
Loss of the Metastasis Suppressor NME1, But Not of Its Highly Related Isoform NME2, Induces a Hybrid Epithelial–Mesenchymal State in Cancer Cells

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibodies
2.3. RNA Interference
2.4. CRISPR–Cas9 Gene Editing
2.5. DNA Extraction, PCR Amplification, DNA Typing of Human Breast Cell Lines
2.6. RT-qPCR Analysis of Human Mammary Cell Lines
2.7. RT-qPCR of Breast Tumor Cell Lines Genetically Modified for NME1 and NME2
2.8. E-Cadherin Cell Surface Expression
2.9. Quantification of E-Cadherin Fragments
2.10. Western Blotting Analysis
2.11. Cell Morphology and Line Scan Profiling
2.12. Permeability Assay
2.13. Cell Adhesion Assay
2.14. Cell Dispersion Assay
2.15. METABRIC and TCGA Databases
2.16. Statistical Analysis
3. Results
3.1. Depletion of NME1 from Epithelial Breast Cancer Cells Induces an Incomplete EMT
3.2. Inactivation of NME1 by CRISPR–Cas9 Gene Editing Reinforces EMT
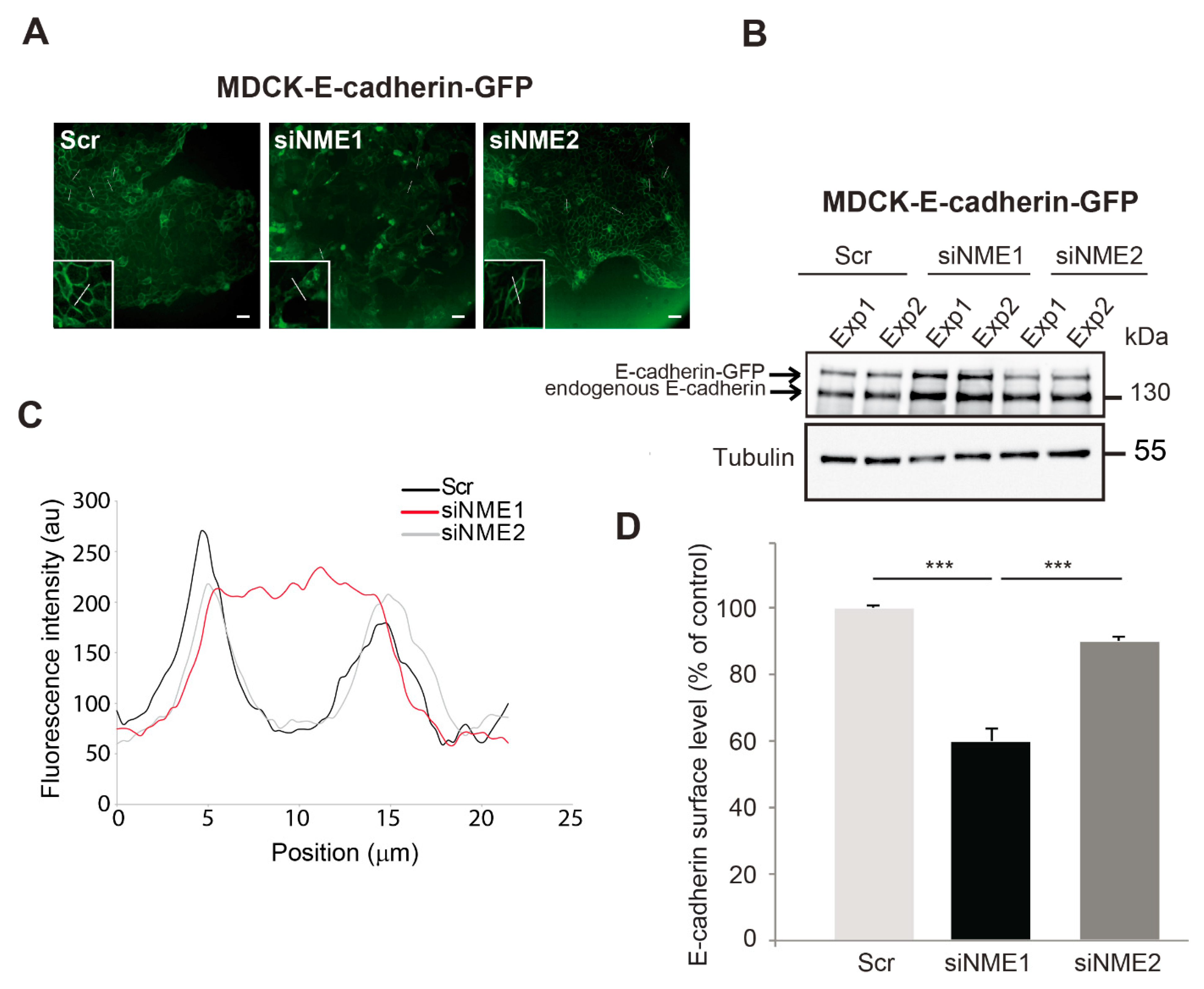
3.3. E-cadherin Distribution Is Altered in NME1-Depleted Cells
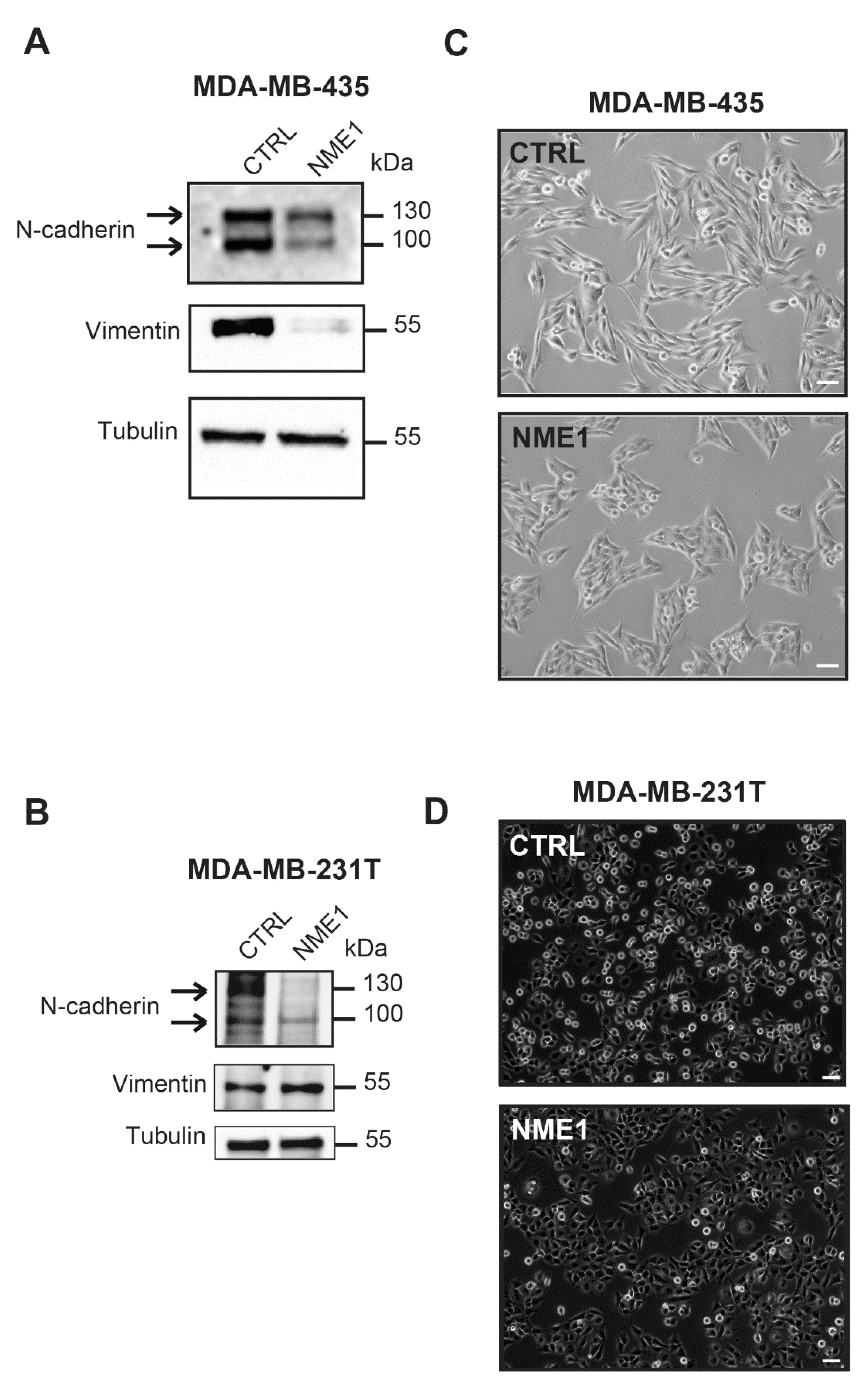
3.4. Stable Overexpression of NME1 Reverses EMT in Mesenchymal Breast Cancer Cells
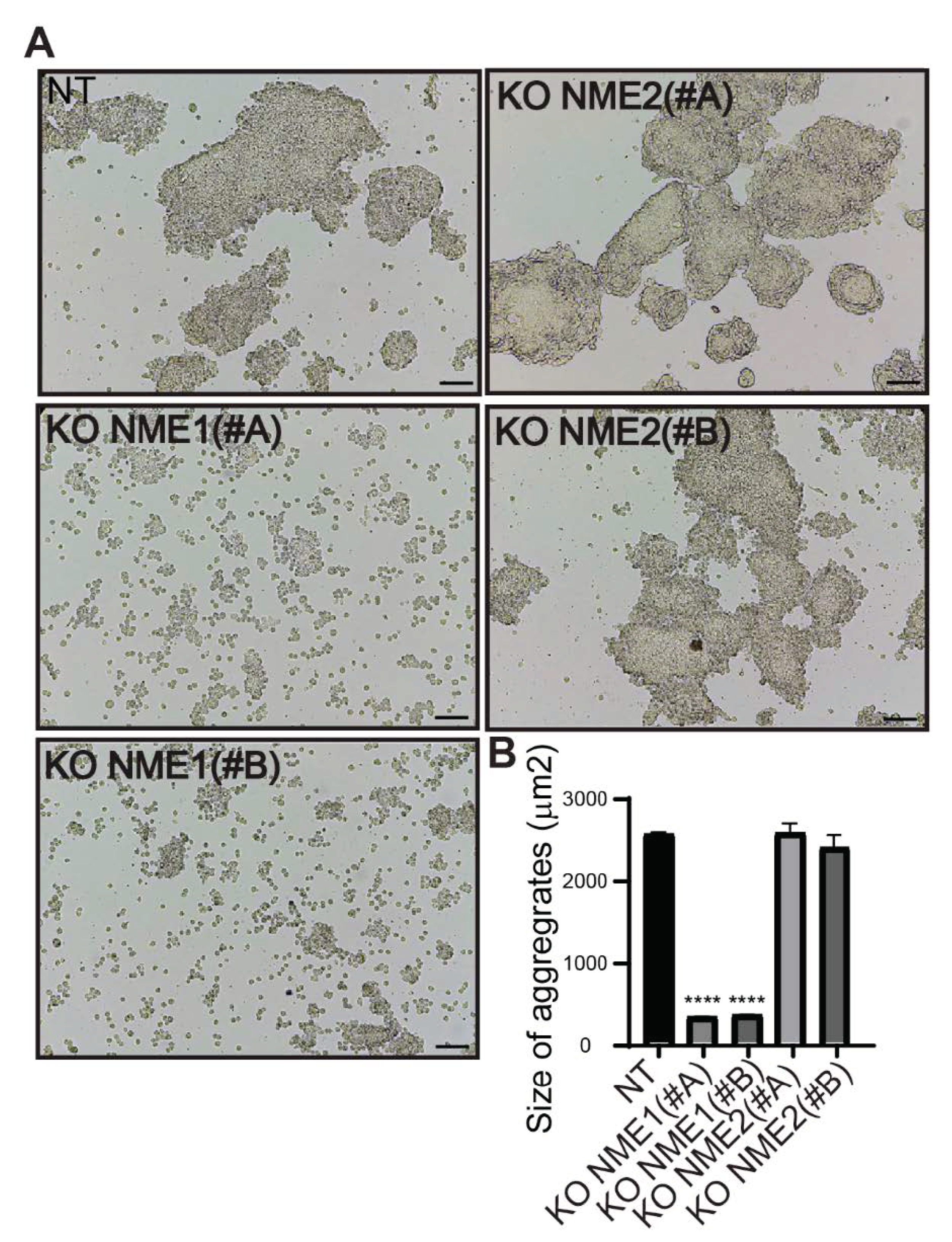
3.5. Inactivation of NME1 Decreases E-Cadherin-Mediated Cell–Cell Adhesion
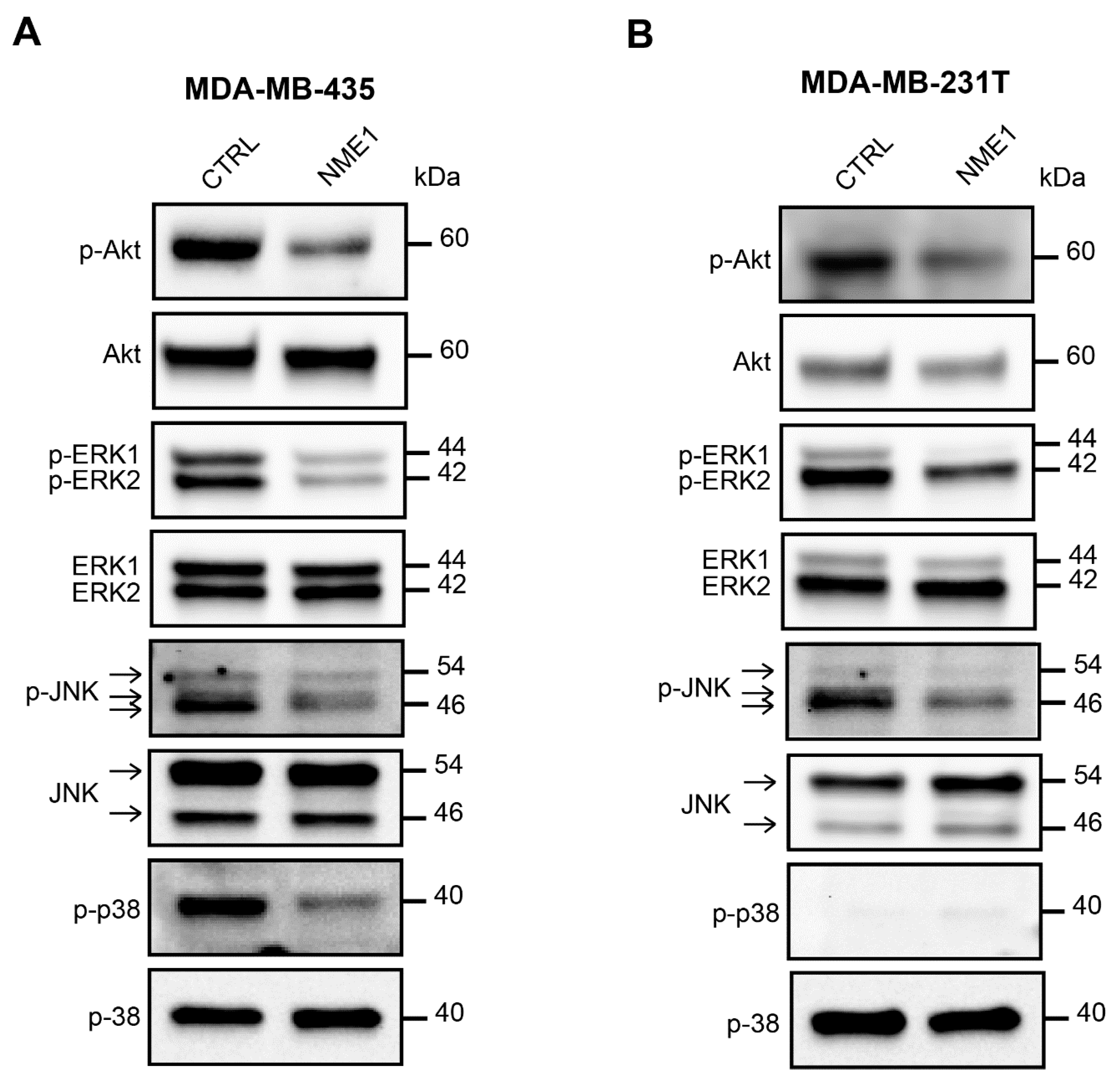
3.6. NME1 Expression Inhibits the Activity of Pro-EMT Signaling Pathways
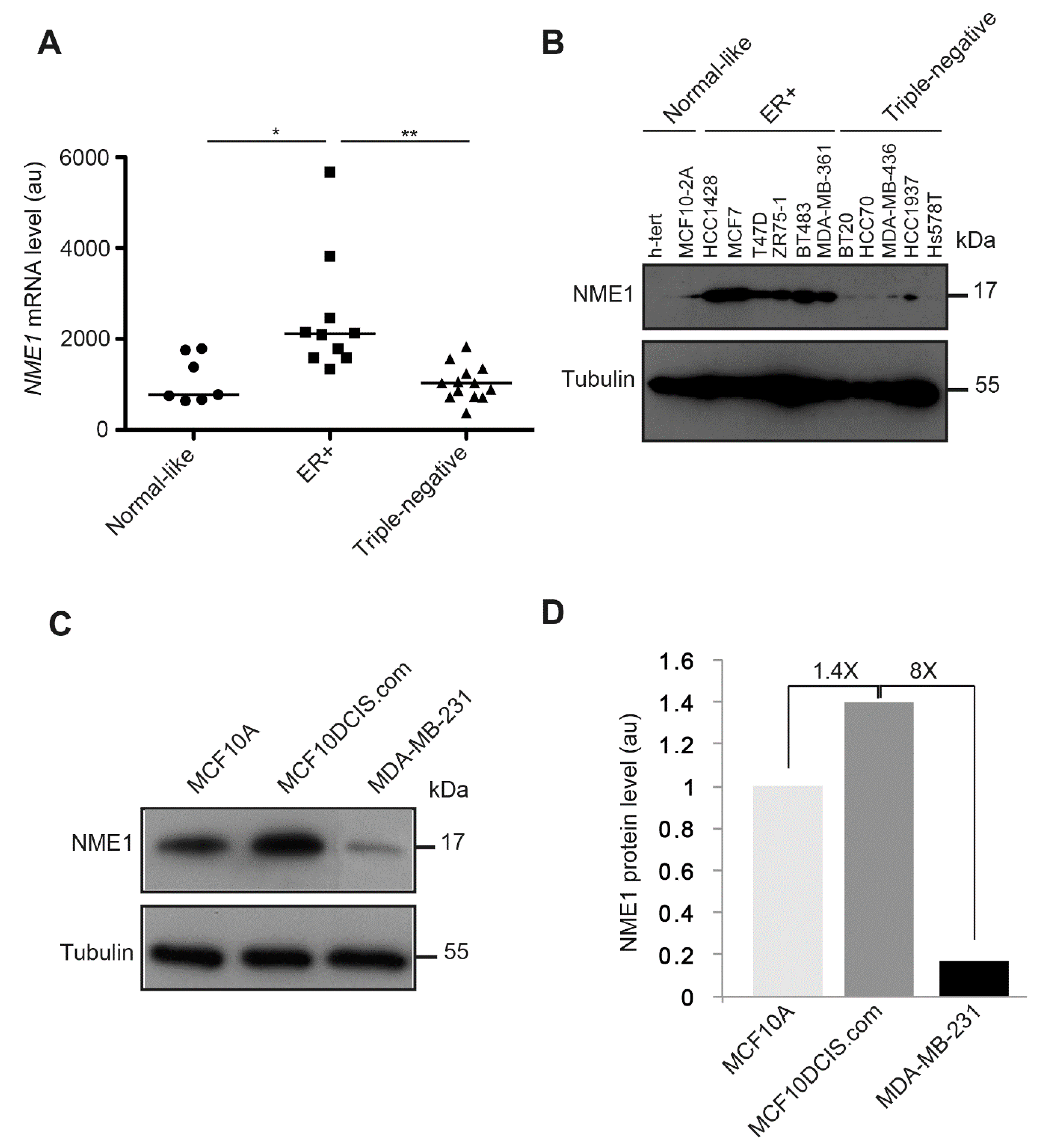
3.7. NME1 Expression Is Reduced in Human Breast Tumor Cell Lines with the Triple-Negative Phenotype
3.8. NME1 Expression Is Negatively Associated with EMT Markers in Human Cancer
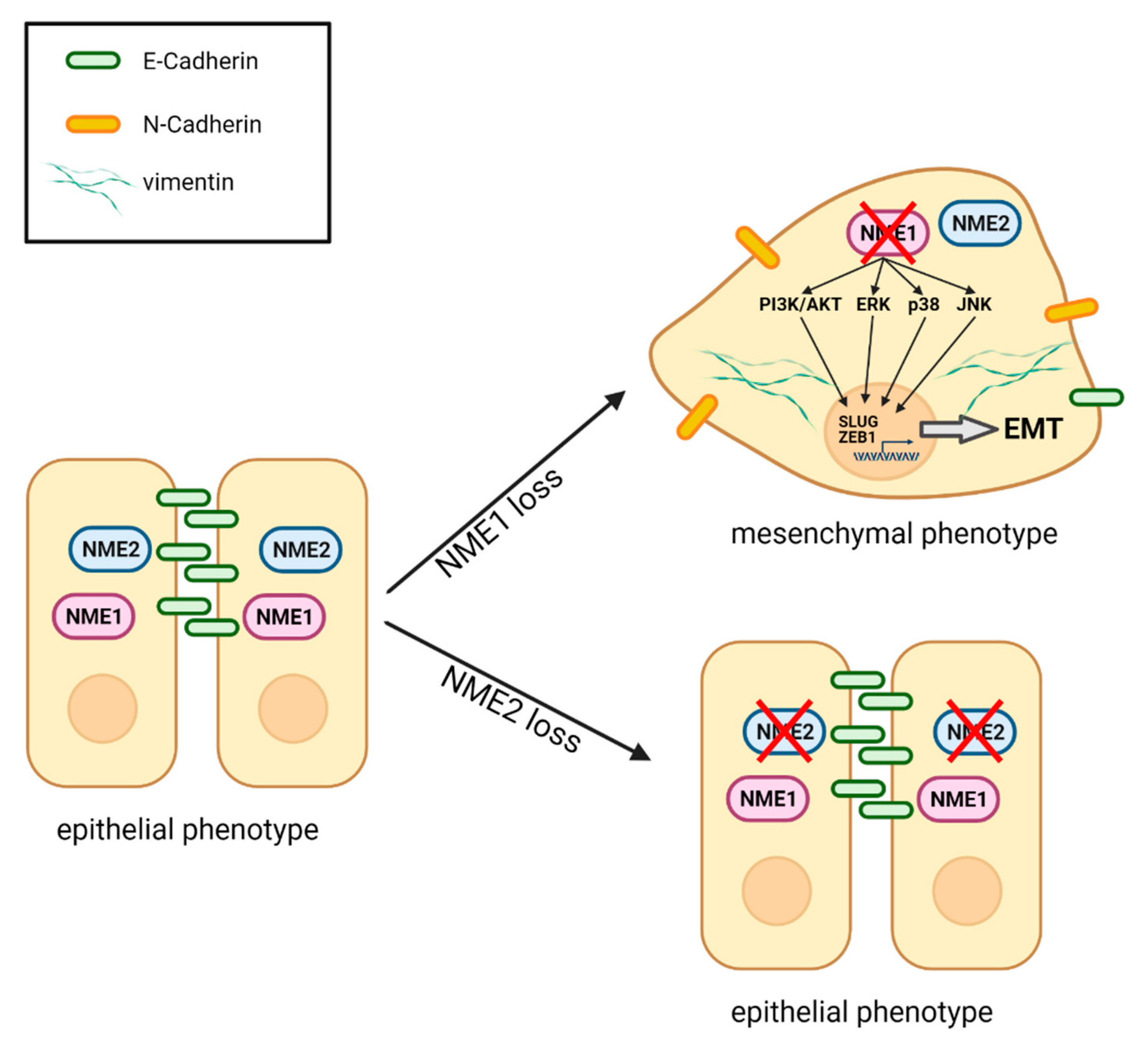
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Simeone, P.; Trerotola, M.; Franck, J.; Cardon, T.; Marchisio, M.; Fournier, I.; Salzet, M.; Maffia, M.; Vergara, D. The multiverse nature of epithelial to mesenchymal transition. Semin. Cancer Biol. 2019, 58, 1–10. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef]
