward right and their right aromatic part toward bottom, further from the BL2 (
Figure 7C,H). Additionally, these potential inhibitors create
–cation interactions with Tyr264 using the left fragment (
Figure 7F), contrarily to the compounds with a more crystal-like binding mode that form the same interactions utilizing the central part of the molecule. When it comes to the other chemical constituents in the left fragment, there are three compounds containing a halogenphenyl group. Interestingly, two of them form
–
interactions with Tyr268 here, instead of the right part, being a third, least often observed binding mode. Additionally, these compounds create
–anion interactions with Asp164. Four other potential inhibitors contain various hydrophilic, noncyclic groups in the left fragment, including ester, amide, ether or amine groups. This set of ligands interacts with this region of the binding site in various manners, e.g., via hydrogen bonds with Gln269 or Gly163. Lastly, there are three compounds with only hydrophobic groups in the left part. They do not form any strong interactions using this fragment, owing their possibly high affinity to the favorable contacts in the other regions of the binding site. Similarly to many inhibitors from crystal structures, there may be a possibility to optimize the structure here.
Summarizing the chemical structure of the 20 analyzed potential inhibitors and their binding modes, a few key characteristics should be emphasized. In general, the structures of selected compounds are similar to those from crystal complexes. In the central part, all compounds possess functional groups forming crucial hydrogen bonds or salt bridges, most importantly with Asp164. In the right fragment, the vast majority of the molecules contain an aromatic ring or a polycyclic aromatic scaffold, and the latter seems to be favored. However, only 12 compounds utilize this part of their structure to form interactions with Tyr268, observed in nearly all crystals. A lesser number of potential inhibitors adopt a slightly distinct binding mode, with the lack of the above mentioned contact, and instead with a bigger role of the left fragment. This is especially valid for compounds with a piperidine in the left, as its nitrogen atom forms strong interactions with amino acids in the central pocket of the binding site. Overall, the left fragment of the selected compounds is most diversified both in terms of the chemical structure and interactions. This part seems to be the most promising one for a potential lead optimization.
2.5.2. Detailed Analysis of Compounds with Best Binding Modes
The visual inspection and the analysis of the binding modes of the top scored compounds show that overall they are placed at the binding site similarly to the inhibitors in the crystals. However, only some of them adopt exactly the same binding mode (
Figure 7G), while others slightly differ or adopt a wider range of binding modes (
Figure 7H). The experimental evidence and knowledge about PLpro inhibitors is still expanding. So far, the in vitro studies have included only a very limited range of structurally relatively similar compounds. Thus, it is difficult to judge whether molecules with different structural features, obtaining in silico slightly varying poses at the binding site, have their predicted binding modes well- or misrepresented. While they are alternative to those from crystals, and as such may be treated as potentially wrong, according to today’s knowledge it is impossible to state that certainly. Hence, if one would want to assess their binding affinity in vitro, it is not an unreasonable choice. Nevertheless, such a direction could be more risky, compared to compounds with binding modes nearly identical to the crystal ones. Therefore, we will focus on such molecules with more conserved poses and will analyze in more detail a few selected, safe proposals.
