
Anti-Angiogenic Therapy: Current Challenges and Future Perspectives




Abstract
1. Introduction
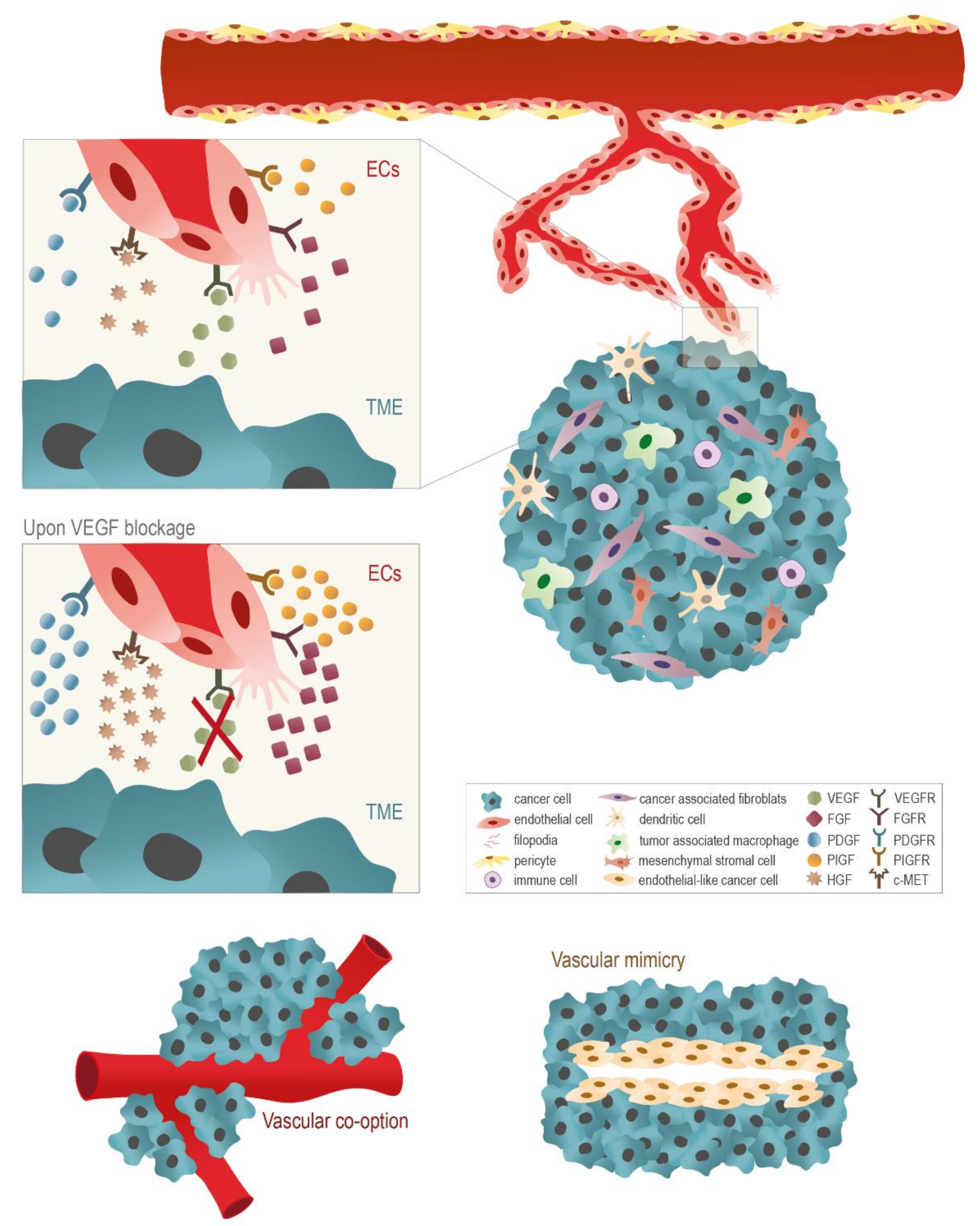
Anti-Angiogenic Therapy, Focused on VEGF, for Cancer Treatment
2. Targeting Alternative Angiogenic Signaling Pathways to Weaken Cancer
2.1. Fibroblast Growth Factors (FGFs)
2.2. Angiopoietins (ANG)
2.3. Platelet-Derived Growth Factor (PDGF)
2.4. Hepatocyte Growth Factor (HGF)/c-MET
2.5. Placental Growth Factor (PIGF)
2.6. Alternative Anti-Angiogenic Factors
3. The Use of Anti-Angiogenic Agents in Cancer: A Disappointing Therapeutic Strategy
4. The Versatile Use of Anti-VEGF Agents to Enhance Immunotherapies
5. The Potential Use of Other Therapeutic Strategies Targeting Cancer Cells and Impacting Angiogenesis
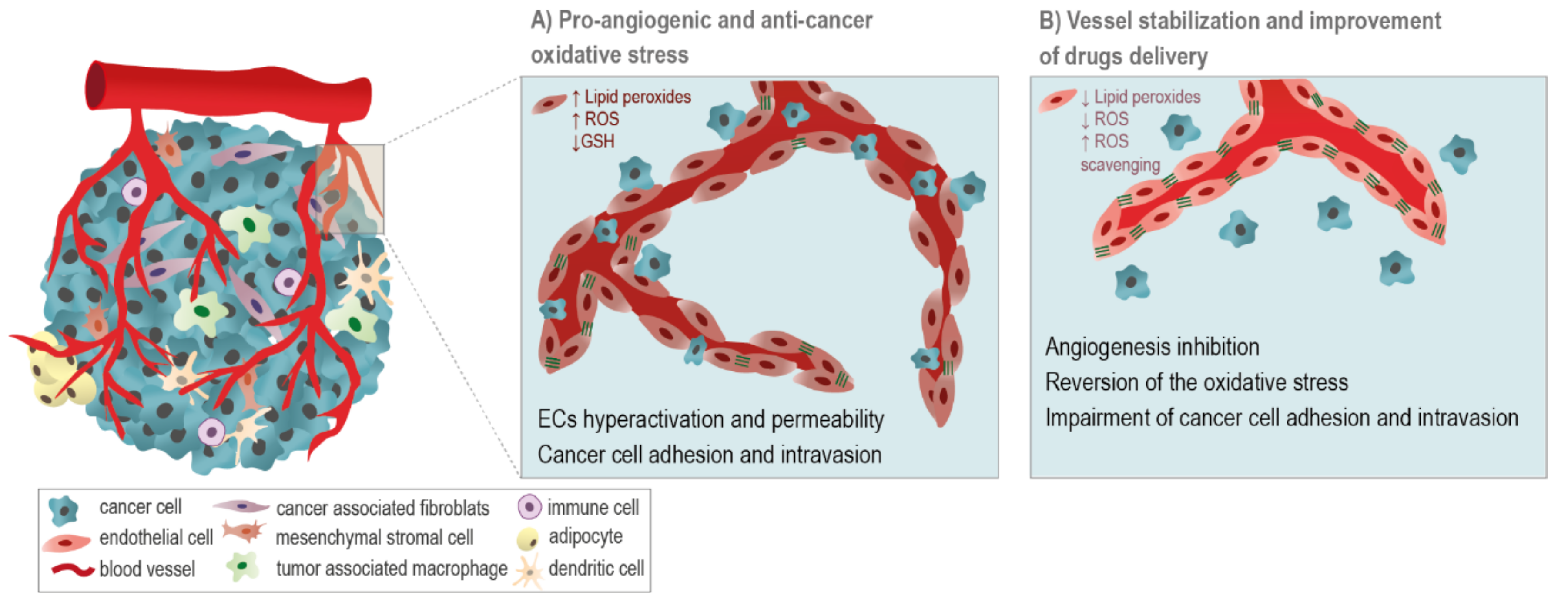
5.1. ROS-Related Drugs: A Double-Edge Sword
5.2. β-Adrenergic Drugs: Repurposing Existing Drugs for Anti-Cancer and Anti-Angiogenic Clinical Purposes
5.3. The Anti-Angiogenic Modulatory Role of Oxidative Stress and DNA Repair Controllers
6. Final Remarks
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug Resistance and the Solid Tumor Microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; di Tomaso, E.; McDonald, D.M.; Jones, R.; Jain, R.K.; Munn, L.L. Mosaic Blood Vessels in Tumors: Frequency of Cancer Cells in Contact with Flowing Blood. Proc. Natl. Acad. Sci. USA 2002, 97, 14608–14613. [Google Scholar] [CrossRef]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef]
- Domingues, G.; Fernandes, S.G.; Serpa, J. Dynamics of VEGF-A and its Receptors in Cancer Vascularization—An Overview. In Understand Cancer: Research and Treatment; iConcept Press: Hong Kong, China, Chapter 3; 2015; ISBN 978-1-922227-386. [Google Scholar]
- Gacche, R.N. Compensatory Angiogenesis and Tumor Refractoriness. Oncogenesis 2015, 4, e153. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus Guidelines for the Use and Interpretation of Angiogenesis Assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef]
- Masiero, M.; Simões, F.C.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.Y.Y.; Pradeep, S.; Li, D.; et al. A Core Human Primary Tumor Angiogenesis Signature Identifies the Endothelial Orphan Receptor ELTD1 as a Key Regulator of Angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef]
- Folkman, J. Successful Treatment of an Angiogenic Disease. N. Engl. J. Med. 1989, 320, 1211–1212. [Google Scholar] [CrossRef] [PubMed]
- Zetter, B.R. The Scientific Contributions of M. Judah Folkman to Cancer Research. Nat. Rev. Cancer 2008, 8, 647–654. [Google Scholar] [CrossRef]
- Vasudev, N.S.; Reynolds, A.R. Anti-Angiogenic Therapy for Cancer: Current Progress, Unresolved Questions and Future Directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Bergers, G.; Javaherian, K.; Lo, K.M.; Folkman, J.; Hanahan, D. Effects of Angiogenesis Inhibitors on Multistage Carcinogenesis in Mice. Science 1999, 284, 808–812. [Google Scholar] [CrossRef]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the Tumour Vasculature: Insights from Physiological Angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor Angiogenesis: Past, Present and the near Future. Carcinogenesis 2000, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-Specific Growth Factors and Blood Vessel Formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhong, Z.; Huber, J.; Bassi, R.; Finnerty, B.; Corcoran, E.; Li, H.; Navarro, E.; Balderes, P.; Jimenez, X.; et al. Anti-Vascular Endothelial Growth Factor Receptor-1 Antagonist Antibody as a Therapeutic Agent for Cancer. Clin. Cancer Res. 2006, 12, 6573–6584. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hooper, A.T.; Zhong, Z.; Witte, L.; Bohlen, P.; Rafii, S.; Hicklin, D.J. The Vascular Endothelial Growth Factor Receptor (VEGFR-1) Supports Growth and Survival of Human Breast Carcinoma. Int. J. Cancer 2006, 119, 1519–1529. [Google Scholar] [CrossRef]
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson, A.B. Bevacizumab in Combination with Oxaliplatin, Fluorouracil, and Leucovorin (FOLFOX4) for Previously Treated Metastatic Colorectal Cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544. [Google Scholar] [CrossRef]
- Taylor, A.P.; Rodriguez, M.; Adams, K.; Goldenberg, D.M.; Blumenthal, R.D. Altered Tumor Vessel Maturation and Proliferation in Placenta Growth Factor-Producing Tumors: Potential Relationship to Post-Therapy Tumor Angiogenesis and Recurrence. Int. J. Cancer 2003, 105, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug Resistance by Evasion of Antiangiogenic Targeting of VEGF Signaling in Late-Stage Pancreatic Islet Tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C Mediates the Angiogenic and Tumorigenic Properties of Fibroblasts Associated with Tumors Refractory to Anti-VEGF Treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, K.; Ding, G.; Li, J.; Chen, K.; Li, H.; Qian, J.; Jiang, C.; Fang, J. Tumor Resistance to Anti-VEGF Therapy through up-Regulation of VEGF-C Expression. Cancer Lett. 2014, 346, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Michaelsen, S.R.; Staberg, M.; Pedersen, H.; Jensen, K.E.; Majewski, W.; Broholm, H.; Nedergaard, M.K.; Meulengracht, C.; Urup, T.; Villingshøj, M.; et al. VEGF-C Sustains VEGFR2 Activation under Bevacizumab Therapy and Promotes Glioblastoma Maintenance. Neuro. Oncol. 2018, 20, 1462–1474. [Google Scholar] [CrossRef]
- Auguste, P.; Lemiere, S.; Larrieu-Lahargue, F.; Bikfalvi, A. Molecular Mechanisms of Tumor Vascularization. Crit. Rev. Oncol. Hematol. 2005, 54, 53–61. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.R.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel Co-Option Mediates Resistance to Anti-Angiogenic Therapy in Liver Metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Angara, K.; Borin, T.F.; Arbab, A.S. Vascular Mimicry: A Novel Neovascularization Mechanism Driving Anti-Angiogenic Therapy (AAT) Resistance in Glioblastoma. Transl. Oncol. 2017, 10, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cortés, M.; Delgado-Bellido, D.; Oliver, F.J. Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much”. Front. Oncol. 2019, 9, 803. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C. Human Endothelial Progenitor Cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006692. [Google Scholar] [CrossRef] [PubMed]
- Haider, K.H.; Aziz, S.; Al-Reshidi, M.A. Endothelial Progenitor Cells for Cellular Angiogenesis and Repair: Lessons Learned from Experimental Animal Models. Regen. Med. 2017, 12, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Pahler, J.C.; Tazzyman, S.; Erez, N.; Chen, Y.Y.; Murdoch, C.; Nozawa, H.; Lewis, C.E.; Hanahan, D. Plasticity in Tumor-Promoting Inflammation: Impairment of Macrophage Recruitment Evokes a Compensatory Neutrophil Response. Neoplasia 2008, 10, 329–340. [Google Scholar] [CrossRef]