- Zhang, J.; Tian, X.J.; Zhang, H.; Teng, Y.; Li, R.; Bai, F.; Elankumaran, S.; Xing, J. TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal 2014, 7, ra91. [Google Scholar] [CrossRef] [PubMed]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.-H.; Wang, H.; et al. Molecular Portraits of Epithelial, Mesenchymal and Hybrid States in Lung Adenocarcinoma and Their Relevance to Survival. Cancer Res. 2015, 75, 1789–1800. [Google Scholar] [CrossRef]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef]
- Harner-Foreman, N.; Vadakekolathu, J.; Laversin, S.A.; Mathieu, M.G.; Reeder, S.; Pockley, A.G.; Rees, R.C.; Boocock, D.J. A novel spontaneous model of epithelial-mesenchymal transition (EMT) using a primary prostate cancer derived cell line demonstrating distinct stem-like characteristics. Sci. Rep. 2017, 7, 40633. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Boissan, M.; Dabernat, S.; Peuchant, E.; Schlattner, U.; Lascu, I.; Lacombe, M.-L. The mammalian Nm23/NDPK family: From metastasis control to cilia movement. Mol. Cell. Biochem. 2009, 329, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgeirsson, U.P.; Talmadge, J.E.; Liotta, L.A.; Sobel, M.E. Evidence for a Novel Gene Associated with Low Tumor Metastatic Potential. J. Natl. Cancer Inst. 1988, 80, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Boissan, M.; Wendum, D.; Arnaud-Dabernat, S.; Munier, A.; Debray, M.; Lascu, I.; Daniel, J.-Y.; Lacombe, M.-L. Increased Lung Metastasis in Transgenic NM23-Null/SV40 Mice with Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2005, 97, 836–845. [Google Scholar] [CrossRef]
- Marino, N.; Nakayama, J.; Collins, J.W.; Steeg, P.S. Insights into the biology and prevention of tumor metastasis provided by the Nm23 metastasis suppressor gene. Cancer Metastasis Rev. 2012, 31, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Le Guelte, A.; Galan-Moya, E.-M.; Dwyer, J.; Treps, L.; Kettler, G.; Hebda, J.K.; Dubois, S.; Auffray, C.; Chneiweiss, H.; Bidere, N.; et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J. Cell Sci. 2012, 125, 4137–4146. [Google Scholar] [CrossRef] [PubMed]