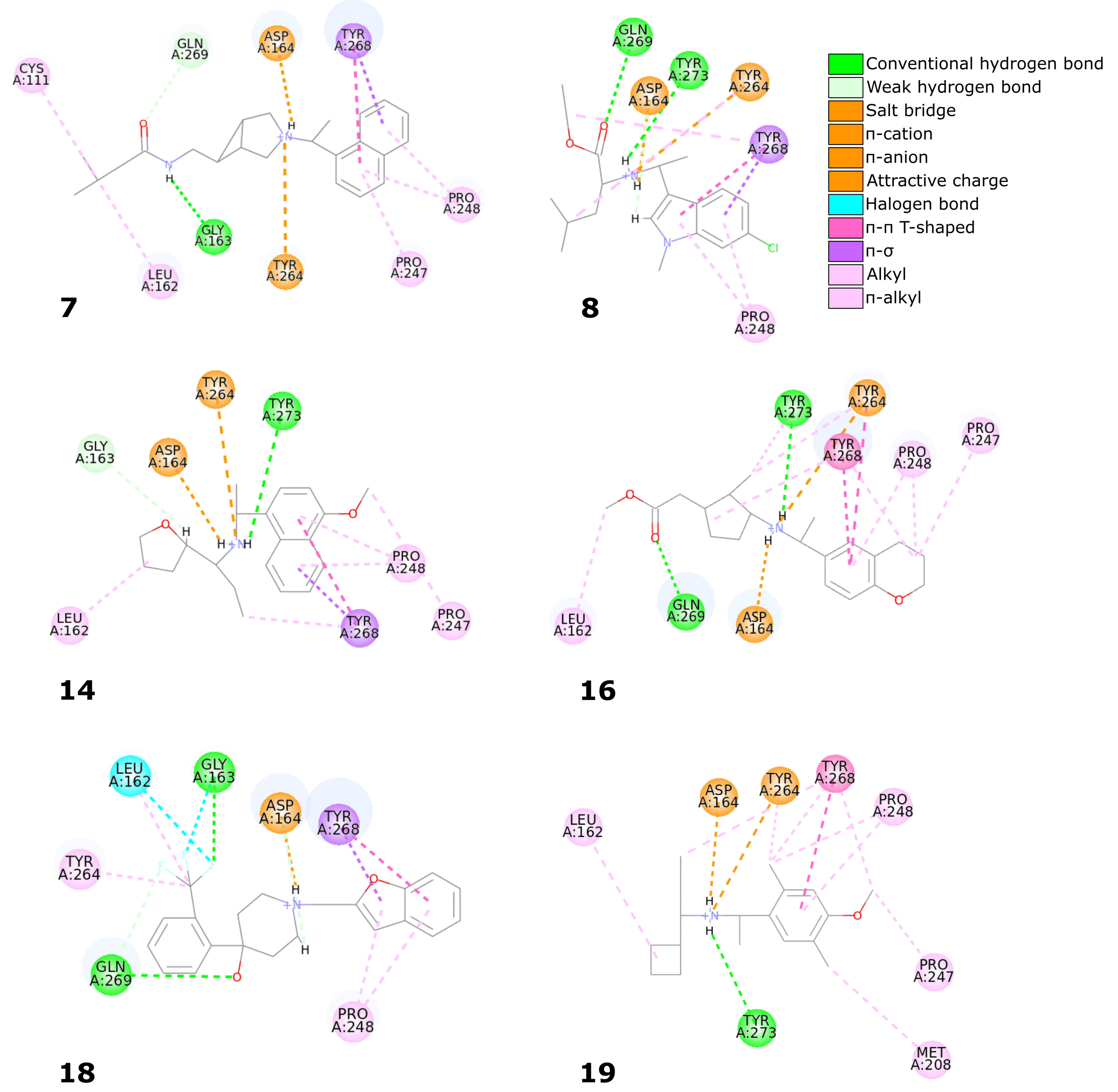
Six compounds from the set of top 20 potential inhibitors (compounds
7,
8,
14,
16,
18, and
19) displayed nearly identical binding mode to the one observed in the SARS-CoV-2 PLpro crystal structures (
Table 1,
Figure 8). Therefore, we conducted a more detailed analysis of the six molecules based on the PLpro complex with PDB ID: 7jn2.