- Li, D.; Xie, K.; Zhang, L.; Yao, X.; Li, H.; Xu, Q.; Wang, X.; Jiang, J.; Fang, J. Dual Blockade of Vascular Endothelial Growth Factor (VEGF) and Basic Fibroblast Growth Factor (FGF-2) Exhibits Potent Anti-Angiogenic Effects. Cancer Lett. 2016, 377, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, F.; He, G.; Li, K.; Zhu, C.; Yu, W.; Zhang, C.; Xie, M.; Lin, J.; Zhang, J.; et al. Discovery of Arylamide-5-Anilinoquinazoline-8-Nitro Derivatives as VEGFR-2 Kinase Inhibitors: Synthesis, in Vitro Biological Evaluation and Molecular Docking. Bioorg. Med. Chem. Lett. 2019, 29, 126711. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of Vascular Endothelial Growth Factor-Induced Angiogenesis Suppresses Tumour Growth in Vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- Kopetz, S.; Hoff, P.M.; Morris, J.S.; Wolff, R.A.; Eng, C.; Glover, K.Y.; Adinin, R.; Overman, M.J.; Valero, V.; Wen, S.; et al. Phase II Trial of Infusional Fluorouracil, Irinotecan, and Bevacizumab for Metastatic Colorectal Cancer: Efficacy and Circulating Angiogenic Biomarkers Associated with Therapeutic Resistance. J. Clin. Oncol. 2010, 28, 453–459. [Google Scholar] [CrossRef]
- Lieu, C.H.; Tran, H.; Jiang, Z.Q.; Mao, M.; Overman, M.J.; Lin, E.; Eng, C.; Morris, J.; Ellis, L.; Heymach, J.V.; et al. The Association of Alternate VEGF Ligands with Resistance to Anti-VEGF Therapy in Metastatic Colorectal Cancer. PLoS ONE 2013, 8, e77117. [Google Scholar]
- Van de Veire, S.; Stalmans, I.; Heindryckx, F.; Oura, H.; Tijeras-Raballand, A.; Schmidt, T.; Loges, S.; Albrecht, I.; Jonckx, B.; Vinckier, S.; et al. Further Pharmacological and Genetic Evidence for the Efficacy of PlGF Inhibition in Cancer and Eye Disease. Cell 2010, 141, 178–190. [Google Scholar] [CrossRef]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast Growth Factor/Fibroblast Growth Factor Receptor System in Angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Fabbro, D.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Catalytic Receptors. Br. J. Pharmacol. 2019, 176, S247–S296. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Cao, R.; Hedlund, E.M. R Regulation of Tumor Angiogenesis and Metastasis by FGF and PDGF Signaling Pathways. J. Mol. Med. 2008, 76, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 Amplification Drives Endocrine Therapy Resistance and Is a Therapeutic Target in Breast Cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Walters, I.B.; Hanahan, D. Brivanib, a Dual FGF/VEGF Inhibitor, Is Active Both First and Second Line against Mouse Pancreatic Neuroendocrine Tumors Developing Adaptive/Evasive Resistance to VEGF Inhibition. Clin. Cancer Res. 2011, 17, 5299–5310. [Google Scholar] [CrossRef]
- André, F.; Bachelot, T.; Campone, M.; Dalenc, F.; Perez-Garcia, J.M.; Hurvitz, S.A.; Turner, N.; Rugo, H.; Smith, J.W.; Deudon, S.; et al. Targeting FGFR with Dovitinib (TKI258): Preclinical and Clinical Data in Breast Cancer. Clin. Cancer Res. 2013, 19, 3693–3702. [Google Scholar] [CrossRef]
- Burbridge, M.F.; Bossard, C.J.; Saunier, C.; Fejes, I.; Bruno, A.; Léonce, S.; Ferry, G.; Da Violante, G.; Bouzom, F.; Cattan, V.; et al. S49076 Is a Novel Kinase Inhibitor of MET, AXL, and FGFR with Strong Preclinical Activity Alone and in Association with Bevacizumab. Mol. Cancer Ther. 2013, 12, 1749–1762. [Google Scholar] [CrossRef]
- Lee, C.K.; Lee, M.E.; Lee, W.S.; Kim, J.M.; Park, K.H.; Kim, T.S.; Lee, K.Y.; Ahn, J.B.; Chung, H.C.; Rha, S.Y. Dovitinib (TKI258), a Multi-Target Angiokinase Inhibitor, Is Effective Regardless of KRAS or BRAF Mutation Status in Colorectal Cancer. Am. J. Cancer Res. 2015, 5, 72–86. [Google Scholar]
- Norden, A.D.; Schiff, D.; Ahluwalia, M.S.; Lesser, G.J.; Nayak, L.; Lee, E.Q.; Rinne, M.L.; Muzikansky, A.; Dietrich, J.; Purow, B.; et al. Phase II Trial of Triple Tyrosine Kinase Receptor Inhibitor Nintedanib in Recurrent High-Grade Gliomas. J. Neurooncol. 2015, 121, 297–302. [Google Scholar] [CrossRef]
- Semrad, T.J.; Kim, E.J.; Tanaka, M.S.; Sands, J.; Roberts, C.; Burich, R.A.; Li, Y.; Gandara, D.R.; Lara, P.; Mack, P.C. Phase II Study of Dovitinib in Patients Progressing on Anti-Vascular Endothelial Growth Factor Therapy. Cancer Treat. Res. Commun. 2017, 10, 21–26. [Google Scholar] [CrossRef]
- Jones, R.L.; Ratain, M.J.; O’Dwyer, P.J.; Siu, L.L.; Jassem, J.; Medioni, J.; DeJonge, M.; Rudin, C.; Sawyer, M.; Khayat, D.; et al. Phase II Randomised Discontinuation Trial of Brivanib in Patients with Advanced Solid Tumours. Eur. J. Cancer 2019, 120, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Therapontos, C.; Erskine, L.; Gardner, E.R.; Figg, W.D.; Vargesson, N. Thalidomide Induces Limb Defects by Preventing Angiogenic Outgrowth during Early Limb Formation. Proc. Natl. Acad. Sci. USA 2009, 106, 8573–8578. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Marriott, J.B.; Macdonald, C.D.; Man, H.-W.; Chen, R.; Muller, G.W.; Stirling, D.; Dalgleish, A.G. Novel Thalidomide Analogues Display Anti-Angiogenic Activity Independently of Immunomodulatory Effects. Br. J. Cancer 2002, 87, 1166–1172. [Google Scholar] [CrossRef]
- Sherbet, G.V. Therapeutic Potential of Thalidomide and Its Analogues in the Treatment of Cancer. Anticancer Res. 2015, 35, 5767–5772. [Google Scholar] [PubMed]
- Kaicker, S.; McCrudden, K.W.; Beck, L.; New, T.; Huang, J.; Frischer, J.S.; Serur, A.; Kadenhe-Chiweshe, A.; Yokoi, A.; Kandel, J.J.; et al. Thalidomide Is Anti-Angiogenic in a Xenograft Model of Neuroblastoma. Int. J. Oncol. 2003, 23, 1651–1655. [Google Scholar] [CrossRef]
- Maria de Souza, C.; Fonseca de Carvalho, L.; da Silva Vieira, T.; Cândida Araújo e Silva, A.; Teresa Paz Lopes, M.; Alves Neves Diniz Ferreira, M.; Passos Andrade, S.; Dantas Cassali, G. Thalidomide Attenuates Mammary Cancer Associated-Inflammation, Angiogenesis and Tumor Growth in Mice. Biomed. Pharmacother. 2012, 66, 491–498. [Google Scholar] [CrossRef]
- De Souza, C.M.; Araújo e Silva, A.C.; De Jesus Ferraciolli, C.; Moreira, G.V.; Campos, L.C.; Dos Reis, D.C.; Lopes, M.T.P.; Ferreira, M.A.N.D.; Andrade, S.P.; Cassali, G.D. Combination Therapy with Carboplatin and Thalidomide Suppresses Tumor Growth and Metastasis in 4T1 Murine Breast Cancer Model. Biomed. Pharmacother. 2014, 68, 51–57. [Google Scholar] [CrossRef]
- Shen, Y.; Li, S.; Wang, X.; Wang, M.; Tian, Q.; Yang, J.; Wang, J.; Wang, B.; Liu, P.; Yang, J. Tumor Vasculature Remolding by Thalidomide Increases Delivery and Efficacy of Cisplatin. J. Exp. Clin. Cancer Res. 2019, 38, 427. [Google Scholar] [CrossRef]
- Stephens, T.D.; Bunde, C.J.W.; Fillmore, B.J. Mechanism of Action in Thalidomide Teratogenesis. Biochem. Pharmacol. 2000, 59, 1489–1499. [Google Scholar] [CrossRef]
- Mercurio, A.; Adriani, G.; Catalano, A.; Carocci, A.; Rao, L.; Lentini, G.; Cavalluzzi, M.M.; Franchini, C.; Vacca, A.; Corbo, F. A Mini-Review on Thalidomide: Chemistry, Mechanisms of Action, Therapeutic Potential and Anti-Angiogenic Properties in Multiple Myeloma. Curr. Med. Chem. 2017, 24, 2736–2744. [Google Scholar] [CrossRef]