- Polette, M.; Gilles, C.; Clavel, C.; Strumane, K.; Matos, M.; Zahm, J.-M.; Van Roy, F.; Birembaut, P. Quantitative cell dispersion analysis: New test to measure tumor cell aggressiveness. Int. J. Cancer 2001, 93, 644–652. [Google Scholar] [CrossRef]
- Awadelkarim, K.D.; Callens, C.; Rosse, C.; Susini, A.; Vacher, S.; Rouleau, E.; Lidereau, R.; Bieche, I. Quantification of PKC family genes in sporadic breast cancer by qRT-PCR: Evidence that PKCiota/lambda overexpression is an independent prognostic factor. Int J. Cancer 2012, 131, 2852–2862. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Vilchis-Nestor, C.A.; Roldan, M.L.; Leonardi, A.; Navea, J.G.; Padilla-Benavides, T.; Shoshani, L. Ouabain Enhances Cell-Cell Adhesion Mediated by beta1 Subunits of the Na (+) K (+)-ATPase in CHO Fibroblasts. Int. J. Mol. Sci. 2019, 20, 2111. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.A.; Russell, R.; Sammut, S.-J.; et al. The somatic mutation profiles of 2433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef]
- Lodillinsky, C.; Infante, E.; Guichard, A.; Chaligne, R.; Fuhrmann, L.; Cyrta, J.; Irondelle, M.; Lagoutte, E.; Vacher, S.; Bonsang-Kitzis, H.; et al. p63/MT1-MMP axis is required for in situ to invasive transition in basal-like breast cancer. Oncogene 2016, 35, 344–357. [Google Scholar] [CrossRef]
- Guo, H.-B.; Lee, I.; Kamar, M.; Pierce, M. N-Acetylglucosaminyltransferase V Expression Levels Regulate Cadherin-associated Homotypic Cell-Cell Adhesion and Intracellular Signaling Pathways. J. Biol. Chem. 2003, 278, 52412–52424. [Google Scholar] [CrossRef]
- Guo, H.-B.; Johnson, H.; Randolph, M.; Pierce, M. Regulation of Homotypic Cell-Cell Adhesion by Branched N-Glycosylation of N-cadherin Extracellular EC2 and EC3 Domains. J. Biol. Chem. 2009, 284, 34986–34997. [Google Scholar] [CrossRef]
- Adams, C.L.; Chen, Y.-T.; Smith, S.J.; Nelson, W.J. Mechanisms of Epithelial Cell–Cell Adhesion and Cell Compaction Revealed by High-resolution Tracking of E-Cadherin-Green Fluorescent Protein. J. Cell Biol. 1998, 142, 1105–1119. [Google Scholar] [CrossRef]
- David, J.M.; Rajasekaran, A.K. Dishonorable Discharge: The Oncogenic Roles of Cleaved E-Cadherin Fragments: Figure 1. Cancer Res. 2012, 72, 2917–2923. [Google Scholar] [CrossRef]