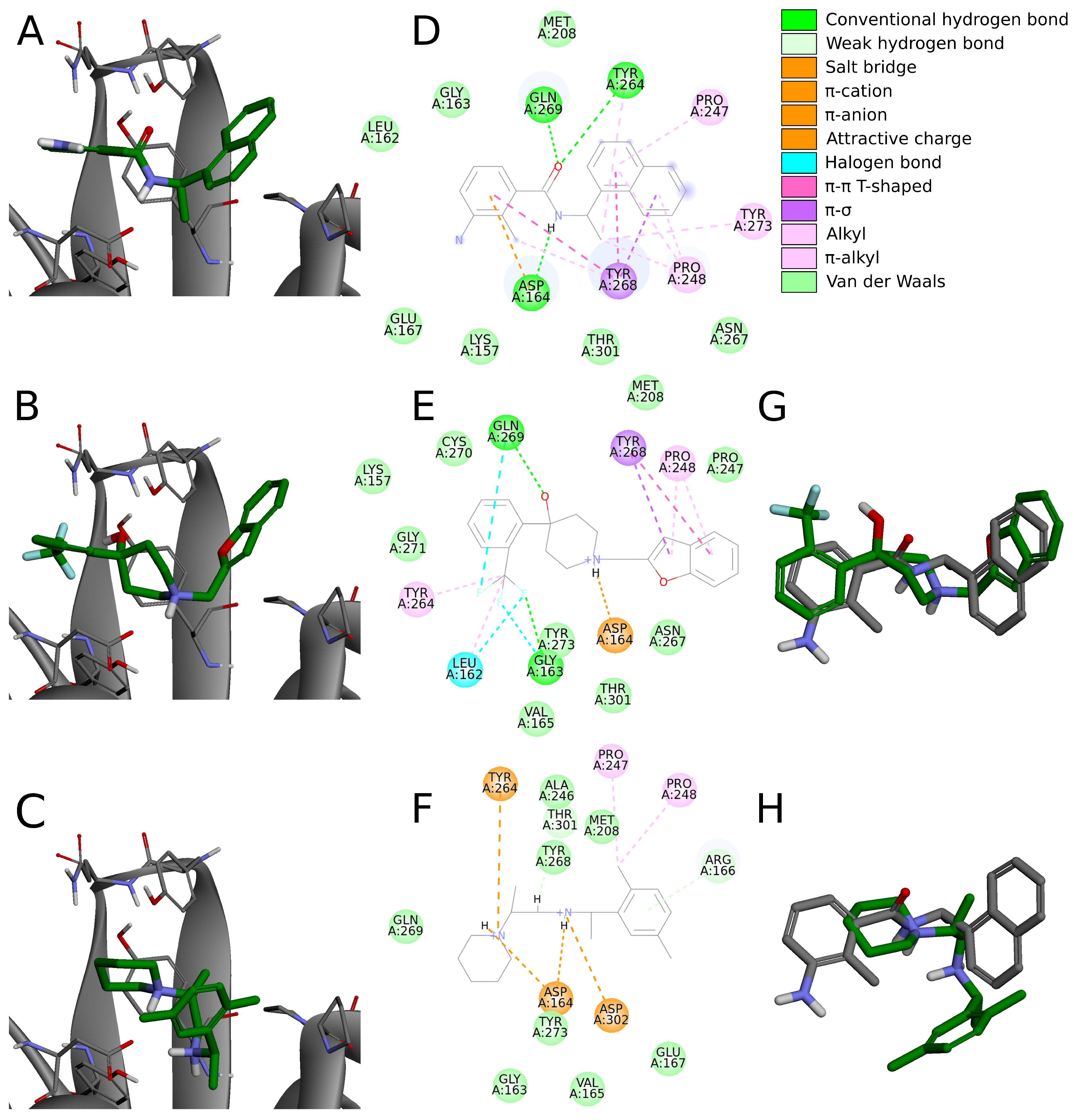
The biggest similarity to the binding of the cocrystallized ligands was observed for the right and central fragments of the compounds. The right part of all six molecules was composed of one or two fused aromatic rings occupying a hydrophobic cavity hedged by Pro247 and Pro248. This fragment of the selected compounds was buried nearly in the same position as the naphthyl group from the cocrystallized ligand, thereby creating similar interactions. The aromatic rings of the inhibitor from the PDB structure formed – T-shaped interaction with Tyr268 and alkyl interactions with Pro247 and Pro248. The first one played the main role in stabilizing the right part of the compound and was observed in all complexes with the six potential inhibitors. The latter interactions were maintained in most cases.
Compounds 7 and 14 showed the biggest similarity in binding of the right part of the molecules. It was due to the fact that these were the only compounds composed of a naphthalene (compound 7) or its derivative (compound 14). The substituent in the naphthyl group of the latter compound had no significant impact on its binding beside the additional alkyl interaction with Pro247. The right fragment of compounds 18 and 16 showed a slightly bigger shift from the naphthyl group of the ligand from the crystal structure than the other four potential inhibitors. The benzofuran rings of the compound 18 were more shifted toward the BL2, causing the loss of an alkyl interaction with Pro247, observed in the rest of the complexes. The benzene ring from the chromane forming the right part of the compound 16 was accommodated higher than one of the naphthalene rings from the cocrystallized inhibitor, enabling the molecule to form – T-shaped interactions not only with Tyr268 but also with Tyr264.