- Du, W.; Hattori, Y.; Hashiguchi, A.; Kondoh, K.; Hozumi, N.; Ikeda, Y.; Sakamoto, M.; Hata, J.I.; Yamada, T. Tumor Angiogenesis in the Bone Marrow of Multiple Myeloma Patients and Its Alterations by Thalidomide Treatment. Pathol. Int. 2004, 54, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Augustin, H.G. The Role of the Angiopoietins in Vascular Morphogenesis. Angiogenesis 2009, 12, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Eklund, L.; Kangas, J.; Saharinen, P. Angiopoietin-Tie Signalling in the Cardiovascular and Lymphatic Systems. Clin. Sci. 2017, 131, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.A.; Saharinen, P. Anti-angiogenic Targets: Angiopoietin and Angiopoietin Receptors. Tumor Angiogenesis 2019. [Google Scholar] [CrossRef]
- Metheny-Barlow, L.J.; Li, L.Y. The Enigmatic Role of Angiopoietin-1 in Tumor Angiogenesis. Cell Res. 2003, 13, 309–317. [Google Scholar] [CrossRef]
- Cascone, T.; Heymach, J.V. Targeting the Angiopoietin/Tie2 Pathway: Cutting Tumor Vessels with a Double-Edged Sword? J. Clin. Oncol. 2012, 30, 441–444. [Google Scholar] [CrossRef]
- Fagiani, E.; Lorentz, P.; Kopfstein, L.; Christofori, G. Angiopoietin-1 and -2 Exert Antagonistic Functions in Tumor Angiogenesis, yet Both Induce Lymphangiogenesis. Cancer Res. 2011, 71, 5717–5727. [Google Scholar] [CrossRef]
- Tait, C.R.; Jones, P.F. Angiopoietins in Tumours: The Angiogenic Switch. J. Pathol. 2004, 204, 1–10. [Google Scholar] [CrossRef]
- Murdoch, C.; Tazzyman, S.; Webster, S.; Lewis, C.E. Expression of Tie-2 by Human Monocytes and Their Responses to Angiopoietin-2. J. Immunol. 2007, 178, 7405–7411. [Google Scholar] [CrossRef]
- Huang, H.; Bhat, A.; Woodnutt, G.; Lappe, R. Targeting the ANGPT-TIE2 Pathway in Malignancy. Nat. Rev. Cancer 2010, 10, 575–585. [Google Scholar] [CrossRef]
- Goede, V.; Coutelle, O.; Neuneier, J.; Reinacher-Schick, A.; Schnell, R.; Koslowsky, T.C.; Weihrauch, M.R.; Cremer, B.; Kashkar, H.; Odenthal, M.; et al. Identification of Serum Angiopoietin-2 as a Biomarker for Clinical Outcome of Colorectal Cancer Patients Treated with Bevacizumab-Containing Therapy. Br. J. Cancer 2010, 103, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF Bispecific Antibody Reprograms Macrophages and Resident Microglia to Anti-Tumor Phenotype and Prolongs Glioblastoma Survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual Inhibition of Ang-2 and VEGF Receptors Normalizes Tumor Vasculature and Prolongs Survival in Glioblastoma by Altering Macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Rmili, C.W.; Leow, C.C.; de Palma, M. Role of Angiopoietin-2 in Adaptive Tumor Resistance to VEGF Signaling Blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Martinez-Garcia, M.; Le Tourneau, C.; Massard, C.; Garralda, E.; Boni, V.; Taus, A.; Albanell, J.; Sablin, M.P.; Alt, M.; et al. First-in-Human Phase i Study of Single-Agent Vanucizumab, a First-in-Class Bispecific Anti-Angiopoietin-2/Anti-Vegf-a Antibody, in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 1536–1545. [Google Scholar] [CrossRef]
- Heil, F.; Babitzki, G.; Julien-Laferriere, A.; Ooi, C.H.; Hidalgo, M.; Massard, C.; Martinez-Garcia, M.; Le Tourneau, C.; Kockx, M.; Gerber, P.; et al. Vanucizumab Mode of Action: Serial Biomarkers in Plasma, Tumor, and Skin-Wound-Healing Biopsies. Transl. Oncol. 2021, 14, 100984. [Google Scholar] [CrossRef]
- Cao, Y. Multifarious Functions of PDGFs and PDGFRs in Tumor Growth and Metastasis. Trends Mol. Med. 2013, 19, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus Paclitaxel versus Placebo plus Paclitaxel in Patients with Previously Treated Advanced Gastric or Gastro-Oesophageal Junction Adenocarcinoma (RAINBOW): A Double-Blind, Randomised Phase 3 Trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Cantanhede, I.G.; De Oliveira, J.R.M. PDGF Family Expression in Glioblastoma Multiforme: Data Compilation from Ivy Glioblastoma Atlas Project Database. Sci. Rep. 2017, 7, 15271. [Google Scholar] [CrossRef]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-Mediated Mesenchymal Transformation Renders Endothelial Resistance to Anti-VEGF Treatment in Glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef]
- Gover-Proaktor, A.; Granot, G.; Shapira, S.; Raz, O.; Pasvolsky, O.; Nagler, A.; Lev, D.L.; Inbal, A.; Lubin, I.; Raanani, P.; et al. Ponatinib Reduces Viability, Migration, and Functionality of Human Endothelial Cells. Leuk. Lymphoma 2017, 58, 1455–1467. [Google Scholar] [CrossRef]
- Massaro, F.; Molica, M.; Breccia, M. Ponatinib: A Review of Efficacy and Safety. Curr. Cancer Drug Targets 2018, 18, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A Novel Multi-Tyrosine Kinase Inhibitor against Human Malignancies. Onco. Targets. Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- You, W.K.; McDonald, D.M. The Hepatocyte Growth Factor/c-Met Signaling Pathway as a Therapeutic Target to Inhibit Angiogenesis. J. Biochem. Mol. Biol. 2008, 41, 833–839. [Google Scholar] [CrossRef]
- Wojta, J.; Kaun, C.; Breuss, J.M.; Koshelnick, Y.; Beckmann, R.; Hattey, E.; Mildner, M.; Weninger, W.; Nakamura, T.; Tschachler, E.; et al. Hepatocyte Growth Factor Increases Expression of Vascular Endothelial Growth Factor and Plasminogen Activator Inhibitor-1 in Human Keratinocytes and the Vascular Endothelial Growth Factor Receptor Flk-1 in Human Endothelial Cells. Lab. Investig. 1999, 79, 427–438. [Google Scholar] [PubMed]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Shawn Carbonell, W.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene Expression Profile Identifies Tyrosine Kinase C-Met as a Targetable Mediator of Antiangiogenic Therapy Resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Lee, J.H.; Simmons, B.H.; Wong, A.; Esparza, C.O.; Plumlee, P.A.; Feng, J.; Stewart, A.E.; Hu-Lowe, D.D.; Christensen, J.G. HGF/c-Met Acts as an Alternative Angiogenic Pathway in Sunitinib-Resistant Tumors. Cancer Res. 2010, 70, 10090–10100. [Google Scholar] [CrossRef]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition through a MET/VEGFR2 Complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab plus Bevacizumab versus Placebo plus Bevacizumab in Patients with Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O6-Methylguanine-DNA Methy. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef]
- Kim, B.; Kim, D.; Kim, J.; Kim, H.; Jang, H. The Efficacy and Safety of Onartuzumab in Patients with Solid Cancers: A Meta-Analysis of Randomized Trials. Indian J. Cancer 2020. [Google Scholar] [CrossRef]
- Rini, B.I.; Michaelson, M.D.; Rosenberg, J.E.; Bukowski, R.M.; Sosman, J.A.; Stadler, W.M.; Hutson, T.E.; Margolin, K.; Harmon, C.S.; DePrimo, S.E.; et al. Antitumor Activity and Biomarker Analysis of Sunitinib in Patients with Bevacizumab-Refractory Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2008, 26, 3743–3748. [Google Scholar] [CrossRef]
- Willett, C.G.; Duda, D.G.; Di Tomaso, E.; Boucher, Y.; Ancukiewicz, M.; Sahani, D.V.; Lahdenranta, J.; Chung, D.C.; Fischman, A.J.; Lauwers, G.Y.; et al. Efficacy, Safety, and Biomarkers of Neoadjuvant Bevacizumab, Radiation Therapy, and Fluorouracil in Rectal Cancer: A Multidisciplinary Phase II Study. J. Clin. Oncol. 2009, 27, 3020–3026. [Google Scholar] [CrossRef]