- Iyer, S.V.; Dange, P.P.; Alam, H.; Sawant, S.S.; Ingle, A.D.; Borges, A.M.; Shirsat, N.V.; Dalal, S.N.; Vaidya, M.M. Understanding the Role of Keratins 8 and 18 in Neoplastic Potential of Breast Cancer Derived Cell Lines. PLoS ONE 2013, 8, e53532. [Google Scholar] [CrossRef]
- Bozza, W.P.; Zhang, Y.; Zhang, B. Cytokeratin 8/18 protects breast cancer cell lines from TRAIL-induced apoptosis. Oncotarget 2018, 9, 23264–23273. [Google Scholar] [CrossRef] [PubMed]
- Boissan, M.; De Wever, O.; Lizarraga, F.; Wendum, D.; Poincloux, R.; Chignard, N.; Desbois-Mouthon, C.; Dufour, S.; Nawrocki-Raby, B.; Birembaut, P.; et al. Implication of Metastasis Suppressor NM23-H1 in Maintaining Adherens Junctions and Limiting the Invasive Potential of Human Cancer Cells. Cancer Res. 2010, 70, 7710–7722. [Google Scholar] [CrossRef]
- Mahler-Araujo, B.; Savage, K.; Parry, S.; Reis-Filho, J.S. Reduction of E-cadherin expression is associated with non-lobular breast carcinomas of basal-like and triple negative phenotype. J. Clin. Pathol. 2008, 61, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Wilde, A.; Fouquier d’Herouel, A.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; Del Sol, A.; Walters, K.A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.L.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.M.; Panzilius, E.; Bartsch, H.S.; Irmler, M.; Beckers, J.; Kari, V.; Linnemann, J.R.; Dragoi, D.; Hirschi, B.; Kloos, U.J.; et al. Stem-Cell-like Properties and Epithelial Plasticity Arise as Stable Traits after Transient Twist1 Activation. Cell Rep. 2015, 10, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G.; et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol. Oncol. 2016, 10, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Celia-Terrassa, T.; Bastian, C.; Liu, D.D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef]