The central fragment of the inhibitor from the crystal structure consisted of an amide group, which was stabilized by the hydrogen bonds formed with Gln269, Tyr264, and Asp164. Furthermore, multiple alkyl interactions were established between the target protein and a methylene group connecting the right and central part of the ligand. Four out of six potential inhibitors (compounds 8, 14, 16, and 19) possessed an acyclic secondary amine group in the central part of the molecule that, similarly to the crystal structure, was connected to the right part with a methylene group. All compounds were stabilized by the salt bridges formed between the amine nitrogen and Asp164, which were analogous to the hydrogen bond established in the crystal complex. Although the four compounds lacked the oxygen atom, which was a hydrogen bond acceptor in the crystal structure, the interaction with Tyr264 was still established in the form of the –cation interaction. We observed that the amine nitrogen atoms from the central part of these four molecules were positioned deeper in the binding site than the amide nitrogen from the cocrystallized ligand. It allowed the central fragment of these potential inhibitors to be additionally stabilized by the hydrogen bond formed with Tyr273.
Unlike the previous four molecules, the central fragment of the compounds 7 and 18 was composed of a tertiary heterocyclic amine. Although both molecules also possessed a methylene group connecting their right and central part, the interactions formed by these two compounds were slightly different. Similarly to the crystal ligand, compound 7 formed the salt bridge and –cation interactions with Asp164 and Tyr264, respectively. Compound 18 established an additional interaction. The central part of the molecule consisted of a 4-hydroxypiperidine ring. After superimposing the complex with compound 18 onto the crystal structure, we observed that the oxygen atoms from both ligands were localized in the similar position, allowing the potential inhibitor to establish an additional hydrogen bond with Gln269, apart from the salt bridge formed with Asp164.
The left fragment was the most diverse among the compounds. The ligand from the crystal structure possessed the 2-amine-1-methylphenyl group in its left part, which was mainly stabilized by the – T-shaped interaction with Tyr268 and –anion with Asp164. The left fragment of potential inhibitors was composed of various groups. However, not only the chemical properties affected the binding of the compounds in the left part of the PLpro binding site, but also their size. Compound 7 was longer than the cocrystallized ligand and did not bind in the bent conformation around the BL2. Therefore, the molecule established a hydrogen bond with Gly163 and alkyl interactions with Cys111 and Leu162. The other potential inhibitors were of similar length to the ligand from the crystal structure. The left terminal part of the compounds 8 and 16 consisted of the ethoxycarbonyl and methoxycarbonyl groups, respectively. Both compounds were stabilized by a hydrogen bond formed between the carbonyl oxygen and Gln269. In terms of the left fragment, compound 18 stood out the most from the rest of potential inhibitors. The molecule possessed the trifluoromethylphenyl group, which was accommodated in the similar position as the benzene ring from the cocrystallized inhibitor. The compound established multiple strong interactions with the target protein—four halogen bonds between the fluorine atoms and the residues Gln269, Leu162, Gly163, and also a hydrogen bond with the latter. All molecules were additionally stabilized by a few alkyl interactions.
In conclusion, the six potential PLpro inhibitors displayed similar binding mode to the noncovalent ligands from the crystal structures and in some cases they also formed additional interactions, which is one of the main factors of their potentially very high binding affinity. Most interactions stabilizing the naphthyl group of the cocrystallized inhibitor were maintained in the analogous groups of all six compounds. The central fragment of the analyzed molecules formed several interactions similar to the ones observed in the compared crystal structure, but it also established some additional ones, in most cases strong hydrogen bonds. The fragment located the closest to the catalytic triad was the most diverse among the compounds. Although in some cases this part of the molecules formed strong interactions with the PLpro binding site, several potential inhibitors were only stabilized by the alkyl interactions. Therefore, optimization of this fragment of the compounds may lead to the enhancement of binding affinity.