- Bagley, R.G.; Ren, Y.; Weber, W.; Yao, M.; Kurtzberg, L.; Pinckney, J.; Bangari, D.; Nguyen, C.; Brondyk, W.; Kaplan, J.; et al. Placental Growth Factor Upregulation Is a Host Response to Antiangiogenic Therapy. Clin. Cancer Res. 2011, 17, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Chiron, M.; Bagley, R.G.; Pollard, J.; Mankoo, P.K.; Henry, C.; Vincent, L.; Geslin, C.; Baltes, N.; Bergstrom, D.A. Differential Antitumor Activity of Aflibercept and Bevacizumab in Patient-Derived Xenograft Models of Colorectal Cancer. Mol. Cancer Ther. 2014, 13, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lawler, J. Thrombospondin-Based Antiangiogenic Therapy. Microvasc. Res. 2007, 74, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, P. Thrombospondins Function as Regulators of Angiogenesis. J. Cell Commun. Signal. 2009, 3, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, R.; de Vos, F.Y.F.L.; Eskens, F.A.L.M.; Gietema, J.A.; van der Gaast, A.; Groen, H.J.M.; Knight, R.A.; Carr, R.A.; Humerickhouse, R.A.; Verweij, J.; et al. Phase I Safety, Pharmacokinetic, and Pharmacodynamic Study of the Thrombospondin-1-Mimetic Angiogenesis Inhibitor ABT-510 in Patients with Advanced Cancer. J. Clin. Oncol. 2005, 23, 5188–5197. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Fiveash, J.B.; Markert, J.M.; Kekan, M.S.; Gillespie, G.Y.; Huang, Z.; Johnson, M.J.; Meleth, S.; Kuo, H.; Gladson, C.L.; et al. A Phase 1 Trial of ABT-510 Concurrent with Standard Chemoradiation for Patients with Newly Diagnosed Glioblastoma. Arch. Neurol. 2010, 67, 313–319. [Google Scholar] [CrossRef]
- Lawler, J. Thrombospondin-1 as an Endogenous Inhibitor of Angiogenesis and Tumor Growth. J. Cell. Mol. Med. 2002, 2, a006627. [Google Scholar] [CrossRef] [PubMed]
- Markovic, S.N.; Suman, V.J.; Rao, R.A.; Ingle, J.N.; Kaur, J.S.; Erickson, L.A.; Pitot, H.C.; Croghan, G.A.; McWilliams, R.R.; Merchan, J.; et al. A Phase II Study of ABT-510 (Thrombospondin-1 Analog) for the Treatment of Metastatic Melanoma. Am. J. Clin. Oncol. Cancer Clin. Trials 2007, 30, 303–309. [Google Scholar] [CrossRef]
- Baker, L.H.; Rowinsky, E.K.; Mendelson, D.; Humerickhouse, R.A.; Knight, R.A.; Qian, J.; Carr, R.A.; Gordon, G.B.; Demetri, G.D. Randomized, Phase II Study of the Thrombospondin-1-Mimetic Angiogenesis Inhibitor ABT-510 in Patients with Advanced Soft Tissue Sarcoma. J. Clin. Oncol. 2008, 26, 5583–5588. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Duquette, M.; Liu, J.; Drapkin, R.; Lawler, J.; Petrik, J. Combined Therapy with Thrombospondin-1 Type I Repeats (3TSR) and Chemotherapy Induces Regression and Significantly Improves Survival in a Preclinical Model of Advanced Stage Epithelial Ovarian Cancer. FASEB J. 2015, 29, 576–588. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Cheng, R.; Benyajati, S.; Ma, J.X. PEDF and Its Roles in Physiological and Pathological Conditions: Implication in Diabetic and Hypoxia-Induced Angiogenic Diseases. Clin. Sci. 2015, 129, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Maik-Rachline, G.; Shaltiel, S.; Seger, R. Extracellular Phosphorylation Converts Pigment Epithelium-Derived Factor from a Neurotrophic to an Antiangiogenic Factor. Blood 2005, 105, 670–678. [Google Scholar] [CrossRef][Green Version]
- Maik-Rachline, G.; Seger, R. Variable Phosphorylation States of Pigment-Epithelium-Derived Factor Differentially Regulate Its Function. Blood 2006, 107, 2745–2752. [Google Scholar] [CrossRef]
- Duh, E.J.; Yang, H.S.; Suzuma, I.; Miyagi, M.; Youngman, E.; Mori, K.; Katai, M.; Yan, L.; Suzuma, K.; West, K.; et al. Pigment Epithelium-Derived Factor Suppresses Ischemia-Induced Retinal Neovascularization and VEGF-Induced Migration and Growth. Investig. Ophthalmol. Vis. Sci. 2002, 43, 821–829. [Google Scholar]
- Subramanian, P.; Deshpande, M.; Locatelli-Hoops, S.; Moghaddam-Taaheri, S.; Gutierrez, D.; Fitzgerald, D.P.; Guerrier, S.; Rapp, M.; Notario, V.; Becerra, S.P. Identification of Pigment Epithelium-Derived Factor Protein Forms with Distinct Activities on Tumor Cell Lines. J. Biomed. Biotechnol. 2012, 2012, 42590. [Google Scholar] [CrossRef]
- Bhutto, I.A.; McLeod, D.S.; Hasegawa, T.; Kim, S.Y.; Merges, C.; Tong, P.; Lutty, G.A. Pigment Epithelium-Derived Factor (PEDF) and Vascular Endothelial Growth Factor (VEGF) in Aged Human Choroid and Eyes with Age-Related Macular Degeneration. Exp. Eye Res. 2006, 82, 99–110. [Google Scholar] [CrossRef]
- Michalczyk, E.R.; Chen, L.; Fine, D.; Zhao, Y.; Mascarinas, E.; Grippo, P.J.; DiPietro, L.A. Pigment Epithelium-Derived Factor (PEDF) as a Regulator of Wound Angiogenesis. Sci. Rep. 2018, 8, 11142. [Google Scholar] [CrossRef]
- Becerra, S.P.; Notario, V. The Effects of PEDF on Cancer Biology: Mechanisms of Action and Therapeutic Potential. Nat. Rev. Cancer 2013, 13, 258–271. [Google Scholar] [CrossRef]
- Belkacemi, L.; Zhang, S.X. Anti-Tumor Effects of Pigment Epithelium-Derived Factor (PEDF): Implication for Cancer Therapy. A Mini-Review. J. Exp. Clin. Cancer Res. 2016, 35, 4. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An Endogenous Inhibitor of Angiogenesis and Tumor Growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef]
- Walia, A.; Yang, J.F.; Huang, Y.H.; Rosenblatt, M.I.; Chang, J.H.; Azar, D.T. Endostatin’s Emerging Roles in Angiogenesis, Lymphangiogenesis, Disease, and Clinical Applications. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 2422–2438. [Google Scholar] [CrossRef] [PubMed]
- Biaoxue, R.; Shuanying, Y.; Wei, L.; Wei, Z.; Zongjuan, M. Systematic Review and Meta-Analysis of Endostar (Rh-Endostatin) Combined with Chemotherapy versus Chemotherapy Alone for Treating Advanced Non-Small Cell Lung Cancer. World J. Surg. Oncol. 2012, 10, 170. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kang, G.; Wang, T.; Huang, H. Tumor Angiogenesis and Anti-Angiogenic Gene Therapy for Cancer (Review). Oncol. Lett. 2018, 16, 687–702. [Google Scholar] [CrossRef]
- Li, K.; Shi, M.; Qin, S. Current Status and Study Progress of Recombinant Human Endostatin in Cancer Treatment. Oncol. Ther. 2018, 6, 21–43. [Google Scholar] [CrossRef]
- Sorensen, D.R.; Read, T.A. Delivery of Endostatin in Experimental Cancer Therapy. Int. J. Exp. Pathol. 2002, 83, 265–274. [Google Scholar] [CrossRef]
- Bellon, J.R.; Come, S.E.; Gelman, R.S.; Henderson, I.C.; Shulman, L.N.; Silver, B.J.; Harris, J.R.; Recht, A. Sequencing of Chemotherapy and Radiation Therapy in Early-Stage Breast Cancer: Updated Results of a Prospective Randomized Trial. J. Clin. Oncol. 2005, 23, 1934–1940. [Google Scholar] [CrossRef]
- Biondi, A.; Persiani, R.; Cananzi, F.; Zoccali, M.; Vigorita, V.; Tufo, A.; D’Ugo, D. R0 Resection in the Treatment of Gastric Cancer: Room for Improvement. World J. Gastroenterol. 2010, 16, 3358–3370. [Google Scholar] [CrossRef]