- Jiao, G.-J.; Zhang, S.-J.; Li, Y.; Wu, W.-L.; Liu, H.-C. MicroRNA-645 promotes metastasis of osteosarcoma via targeting tumor suppressor NM23 nucleoside diphosphate kinase 2. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Polański, R.; Maguire, M.; Nield, P.C.; Jenkins, R.E.; Park, B.; Krawczyńska, K.; Devling, T.; Ray-Sinha, A.; Rubbi, C.P.; Vlatković, N.; et al. MDM2 interacts with NME2 (non-metastatic cells 2, protein) and suppresses the ability of NME2 to negatively regulate cell motility. Carcinogenesis 2011, 32, 1133–1142. [Google Scholar] [CrossRef]
- Khan, I.; Gril, B.; Steeg, P.S. Metastasis Suppressors NME1 and NME2 Promote Dynamin 2 Oligomerization and Regulate Tumor Cell Endocytosis, Motility and Metastasis. Cancer Res. 2019, 79, 4689–4702. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Yang, A.; Liu, W.; Wang, C.; Wang, M.; Zhang, L.; Wang, D.; Dong, J.-F.; Lihan, Z. NME2 Reduces Proliferation, Migration and Invasion of Gastric Cancer Cells to Limit Metastasis. PLoS ONE 2015, 10, e0115968. [Google Scholar] [CrossRef] [PubMed]
- McDermott, W.G.; Boissan, M.; Lacombe, M.-L.; Steeg, P.S.; Horak, C.E. Nm23-H1 homologs suppress tumor cell motility and anchorage independent growth. Clin. Exp. Metastasis 2007, 25, 131–138. [Google Scholar] [CrossRef]
- Thakur, R.K.; Yadav, V.K.; Kumar, A.; Singh, A.; Pal, K.; Hoeppner, L.; Saha, D.; Purohit, G.; Basundra, R.; Kar, A.; et al. Non-metastatic 2 (NME2)-mediated suppression of lung cancer metastasis involves transcriptional regulation of key cell adhesion factor vinculin. Nucleic Acids Res. 2014, 42, 11589–11600. [Google Scholar] [CrossRef]
- Miyazaki, H.; Fukuda, M.; Ishijima, Y.; Takagi, Y.; Iimura, T.; Negishi, A.; Hirayama, R.; Ishikawa, N.; Amagasa, T.; Kimura, N. Overexpression of nm23-H2/NDP kinase B in a human oral squamous cell carcinoma cell line results in reduced metastasis, differentiated phenotype in the metastatic site, and growth factor-independent proliferative activity in culture. Clin. Cancer Res. 1999, 5, 4301–4307. [Google Scholar]