2.5.3. Binding Modes from the Perspective of the Protein
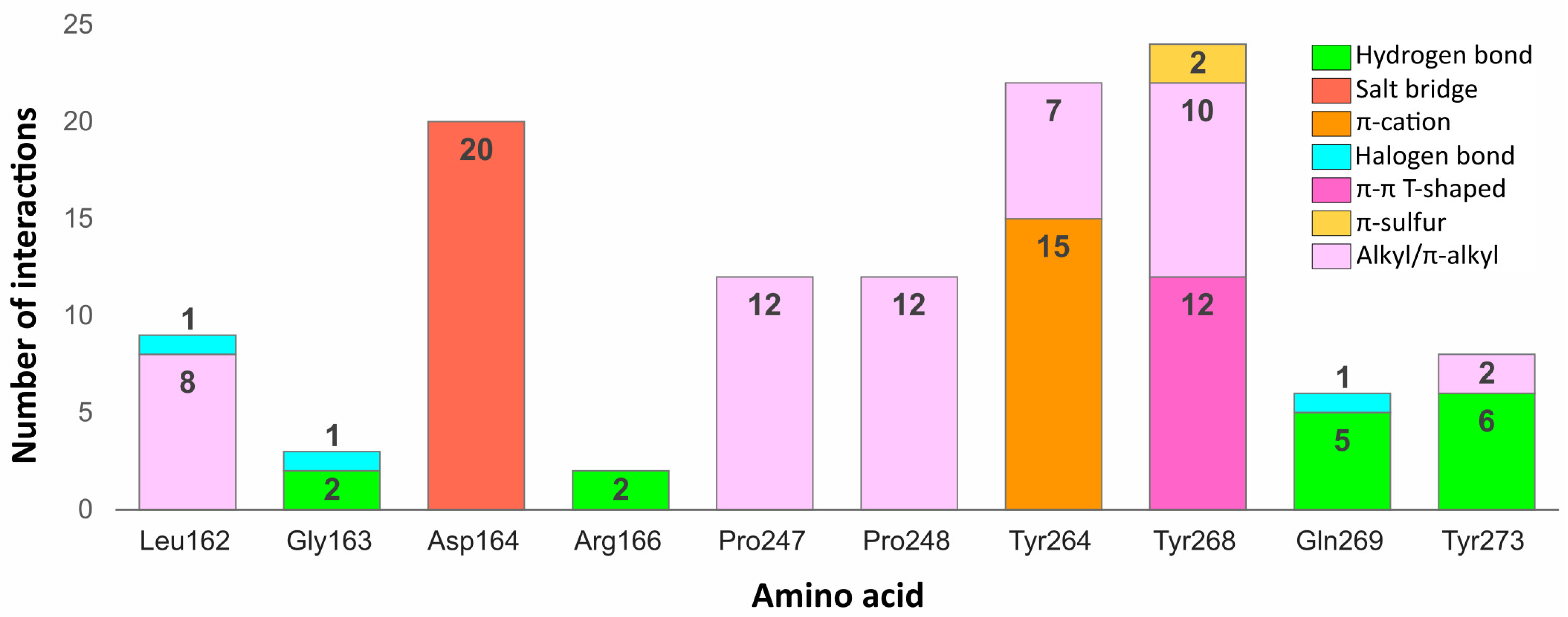
Some residues in the PLpro binding site were especially important in forming interactions with the potential inhibitors (
Figure 9). Two of the key amino acids were Tyr268 and Gln269 placed in the flexible BL2. The first residue stabilized the ligand poses by forming interactions with the aromatic rings mainly located in a hydrophobic cavity hedged by Pro248, Pro247, and Met208. The side chain of Tyr268 established
–
T-shaped interactions with 12 ligands and
-S with two ligands from the set of top 20 potential inhibitors. Gln269, however, was involved in stabilizing the central part and the other end of the compounds by creating hydrogen bonds with five potential inhibitors, three of which interacted with the backbone and two with the side chain of the amino acid. The interactions formed between the molecules and the residues Tyr268 and Gln269 were similar to those observed in most inhibitor-bound SARS-CoV-2 PLpro crystal structures.