- Kajiyama, H.; Suzuki, S.; Yoshihara, M.; Nishino, K.; Yoshikawa, N.; Utsumi, F.; Niimi, K.; Mizuno, M.; Kawai, M.; Oguchi, H.; et al. The Possible Existence of Occult Metastasis in Patients with Ovarian Clear-Cell Carcinoma Who Underwent Complete Resection without Any Residual Tumours. Oncotarget 2018, 9, 6298–6630. [Google Scholar] [CrossRef][Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and Mechanisms of Vessel Normalization for Cancer and Other Angiogenic Diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Jain, R.K. Tumor Microvasculature and Microenvironment: Targets for Anti-Angiogenesis and Normalization. Microvasc. Res. 2007, 74, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor Refractoriness to Anti-VEGF Treatment Is Mediated by CD11b +Gr1+ Myeloid Cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of Vascular Endothelial Growth Factor by Human Tumors Inhibits the Functional Maturation of Dendritic Cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Ran, S.; Ishida, T.; Nadaf, S.; Kerr, L.; Carbone, D.P.; Gabrilovich, D.I. Vascular Endothelial Growth Factor Affects Dendritic Cell Maturation through the Inhibition of Nuclear Factor-Kappa B Activation in Hemopoietic Progenitor Cells. J. Immunol. 1998, 160, 1224–1232. [Google Scholar]
- Huang, Y.; Chen, X.; Dikov, M.M.; Novitskiy, S.V.; Mosse, C.A.; Yang, L.; Carbone, D.P. Distinct Roles of VEGFR-1 and VEGFR-2 in the Aberrant Hematopoiesis Associated with Elevated Levels of VEGF. Blood 2007, 110, 624–631. [Google Scholar] [CrossRef]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 Improves Myeloid Dendritic Cell-Mediated Antitumor Immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef]
- Alfaro, C.; Suarez, N.; Gonzalez, A.; Solano, S.; Erro, L.; Dubrot, J.; Palazon, A.; Hervas-Stubbs, S.; Gurpide, A.; Lopez-Picazo, J.M.; et al. Influence of Bevacizumab, Sunitinib and Sorafenib as Single Agents or in Combination on the Inhibitory Effects of VEGF on Human Dendritic Cell Differentiation from Monocytes. Br. J. Cancer 2009, 100, 1111–1119. [Google Scholar] [CrossRef]
- Motz, G.T.; Coukos, G. The Parallel Lives of Angiogenesis and Immunosuppression: Cancer and Other Tales. Nat. Rev. Immunol. 2011, 11, 702–711. [Google Scholar] [CrossRef]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.A.; Mantovani, A.; Marme, D. Migration of Human Monocytes in Response to Vascular Endothelial Growth Factor (VEGF) Is Mediated via the VEGF Receptor Flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Coelho, F.; Silva, F.; Gouveia-Fernandes, S.; Martins, C.; Lopes, N.; Domingues, G.; Brito, C.; Almeida, A.M.; Pereira, S.A.; Serpa, J. Monocytes as Endothelial Progenitor Cells (EPCs), Another Brick in the Wall to Disentangle Tumor Angiogenesis. Cells 2020, 9, 107. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; Gabrilovich, D.I.; Sempowski, G.D.; Kisseleva, E.; Parman, K.S.; Nadaf, S.; Carbone, D.P. VEGF Inhibits T-Cell Development and May Contribute to Tumor-Induced Immune Suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef] [PubMed]
- Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Ioannou, K.; Ziogas, A.C.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF Directly Suppresses Activation of T Cells from Ascites Secondary to Ovarian Cancer via VEGF Receptor Type 2. Br. J. Cancer 2012, 107, 1869–1875. [Google Scholar] [CrossRef]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory Activity of VEGF in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 295–342. [Google Scholar] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T Cells in Tumor Immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Kerbel, R.S. Improving Immunotherapy Outcomes with Anti-Angiogenic Treatments and Vice Versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef]
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The Contribution of Vascular Endothelial Growth Factor to the Induction of Regulatory T- Cells in Malignant Effusions. Anticancer Res. 2009, 29, 881–888. [Google Scholar] [PubMed]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR Pathway Blockade Inhibits Tumor-Induced Regulatory T-Cell Proliferation in Colorectal Cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 Deficiency on CD4+Foxp3+ Regulatory T Cells Impairs Mouse Melanoma Growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef]
- Hansen, W. Neuropilin 1 Guides Regulatory T Cells into Vegf-Producing Melanoma. Oncoimmunology 2013, 2, e2303. [Google Scholar] [CrossRef] [PubMed]
- De Aguiar, R.B.; De Moraes, J.Z. Exploring the Immunological Mechanisms Underlying the Anti-Vascular Endothelial Growth Factor Activity in Tumors. Front. Immunol. 2019, 10, 1023. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic Effect of Immune Checkpoint Blockade and Anti-Angiogenesis in Cancer Treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-Angiogenesis for Cancer Revisited: Is There a Role for Combinations with Immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef]
- Procaccio, L.; Damuzzo, V.; Di Sarra, F.; Russi, A.; Todino, F.; Dadduzio, V.; Bergamo, F.; Prete, A.A.; Lonardi, S.; Prenen, H.; et al. Safety and Tolerability of Anti-Angiogenic Protein Kinase Inhibitors and Vascular-Disrupting Agents in Cancer: Focus on Gastrointestinal Malignancies. Drug Saf. 2019, 42, 159–179. [Google Scholar] [CrossRef] [PubMed]
- Bluthgen, M.V.; Basté, N.; Recondo, G. Immunotherapy Combinations for the Treatment of Patients with Solid Tumors. Future Oncol. 2020, 16, 1715–1736. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy—Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef]
- Haanen, J.B.A.G.; Robert, C. Immune Checkpoint Inhibitors. Prog. Tumor Res. 2015, 42, 55–66. [Google Scholar]
- Duan, J.; Cui, L.; Zhao, X.; Bai, H.; Cai, S.; Wang, G.; Zhao, Z.; Zhao, J.; Chen, S.; Song, J.; et al. Use of Immunotherapy with Programmed Cell Death 1 vs Programmed Cell Death Ligand 1 Inhibitors in Patients with Cancer: A Systematic Review and Meta-Analysis. JAMA Oncol. 2020, 6, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, L.; Yu, J.; Zhang, Y.; Pang, X.; Ma, C.; Shen, M.; Ruan, S.; Wasan, H.S.; Qiu, S. Clinical Efficacy and Safety of Anti-PD-1/PD-L1 Inhibitors for the Treatment of Advanced or Metastatic Cancer: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 2083. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Bellmunt, J.; Clancy, J.; Wang, K.; Niethammer, A.G.; Hariharan, S.; Escudier, B. Randomized Phase III Trial of Temsirolimus and Bevacizumab versus Interferon Alfa and Bevacizumab in Metastatic Renal Cell Carcinoma: INTORACT Trial. J. Clin. Oncol. 2014, 32, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 308, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; Solimando, A.G.; Krebs, M.; Leone, P.; Susca, N.; Brunetti, O.; Racanelli, V.; Vacca, A.; Silvestris, N. Anti-Angiogenesis and Immunotherapy: Novel Paradigms to Envision Tailored Approaches in Renal Cell-Carcinoma. J. Clin. Med. 2020, 9, 1594. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in Combination with Bevacizumab Enhances Antigen-Specific T-Cell Migration in Metastatic Renal Cell Carcinoma. Nat. Commun. 2016, 7, 2624. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Ipilimumab with or without Bevacizumab in Treating Patients with Stage III-IV Melanoma That Cannot Be Removed by Surgery (NCT01950390); ClinicalTrials.gov: Bethesda, MD, USA, 2013.