- Aktary, Z.; Chapman, K.; Lam, L.; Lo, A.; Ji, C.; Graham, K.; Cook, L.; Li, L.; Mackey, J.R.; Pasdar, M. Plakoglobin interacts with and increases the protein levels of metastasis suppressor Nm23-H2 and regulates the expression of Nm23-H1. Oncogene 2010, 29, 2118–2129. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, M.-L.L.; Milon, L.; Munier, A.; Mehus, J.G.; Lambeth, D.O. The Human Nm23/Nucleoside Diphosphate Kinases. J. Bioenerg. Biomembr. 2000, 32, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Caligo, M.A.; Cipollini, G.; Fiore, L.; Calvo, S.; Basolo, F.; Collecchi, P.; Ciardiello, F.; Pepe, S.; Petrini, M.; Bevilacqua, G. NM23 gene expression correlates with cell growth rate and S-phase. Int. J. Cancer 1995, 60, 837–842. [Google Scholar] [CrossRef]
- Keim, D.; Hailat, N.; Melhem, R.; Zhu, X.X.; Lascu, I.; Veron, M.; Strahler, J.; Hanash, S.M. Proliferation-related expression of p19/nm23 nucleoside diphosphate kinase. J. Clin. Investig. 1992, 89, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Sorscher, S.; Steeg, P.; Feramisco, J.; Buckmaster, C.; Boss, G.; Meinkoth, J. Microinjection of an NM23-Specific Antibody Inhibits Cell Division in Rat Embryo Fibroblasts. Biochem. Biophys. Res. Commun. 1993, 195, 336–345. [Google Scholar] [CrossRef]
- Cipollini, G.; Berti, A.; Fiore, L.; Rainaldi, G.; Basolo, F.; Bevilacqua, G.; Caligo, M.A. Down-regulation of thenm23.h1 gene inhibits cell proliferation. Int. J. Cancer 1997, 73, 297–302. [Google Scholar] [CrossRef]
- Lee, H.; Okada, K.; Baba, H.; Furukawa, K.; Chang, S.; Shiku, H. Up-regulation of nm23/NDP kinase expression in regenerating hepatocytes. Int. J. Oncol. 1997, 11, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Arnaud-Dabernat, S.; Bourbon, P.M.; Dierich, A.; Le Meur, M.; Daniel, J.-Y. Knockout mice as model systems for studying nm23/NDP kinase gene functions. Application to the nm23-M1 gene. J. Bioenerg. Biomembr. 2003, 35, 19–30. [Google Scholar] [CrossRef]
- Deplagne, C.; Peuchant, E.; Moranvillier, I.; Dubus, P.; Dabernat, S. The Anti-Metastatic nm23-1 Gene is Needed for the Final Step of Mammary Duct Maturation of the Mouse Nipple. PLoS ONE 2011, 6, e18645. [Google Scholar] [CrossRef]