The flexible BL2 may adopt various conformations among different structures depending on the interacting compound. However, due to the fact that most cocrystallized SARS-CoV-2 PLpro inhibitors adopt nearly identical binding mode, the BL2 of the analyzed crystals, especially the backbone and side chains of two important binding residues—Tyr268 and Gln269, show great conformational similarities. One of the few examples, where the BL2 is differently arranged is the structure with PDB ID: 7e35, bound to the compound with a different chemical structure (derivative of rac3j) and a binding mode compared to the rest of the crystal ligands. In this case, the backbone of Tyr268 and Gln269, and the side chain of the latter residue adopt a more open conformation due to the induced fit mechanism. This might result in a decreased formation of some of the aforementioned interactions, namely the hydrogen bonds established between certain potential PLpro inhibitors and the backbone and side chain of Gln269. However, the carbonyl oxygen of Tyr268 in the structure with PDB ID: 7e35 is shifted toward the binding site, which may potentially induce the formation of new hydrogen bonds. Additionally, considering that the aromatic side chain of Tyr268, engaged in forming important – contacts, adopts an almost identical conformation in all of the analyzed inhibitor-bound SARS-CoV-2 PLpro crystals, it is highly probable that the overall interaction profile would be maintained in various structures, regardless of their BL2 arrangement.
Another residue, which played an important role in stabilizing both the cocrystallized inhibitors and selected compounds was Asp164. The amino acid occupying the central part of the PLpro binding site, formed salt bridges with all 20 potential inhibitors, which were analogous to the hydrogen bonds observed in the SARS-CoV-2 PLpro crystal complexes.
Two tyrosine residues located close to the BL2 were also engaged in forming relevant interactions with selected compounds. Tyr264 created -cation interactions with the protonated nitrogen atoms from the central or left fragment of 15 molecules. The second tyrosine residue—Tyr273, formed hydrogen bonds with amine groups of six compounds from the set. Although the latter amino acid did not form relevant interactions in the PLpro crystal complexes, it turned out to be an important residue for binding some of the top 20 potential inhibitors.
Apart from the mentioned tyrosine, there were a few more amino acids, which formed relevant interactions with the PLpro potential inhibitors, despite not playing any important role in stabilizing the cocrystallized ligands. These were mainly residues occupying the left part of the binding site, which was localized closer to the catalytic triad. Arg166 and Gly163 were both engaged in binding two compounds by forming the hydrogen bonds with each. In one of the newly obtained complexes, we observed the appearance of a type of interaction, which was not present in the crystal structures, namely the halogen bond. The interaction was formed with Leu162, Gly163, and previously analyzed Gln269 from the BL2.
We noticed some similarities between the weak interactions established in the PLpro crystal structures and the complexes obtained from the virtual screening workflow. The right part of the molecules occupied a hydrophobic cavity formed by Pro248, Pro247, and Met208. Sixteen out of 20 analyzed ligands were engaged in forming alkyl or -alkyl interactions with one or both prolines from the pocket, which resembled the binding of the naphthyl group in the cocrystallized inhibitors. The amino acids from the central part of the PLpro binding site—Tyr264, Tyr268, and Tyr273 also showed an analogous tendency of forming -alkyl interactions. Leu162, which was one of the most often encountered residues stabilizing the left part of potential inhibitors with alkyl interactions, was not involved in binding the ligands from the crystal structures.
In conclusion, most of the interactions formed between potential inhibitors and the target protein were analogous to those observed in the SARS-CoV-2 PLpro crystal structures. However, some amino acids, which did not seem to be relevant in binding the cocrystallized inhibitors, turned out to be engaged in forming important interactions in the complexes obtained as a result of our research. Thus, there is a high possibility that more PLpro binding site residues could be engaged in forming relevant interactions than it may appear from the analysis of the crystal structures. Therefore, it is feasible to design new, potent PLpro inhibitors, which would interact with a greater number of amino acids than the cocrystallized compounds reported so far.
2.5.4. UCH-L1 Binding Modes
We examined the protein–ligand interactions of UCH-L1 using the top 20 potential PLpro inhibitors from the screening. For those compounds, obtained docking scores suggest a low probability of binding to UCH-L1. To support this result, we analyzed the complexes obtained from docking to the UCH-L1 from the PDB ID: 4jkj crystal structure.