- ClinicalTrials.gov. Bevacizumab Plus Ipilimumab in Patients with Unresectable Stage III or IV Melanoma (NCT00790010); ClinicalTrials.gov: Bethesda, MD, USA, 2008.
- Du Four, S.; Maenhout, S.K.; Niclou, S.P.; Thielemans, K.; Neyns, B.; Aerts, J.L. Combined VEGFR and CTLA-4 Blockade Increases the Antigen-Presenting Function of Intratumoral DCs and Reduces the Suppressive Capacity of Intratumoral MDSCs. Am. J. Cancer Res. 2016, 6, 2514–2531. [Google Scholar]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Qiang, H.; Chang, Q.; Xu, J.; Qian, J.; Zhang, Y.; Lei, Y.; Han, B.; Chu, T. New Advances in Antiangiogenic Combination Therapeutic Strategies for Advanced Non-Small Cell Lung Cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 631–645. [Google Scholar] [CrossRef]
- Hashimoto, H.; Sudo, T.; Maruta, H.; Nishimura, R. The Direct PAK1 Inhibitor, TAT-PAK18, Blocks Preferentially the Growth of Human Ovarian Cancer Cell Lines in Which PAK1 Is Abnormally Activated by Autophosphorylation at Thr 423. Drug Discov. Ther. 2010, 4, 1–4. [Google Scholar]
- Draganov, D.; Gopalakrishna-Pillai, S.; Chen, Y.R.; Zuckerman, N.; Moeller, S.; Wang, C.; Ann, D.; Lee, P.P. Modulation of P2X4/P2X7/Pannexin-1 Sensitivity to Extracellular ATP via Ivermectin Induces a Non-Apoptotic and Inflammatory Form of Cancer Cell Death. Sci. Rep. 2015, 5, 16222. [Google Scholar] [CrossRef]
- Dou, Q.; Chen, H.N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N.; et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fang, S.; Sun, Q.; Liu, B. Anthelmintic Drug Ivermectin Inhibits Angiogenesis, Growth and Survival of Glioblastoma through Inducing Mitochondrial Dysfunction and Oxidative Stress. Biochem. Biophys. Res. Commun. 2016, 480, 415–421. [Google Scholar] [CrossRef] [PubMed]
- López-Jiménez, A.; García-Caballero, M.; Medina, M.Á.; Quesada, A.R. Anti-Angiogenic Properties of Carnosol and Carnosic Acid, Two Major Dietary Compounds from Rosemary. Eur. J. Nutr. 2013, 52, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Rui, X.; Pan, H.F.; Shao, S.L.; Xu, X.M. Anti-Tumor and Anti-Angiogenic Effects of Fucoidan on Prostate Cancer: Possible JAK-STAT3 Pathway. BMC Complement. Altern. Med. 2017, 17, 378. [Google Scholar] [CrossRef]
- Jin, W.; Wu, W.; Tang, H.; Wei, B.; Wang, H.; Sun, J.; Zhang, W.; Zhong, W. Structure Analysis and Anti-Tumor and Anti-Angiogenic Activities of Sulfated Galactofucan Extracted from Sargassum Thunbergii. Mar. Drugs 2019, 17, 52. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of Large Amounts of Hydrogen Peroxide by Human Tumor Cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Josson, S.; Matsuoka, Y.; Chung, L.W.K.; Zhau, H.E.; Wang, R. Tumor–Stroma Co-Evolution in Prostate Cancer Progression and Metastasis. Semin. Cell Dev. Biol. 2010, 21, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer Cells Metabolically “Fertilize” the Tumor Microenvironment with Hydrogen Peroxide, Driving the Warburg Effect: Implications for PET Imaging of Human Tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling Hypoxia and Free Radicals Regulate Angiogenesis and Radiotherapy Response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, Y.; Shi, M.; Zhang, Q.; Wang, Q.; Chen, X. Deferoxamine Promotes MDA-MB-231 Cell Migration and Invasion through Increased Ros-Dependent HIF-1α Accumulation. Cell. Physiol. Biochem. 2014, 33, 1036–1046. [Google Scholar] [CrossRef]
- Zhu, D.; Shen, Z.; Liu, J.; Chen, J.; Liu, Y.; Hu, C.; Li, Z.; Li, Y. The ROS-Mediated Activation of STAT-3/VEGF Signaling Is Involved in the 27-Hydroxycholesterol-Induced Angiogenesis in Human Breast Cancer Cells. Toxicol. Lett. 2016, 264, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Khromova, N.V.; Kopnin, P.B.; Stepanova, E.V.; Agapova, L.S.; Kopnin, B.P. P53 Hot-Spot Mutants Increase Tumor Vascularization via ROS-Mediated Activation of the HIF1/VEGF-A Pathway. Cancer Lett. 2009, 276, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Assi, M. The Differential Role of Reactive Oxygen Species in Early and Late Stages of Cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R646–R653. [Google Scholar] [CrossRef]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-Modulated Therapeutic Approaches in Cancer Treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R.R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, A.; Sahu, R.P. Potential Contributions of Antioxidants to Cancer Therapy: Immunomodulation and Radiosensitization. Integr. Cancer Ther. 2018, 17, 210–216. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Alexandre, J.; Batteux, F.; Nicco, C.; Chéreau, C.; Laurent, A.; Guillevin, L.; Weill, B.; Goldwasser, F. Accumulation of Hydrogen Peroxide Is an Early and Crucial Step for Paclitaxel-Induced Cancer Cell Death Both in Vitro and in Vivo. Int. J. Cancer 2006, 119, 41–48. [Google Scholar] [CrossRef]
- Fukui, M.; Yamabe, N.; Zhu, B.T. Resveratrol Attenuates the Anticancer Efficacy of Paclitaxel in Human Breast Cancer Cells in Vitro and in Vivo. Eur. J. Cancer 2010, 46, 1882–1891. [Google Scholar] [CrossRef]
- Subramani, T.; Yeap, S.K.; Ho, W.Y.; Ho, C.L.; Omar, A.R.; Aziz, S.A.; Rahman, N.M.A.N.A.; Alitheen, N.B. Vitamin C Suppresses Cell Death in MCF-7 Human Breast Cancer Cells Induced by Tamoxifen. J. Cell. Mol. Med. 2014, 18, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Zhang, R.; Piao, M.J.; Chae, S.; Kim, H.S.; Park, J.H.; Jung, K.S.; Hyun, J.W. Baicalein Inhibits Oxidative Stress-Induced Cellular Damage via Antioxidant Effects. Toxicol. Ind. Health 2012, 28, 412–421. [Google Scholar] [CrossRef]
- Park, Y.G.; Choi, J.; Jung, H.K.; Kim, B.; Kim, C.; Park, S.Y.; Seol, J.W. Baicalein Inhibits Tumor Progression by Inhibiting Tumor Cell Growth and Tumor Angiogenesis. Oncol. Rep. 2017, 38, 3011–3018. [Google Scholar] [CrossRef]
- Park, C.; Choi, E.O.; Kim, G.Y.; Hwang, H.J.; Kim, B.W.; Yoo, Y.H.; Park, H.T.; Choi, Y.H. Protective Effect of Baicalein on Oxidative Stress-Induced DNA Damage and Apoptosis in RT4-D6P2T Schwann Cells. Int. J. Med. Sci. 2019, 16, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, D.; Sen, C.K.; Bagchi, M.; Atalay, M. Anti-Angiogenic, Antioxidant, and Anti-Carcinogenic Properties of a Novel Anthocyanin-Rich Berry Extract Formula. Biochemistry 2004, 69, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.H.M.K.; Hossain, A.S.M.S.; Khan, M.A.; Kabir, S.R.; Reza, M.A.; Rahman, M.M.; Islam, M.S.; Rahman, M.A.A.; Rashid, M.; Sadik, M.G. The Antioxidative Fraction of White Mulberry Induces Apoptosis through Regulation of P53 and NFκB in EAC Cells. PLoS ONE 2016, 11, e0167536. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative Stress and Reactive Oxygen Species in Endothelial Dysfunction Associated with Cardiovascular and Metabolic Diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, G.; Bao, K.; Liu, W.; Zhang, Y.; Ting, W. Ropivacaine Inhibits Tumor Angiogenesis via Sodium-Channel-Independent Mitochondrial Dysfunction and Oxidative Stress. J. Bioenerg. Biomembr. 2019, 51, 231–238. [Google Scholar] [CrossRef]
- Hulsurkar, M.; Li, Z.; Zhang, Y.; Li, X.; Zheng, D.; Li, W. Beta-Adrenergic Signaling Promotes Tumor Angiogenesis and Prostate Cancer Progression through HDAC2-Mediated Suppression of Thrombospondin-1. Oncogene 2017, 36, 1525–1536. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, W. Beta-Adrenergic Signaling on Neuroendocrine Differentiation, Angiogenesis, and Metastasis in Prostate Cancer Progression. Asian J. Androl. 2019, 21, 253–259. [Google Scholar]