- Di, L.; Srivastava, S.; Zhdanova, O.; Sun, Y.; Li, Z.; Skolnik, E.Y. Nucleoside Diphosphate Kinase B Knock-out Mice Have Impaired Activation of the K+ Channel KCa3.1, Resulting in Defective T Cell Activation. J. Biol. Chem. 2010, 285, 38765–38771. [Google Scholar] [CrossRef]
- Hartsough, M.T.; Clare, S.E.; Mair, M.; Elkahloun, A.G.; Sgroi, D.; Osborne, C.K.; Clark, G.; Steeg, P.S. Elevation of breast carcinoma Nm23-H1 metastasis suppressor gene expression and reduced motility by DNA methylation inhibition. Cancer Res. 2001, 61, 2320–2327. [Google Scholar]
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-Associated PRMT5 Methylates Histone H3 Arginine 8 and Negatively Regulates Expression of ST7 and NM23 Tumor Suppressor Genes. Mol. Cell. Biol. 2004, 24, 9630–9645. [Google Scholar] [CrossRef]
- Almeida, M.I.; Nicoloso, M.S.; Zeng, L.; Ivan, C.; Spizzo, R.; Gafà, R.; Xiao, L.; Zhang, X.; Vannini, I.; Fanini, F.; et al. Strand-Specific miR-28-5p and miR-28-3p Have Distinct Effects in Colorectal Cancer Cells. Gastroenterology 2012, 142, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Fiore, L.S.; Ganguly, S.S.; Sledziona, J.; Cibull, M.L.; Wang, C.; Richards, D.L.; Neltner, J.M.; Beach, C.M.; McCorkle, J.R.; Kaetzel, D.M.; et al. c-Abl and Arg induce cathepsin-mediated lysosomal degradation of the NM23-H1 metastasis suppressor in invasive cancer. Oncogene 2014, 33, 4508–4520. [Google Scholar] [CrossRef]
- Chen, W.; Xiong, S.; Li, J.; Li, X.; Liu, Y.; Zou, C.; Mallampalli, R.K. The Ubiquitin E3 Ligase SCF-FBXO24 Recognizes Deacetylated Nucleoside Diphosphate Kinase A to Enhance Its Degradation. Mol. Cell. Biol. 2015, 35, 1001–1013. [Google Scholar] [CrossRef]
- Paul, C.; Khera, L.; Kaul, R. Hepatitis C virus core protein interacts with cellular metastasis suppressor Nm23-H1 and promotes cell migration and invasion. Arch. Virol. 2019, 164, 1271–1285. [Google Scholar] [CrossRef]
- Khera, L.; Paul, C.; Kaul, R. Hepatitis C Virus E1 protein promotes cell migration and invasion by modulating cellular metastasis suppressor Nm23-H1. Virology 2017, 506, 110–120. [Google Scholar] [CrossRef]