We compared protein–ligand interactions to the 4dm9 crystal structure as a reference, as it is the only available UCH-L1 PDB structure with an inhibitor. 4dm9 was cocrystalized with the covalent inhibitor Z-VAE(OMe)-FMK (benzyloxycarbonyl-Val-Ala-Glu(
-methoxy)-fluoromethylketone) [
39]. The compound irreversibly modifies UCH-L1 by binding covalently to Cys90, which forms, along with His161 and Asp176, the catalytic triad [
45].
Despite that 4dm9 may not be an ideal reference structure, due to the different ligand binding type, the similarities are sufficient to provide relevant comparison. The described comparative interaction analysis of the covalently bonded Z-VAE(OMe)-FMK and other noncovalently bonded compounds constitutes a reliable foundation for the prediction of potential toxicity.
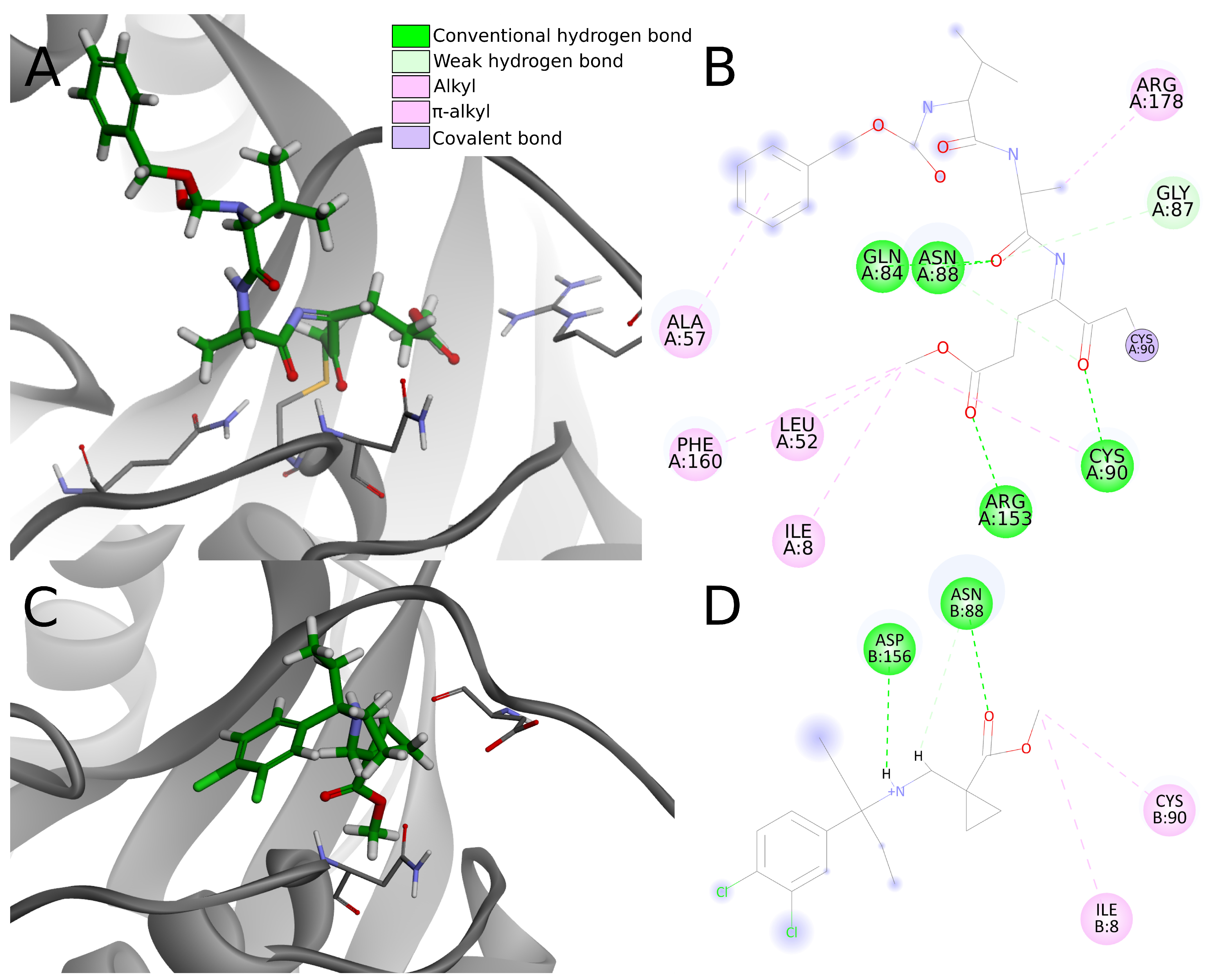
In the analysis of the 4dm9 crystal structure, apart from covalent bonds, noncovalent interactions can be seen: hydrogen bonds with Gln84, Asn88, Cys90, and Arg153, weak carbon–hydrogen bonds with Arg153, alkyl hydrophobic interactions with Ile8, Leu52, Cys90, and Arg178 and
–alkyl hydrophobic interactions with Ala57 and Phe160 (
Figure 10A,B).