- Lutgendorf, S.K.; DeGeest, K.; Dahmoush, L.; Farley, D.; Penedo, F.; Bender, D.; Goodheart, M.; Buekers, T.E.; Mendez, L.; Krueger, G.; et al. Social Isolation Is Associated with Elevated Tumor Norepinephrine in Ovarian Carcinoma Patients. Brain. Behav. Immun. 2011, 25, 250–255. [Google Scholar] [CrossRef]
- Sastry, K.S.R.; Karpova, Y.; Prokopovich, S.; Smith, A.J.; Essau, B.; Gersappe, A.; Carson, J.P.; Weber, M.J.; Register, T.C.; Chen, Y.Q.; et al. Epinephrine Protects Cancer Cells from Apoptosis via Activation of CAMP-Dependent Protein Kinase and BAD Phosphorylation. J. Biol. Chem. 2007, 282, 14094–14100. [Google Scholar] [CrossRef]
- Hermes, G.L.; Delgado, B.; Tretiakova, M.; Cavigelli, S.A.; Krausz, T.; Conzen, S.D.; McClintock, M.K. Social Isolation Dysregulates Endocrine and Behavioral Stress While Increasing Malignant Burden of Spontaneous Mammary Tumors. Proc. Natl. Acad. Sci. USA 2009, 106, 22393–22398. [Google Scholar] [CrossRef]
- Wong, H.P.S.; Ho, J.W.C.; Koo, M.W.L.; Yu, L.; Wu, W.K.K.; Lam, E.K.Y.; Tai, E.K.K.; Ko, J.K.S.; Shin, V.Y.; Chu, K.M.; et al. Effects of Adrenaline in Human Colon Adenocarcinoma HT-29 Cells. Life Sci. 2011, 88, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Al-wadei, H.A.N.; Ullah, M.F.; Plummer, H.K. Regulation of Pancreatic Cancer by Neuropsychological Stress Responses: A Novel Target for Intervention. Carcinogenesis 2012, 33, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, D.W.; Kim, J.; Hong, C.M.; Park, S.M.; Woo, I.A.; Kim, M.Y.; Koo, H.; Namkoong, J.; Kim, J.; et al. Prognostic Role of the Beta-2 Adrenergic Receptor in Clear Cell Renal Cell Carcinoma. Animal Cells Syst. 2019, 23, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.P.S.; Yu, L.; Lam, E.K.Y.; Tai, E.K.K.; Wu, W.K.K.; Cho, C.H. Nicotine Promotes Colon Tumor Growth and Angiogenesis through β-Adrenergic Activation. Toxicol. Sci. 2007, 358, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, H.A.N.; Schuller, H.M. Nicotinic Receptor-Associated Modulation of Stimulatory and Inhibitory Neurotransmitters in NNK-Induced Adenocarcinoma of the Lungs and Pancreas. J. Pathol. 2009, 218, 437–445. [Google Scholar] [CrossRef]
- Chen, H.; Liu, D.; Yang, Z.; Sun, L.; Deng, Q.; Yang, S.; Qian, L.; Guo, L.; Yu, M.; Hu, M.; et al. Adrenergic Signaling Promotes Angiogenesis through Endothelial Cell-Tumor Cell Crosstalk. Endocr. Relat. Cancer 2014, 21, 783–795. [Google Scholar] [CrossRef]
- Garg, J.; Feng, Y.X.; Jansen, S.R.; Friedrich, J.; Lezoualc’h, F.; Schmidt, M.; Wieland, T. Catecholamines Facilitate VEGF-Dependent Angiogenesis via Β2- Adrenoceptor-Induced Epac1 and PKA Activation. Oncotarget 2017, 8, 44732–44748. [Google Scholar] [CrossRef]
- Chakroborty, D.; Goswami, S.; Basu, S.; Sarkar, C. Catecholamines in the Regulation of Angiogenesis in Cutaneous Wound Healing. FASEB J. 2020, 34, 14093–14102. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Arnal-Estapé, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.S.; Frenette, P.S. Adrenergic Nerves Activate an Angio-Metabolic Switch in Prostate Cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef]
- Moreno-Smith, M.; Lee, S.J.; Chunhua, L.; Nagaraja, A.S.; He, G.; Rupaimoole, R.; Han, H.D.; Jennings, N.B.; Roh, J.W.; Nishimura, M.; et al. Biologic Effects of Dopamine on Tumor Vasculature in Ovarian Carcinoma. Neoplasia 2013, 15, 502–510. [Google Scholar] [CrossRef]
- Powe, D.G.; Voss, M.J.; Zänker, K.S.; Habashy, H.O.; Green, A.R.; Ellis, I.O.; Entschladen, F. Beta-Blocker Drug Therapy Reduces Secondary Cancer Formation in Breast Cancer and Improves Cancer Specific Survival. Oncotarget 2010, 1, 628–638. [Google Scholar] [CrossRef]
- Barron, T.I.; Connolly, R.M.; Sharp, L.; Bennett, K.; Visvanathan, K. Beta Blockers and Breast Cancer Mortality: A Population-Based Study. J. Clin. Oncol. 2011, 29, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- Lowenthal, D.T.; Saris, S.D.; Packer, J.; Haratz, A.; Conry, K. Mechanisms of Action and the Clinical Pharmacology of Beta-Adrenergic Blocking Drugs. Am. J. Med. 1984, 77, 119–127. [Google Scholar] [CrossRef]
- Sans, V.; de la Roque, E.D.; Berge, J.; Grenier, N.; Boralevi, F.; Mazereeuw-Hautier, J.; Lipsker, D.; Dupuis, E.; Ezzedine, K.; Vergnes, P.; et al. Propranolol for Severe Infantile Hemangiomas: Follow-Up Report. Pediatrics 2009, 124, e423–e431. [Google Scholar] [CrossRef]
- Chim, H.; Armijo, B.S.; Miller, E.; Gliniak, C.; Serret, M.A.; Gosain, A.K. Propranolol Induces Regression of Hemangioma Cells through HIF-1α-Mediated Inhibition of VEGF-A. Ann. Surg. 2012, 256, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Bai, N.; Bi, J.H.; Liu, X.W.; Xu, G.Q.; Zhang, L.F.; Li, X.Q.; Huo, R. Propranolol Inhibits the Proliferation, Migration and Tube Formation of Hemangioma Cells through HIF-1α Dependent Mechanisms. Brazilian J. Med. Biol. Res. 2017, 50, e6138. [Google Scholar] [CrossRef]
- Park, S.Y.; Kang, J.H.; Jeong, K.J.; Lee, J.; Han, J.W.; Choi, W.S.; Kim, Y.K.; Kang, J.; Park, C.G.; Lee, H.Y. Norepinephrine Induces VEGF Expression and Angiogenesis by a Hypoxia-Inducible Factor-1α Protein-Dependent Mechanism. Int. J. Cancer 2011, 128, 2306–2316. [Google Scholar] [CrossRef]
- Xia, Y.; Wei, Y.; Li, Z.Y.; Cai, X.Y.; Zhang, L.L.; Dong, X.R.; Zhang, S.; Zhang, R.G.; Meng, R.; Zhu, F.; et al. Catecholamines Contribute to the Neovascularization of Lung Cancer via Tumor-Associated Macrophages. Brain. Behav. Immun. 2019, 81, 111–121. [Google Scholar] [CrossRef]
- Pasquier, E.; Street, J.; Pouchy, C.; Carre, M.; Gifford, A.J.; Murray, J.; Norris, M.D.; Trahair, T.; Andre, N.; Kavallaris, M. B-Blockers Increase Response to Chemotherapy via Direct Antitumour and Anti-Angiogenic Mechanisms in Neuroblastoma. Br. J. Cancer 2013, 108, 2485–2494. [Google Scholar] [CrossRef]
- Stati, T.; Musumeci, M.; Maccari, S.; Massimi, A.; Corritore, E.; Strimpakos, G.; Pelosi, E.; Catalano, L.; Marano, G. β-Blockers Promote Angiogenesis in the Mouse Aortic Ring Assay. J. Cardiovasc. Pharmacol. 2014, 64, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian Cancer: Strategies for Overcoming Resistance to Chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Schumaker, L.M.; Egorin, M.J.; Zuhowski, E.G.; Guo, Z.; Cullen, K.J. Cisplatin Preferentially Binds Mitochondrial DNA and Voltage-Dependent Anion Channel Protein in the Mitochondrial Membrane of Head and Neck Squamous Cell Carcinoma: Possible Role in Apoptosis. Clin. Cancer Res. 2006, 12, 5817–5825. [Google Scholar] [CrossRef] [PubMed]
- Mikuła-Pietrasik, J.; Witucka, A.; Pakuła, M.; Uruski, P.; Begier-Krasińska, B.; Niklas, A.; Tykarski, A.; Książek, K. Comprehensive Review on How Platinum- and Taxane-Based Chemotherapy of Ovarian Cancer Affects Biology of Normal Cells. Cell. Mol. Life Sci. 2019, 76, 681–697. [Google Scholar] [CrossRef]