- Bruner, H.C.; Derksen, P.W. Loss of E-Cadherin-Dependent Cell–Cell Adhesion and the Development and Progression of Cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a029330. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; Van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef] [PubMed]
- Boissan, M.; Montagnac, G.; Shen, Q.; Griparic, L.; Guitton, J.; Romao, M.; Sauvonnet, N.; Lagache, T.; Lascu, I.; Raposo, G.; et al. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science 2014, 344, 1510–1515. [Google Scholar] [CrossRef]
- Carey, R.M.; Blusztajn, J.K.; Slack, B.E. Surface expression and limited proteolysis of ADAM10 are increased by a dominant negative inhibitor of dynamin. BMC Cell Biol. 2011, 12, 20. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huna, A.; Nawrocki-Raby, B.; Padilla-Benavides, T.; Gavard, J.; Coscoy, S.; Bernard, D.; Boissan, M. Loss of the Metastasis Suppressor NME1, But Not of Its Highly Related Isoform NME2, Induces a Hybrid Epithelial–Mesenchymal State in Cancer Cells. Int. J. Mol. Sci. 2021, 22, 3718. https://doi.org/10.3390/ijms22073718
Huna A, Nawrocki-Raby B, Padilla-Benavides T, Gavard J, Coscoy S, Bernard D, Boissan M. Loss of the Metastasis Suppressor NME1, But Not of Its Highly Related Isoform NME2, Induces a Hybrid Epithelial–Mesenchymal State in Cancer Cells. International Journal of Molecular Sciences. 2021; 22(7):3718. https://doi.org/10.3390/ijms22073718
Chicago/Turabian StyleHuna, Anda, Béatrice Nawrocki-Raby, Teresita Padilla-Benavides, Julie Gavard, Sylvie Coscoy, David Bernard, and Mathieu Boissan. 2021. "Loss of the Metastasis Suppressor NME1, But Not of Its Highly Related Isoform NME2, Induces a Hybrid Epithelial–Mesenchymal State in Cancer Cells" International Journal of Molecular Sciences 22, no. 7: 3718. https://doi.org/10.3390/ijms22073718
APA StyleHuna, A., Nawrocki-Raby, B., Padilla-Benavides, T., Gavard, J., Coscoy, S., Bernard, D., & Boissan, M. (2021). Loss of the Metastasis Suppressor NME1, But Not of Its Highly Related Isoform NME2, Induces a Hybrid Epithelial–Mesenchymal State in Cancer Cells. International Journal of Molecular Sciences, 22(7), 3718. https://doi.org/10.3390/ijms22073718