Considering interactions appearing in the 4dm9 crystal, among the top 20 compounds docked to 4jkj three create hydrogen bonds with Gln84, eight with Asn88, two with Cys90 and none with Arg153. In terms of hydrophobic interactions, eight compounds create alkyl bonds with Ile8 and 11 with Cys90. Three of the chosen ligands show none of the interactions indicated for the 4dm9 crystal structure and another six—only one of indicated interaction.
The chosen compounds also create other interactions not occurring in the reference crystal structure. Seventeen out of 20 compounds form hydrogen bonds with Asp156 and seven with Val158. Nine compounds create salt bridges with Asp155 and 16 compounds form an attractive charge interaction with Asp155 or Asp156.
As hydrogen bonds are strong interactions and are considered as the most important ligand binding factor, summing them up enables clear comparison. Five compounds (numbers 1, 2, 5, 15, and 18) create one hydrogen bond, nine (3, 4, 6, 7, 9, 11, 13, 19, 20) have two hydrogen bonds, five (8, 10, 12, 14, 17)—three hydrogen bonds and only one, compound 16, creates four. As it can be seen, the vast majority of the compounds (19 out of 20) create fewer hydrogen bonds than are present in the reference crystal structure. Four of the compounds have unfavorable interactions, including compound 16 (donor-donor unfavorable interaction with Ans88), which had been pointed out before as having a greater number of hydrogen bonds.
Differences in binding strength become more pronounced considering the fact that in Z-VAE(OMe)-FMK the strong covalent bond provides stable ligand binding in the active site. Noncovalent interactions have only a supportive function, so there are fewer of them. Compounds chosen in the conducted screening are designed as noncovalent inhibitors, so they bind to the protein thanks to multiple noncovalent, relatively weaker interactions.
Having less or the same number of strong noncovalent interactions as in 4dm9, the top 20 potential PLpro inhibitors have very little chance to create stable binding to UCH-L1. Apart from the lack of a covalent bond, most compounds create less than three conventional hydrogen bonds, one or two electrostatic interactions, less than three hydrophobic interactions and multiple weak interactions such as carbon hydrogen bonds (
Figure 10C,D). Although the overall number of interactions is greater in some cases, this does not necessary imply stronger binding affinity. Lacking a covalent bond and abundant, strong noncovalent interactions, compounds are rather unable to create stable binding to UCH-L1.
Even though some of the compounds create hydrogen bonds and salt bridges with UCH-L1 residues, it is unlikely that they are sufficient to induce strong ligand binding. Together with the docking score results, protein–ligand interaction analysis suggests that the compounds chosen in the screening have a low probability of exhibiting toxicity due to inhibiting UCH-L1.
2.5.5. Toxicity Estimation
For top 20 potential PLpro inhibitors from screening, we conducted an approximative toxicity prediction. Based on the results, only three compounds exhibit mutagenicity. Thirteen compounds are probably developmental toxicants. Most compounds show a relatively high LD
50 value. In general, the top 20 compounds do not appear to be particularly toxic, however most of them may not be suitable for pregnant women and children (
Table 2).


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}