- Kim, E.-K.; Jang, M.; Song, M.-J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants 2019, 8, 471. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA Damage Response in Cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Toth, R.; Warfel, N. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Ji, K.; Liu, Y.; Kong, Y.; Nie, S.; Li, N.; Hao, J.; Xie, Y.; Xu, C.; et al. NRF2 Preserves Genomic Integrity by Facilitating ATR Activation and G2 Cell Cycle Arrest. Nucleic Acids Res. 2020, 48, 9109–9123. [Google Scholar] [CrossRef]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The Balance between NRF2/GSH Antioxidant Mediated Pathway and DNA Repair Modulates Cisplatin Resistance in Lung Cancer Cells. Sci. Rep. 2019, 9, 17639. [Google Scholar] [CrossRef]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 Facilitates Repair of Radiation Induced DNA Damage through Homologous Recombination Repair Pathway in a ROS Independent Manner in Cancer Cells. Mutat. Res. Mol. Mech. Mutagen. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 1–34. [Google Scholar] [CrossRef]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.-Q.; Lin, Z.-X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol Provokes a Rapid and Transient Inhibition of Nrf2 Signaling and Sensitizes Mammalian Cells to Chemical Toxicity—Implications for Therapeutic Targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, B.; Shi, Q.; Wang, X.; Wang, D.; Zhu, L. Brusatol Inhibits HIF-1 Signaling Pathway and Suppresses Glucose Uptake under Hypoxic Conditions in HCT116 Cells. Sci. Rep. 2016, 6, 39123. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about Pharmacological Modulation of Nrf2 for Cancer Therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef]
- Pastukh, V.; Roberts, J.T.; Clark, D.W.; Bardwell, G.C.; Patel, M.; Al-Mehdi, A.B.; Borchert, G.M.; Gillespie, M.N. An Oxidative DNA “Damage” and Repair Mechanism Localized in the VEGF Promoter Is Important for Hypoxia-Induced VEGF MRNA Expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1367–L1375. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.R.; Gueble, S.E.; Liu, Y.; Oeck, S.; Kim, H.; Yun, Z.; Glazer, P.M. Cediranib Suppresses Homology-Directed DNA Repair through down-Regulation of BRCA1/2 and RAD51. Sci. Transl. Med. 2019, 11, eaav4508. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.R.; Glazer, P.M. Impact of Hypoxia on DNA Repair and Genome Integrity. Mutagenesis 2020, 35, 61–68. [Google Scholar] [CrossRef]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef]
- Tentori, L.; Lacal, P.M.; Muzi, A.; Dorio, A.S.; Leonetti, C.; Scarsella, M.; Ruffini, F.; Xu, W.; Min, W.; Stoppacciaro, A.; et al. Poly(ADP-Ribose) Polymerase (PARP) Inhibition or PARP-1 Gene Deletion Reduces Angiogenesis. Eur. J. Cancer 2007, 43, 2124–2133. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.-M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination Cediranib and Olaparib versus Olaparib Alone for Women with Recurrent Platinum-Sensitive Ovarian Cancer: A Randomised Phase 2 Study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Mirza, M.R.; Bergmann, T.K.; Mau-Sørensen, M.; Christensen, R.d.P.; Åvall-Lundqvist, E.; Birrer, M.J.; Jørgensen, M.; Roed, H.; Malander, S.; Nielsen, F.; et al. A Phase I Study of the PARP Inhibitor Niraparib in Combination with Bevacizumab in Platinum-Sensitive Epithelial Ovarian Cancer: NSGO AVANOVA1/ENGOT-OV24. Cancer Chemother. Pharmacol. 2019, 84, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Åvall Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus Bevacizumab versus Niraparib Alone for Platinum-Sensitive Recurrent Ovarian Cancer (NSGO-AVANOVA2/ENGOT-Ov24): A Randomised, Phase 2, Superiority Trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef]
- Yuan, L.; Mao, Y.; Luo, W.; Wu, W.; Xu, H.; Wang, X.L.; Shen, Y.H. Palmitic Acid Dysregulates the Hippo-YAP Pathway and Inhibits Angiogenesis by Inducing Mitochondrial Damage and Activating the Cytosolic DNA Sensor CGAS-STING-IRF3 Signaling Mechanism. J. Biol. Chem. 2017, 292, 15002–15015. [Google Scholar] [CrossRef] [PubMed]
- Boopathy, G.T.K.; Hong, W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49. [Google Scholar] [CrossRef]
- Satoh, M.; Fujimoto, S.; Horike, H.; Ozeki, M.; Nagasu, H.; Tomita, N.; Sasaki, T.; Kashihara, N. Mitochondrial Damage-Induced Impairment of Angiogenesis in the Aging Rat Kidney. Lab. Investig. 2011, 91, 190–202. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, S.; Peng, G.; Yu, J.; Liu, T.; Meng, R.; Li, Z.; Zhao, Y.; Wu, G. Juncendothelial NO Synthase and Reactive Oxygen Species Mediated Effect of Simvastatin on Vessel Structure and Function: Pleiotropic and Dose-Dependent Effect on Tumor Vascular Stabilization. Int. J. Oncol. 2013, 42, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS Signalling in the Biology of Cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trial Identifier | Treatment (Arm of Combination Therapy) | Comparison | Cancer Types | Study Phase | Primary Endpoint | Registration Date | Status * |
|---|---|---|---|---|---|---|---|
| NCT00790010 | ipilimumab + bevacizumab | between cohorts | Melanoma (III-IV) | I | safety, tolerability, max tolerated dose | 12 November 2008 | Active, not recruiting |
| NCT01950390 | ipilimumab + bevacizumab | ipilimumab | Melanoma (III-IV) | II | OS | 23 September 2013 | Active, not recruiting |
| NCT02420821 | atezolizumab + bevacizumab | sunitinib | mRCC | III | PD, PFS, OS | 15 April 2015 | Active, not recruiting |
| NCT02684006 | avelumab + axitinib | sunitinib | mRCC | III | PFS, OS | 25 January 2016 | Active, not recruiting |
| NCT02853331 | pembrolizumab + axitinib | sunitinib | mRCC | III | PFS, OS | 29 July 2016 | Active, not recruiting |
| NCT03914300 | cabozantinib + nivolumab + ipilimumab | - | Thyroid cancer | II | ORR | 11 April 2019 | Recruiting |
| NCT03990571 | axitinib + avelumab | - | Recurrent/metastatic ACC | II | ORR | 17 June 2019 | Recruiting |
| NCT04017455 | atezolizumab + bevacizumab | - | Rectal cancer | II | clinical complete and near-complete response rate | 10 July 2019 | Recruiting |
| NCT04170556 | regorafenib + nivolumab | - | HCC | I/II | Rate of AE | 18 November 2019 | Recruiting |
| NCT04213170 | sintilimab + bevacizumab | - | Brain metastases from NSCLC | II | iPFS, OS, PFS | 25 December 2019 | Recruiting |
| NCT04408118 | atezolizumab + bevacizumab + paclitaxel | - | BC, TNBC | II | PFS | 20 May 2020 | Recruiting |
| NCT04493203 | nivolumab + axitinib | - | Melanoma (III-IV) | II | ORR | 22 July 2020 | Recruiting |
| NCT04727307 | atezolizumab + RFA + bevacizumab + atezolizumab | RFA | Small HCC | II | Recurrence-free survival | 22 January 2021 | Recruiting |
| NCT04732598 | bevacizumab + atezolizumab + paclitaxel | bevacizumab + paclitaxel | BC | III | PFS | 28 January 2021 | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes-Coelho, F.; Martins, F.; Pereira, S.A.; Serpa, J. Anti-Angiogenic Therapy: Current Challenges and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 3765. https://doi.org/10.3390/ijms22073765
Lopes-Coelho F, Martins F, Pereira SA, Serpa J. Anti-Angiogenic Therapy: Current Challenges and Future Perspectives. International Journal of Molecular Sciences. 2021; 22(7):3765. https://doi.org/10.3390/ijms22073765
Chicago/Turabian StyleLopes-Coelho, Filipa, Filipa Martins, Sofia A. Pereira, and Jacinta Serpa. 2021. "Anti-Angiogenic Therapy: Current Challenges and Future Perspectives" International Journal of Molecular Sciences 22, no. 7: 3765. https://doi.org/10.3390/ijms22073765
APA StyleLopes-Coelho, F., Martins, F., Pereira, S. A., & Serpa, J. (2021). Anti-Angiogenic Therapy: Current Challenges and Future Perspectives. International Journal of Molecular Sciences, 22(7), 3765. https://doi.org/10.3390/ijms22073765