
Molecular Mechanism of Cannabinoids in Cancer Progression

 ,
, 

Abstract
1. Introduction
2. ECS in Cancer
2.1. CB Receptor Expression in Cancer
2.2. Endocannabinoid Levels and Degrading Enzymes in Cancer
3. Cannabinoid Ligands
3.1. Endocannabinoids
3.2. Phytocannabinoids
3.3. Synthetic Cannabinoids
3.3.1. Inhibitors of Endocannabinoid Cellular Uptake
3.3.2. Inhibitors FAAH
3.3.3. MAGL Inhibitor
3.3.4. Dual CB1R/CB2R Agonists
3.3.5. Anandamide Analogs
3.3.6. Selective CB1R Agonists
3.3.7. Selective CB2R Agonists
3.3.8. Selective Antagonists/Inverse Agonists for CB1 Receptors
3.3.9. Selective Antagonists/Inverse Agonists for CB2 Receptors
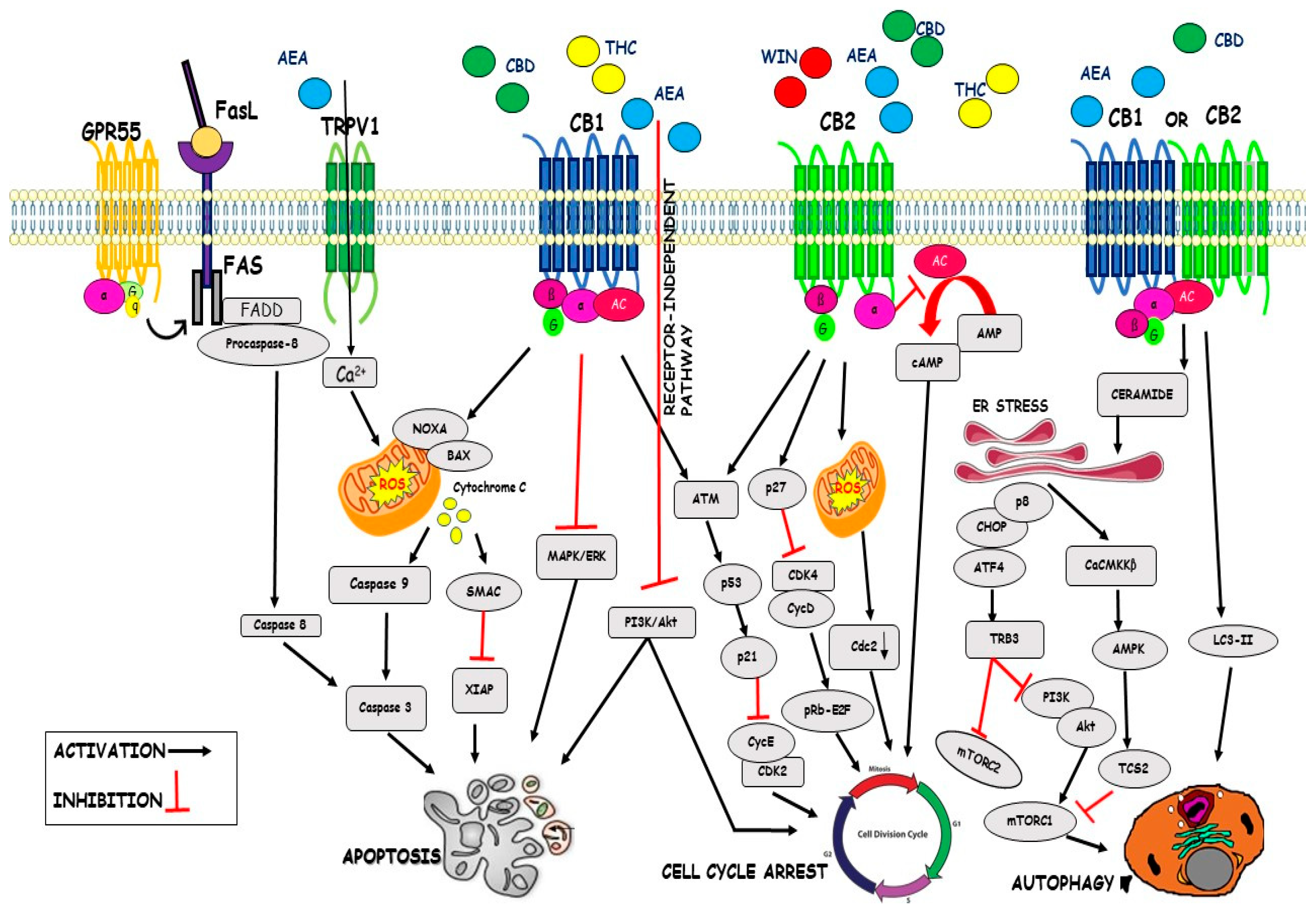
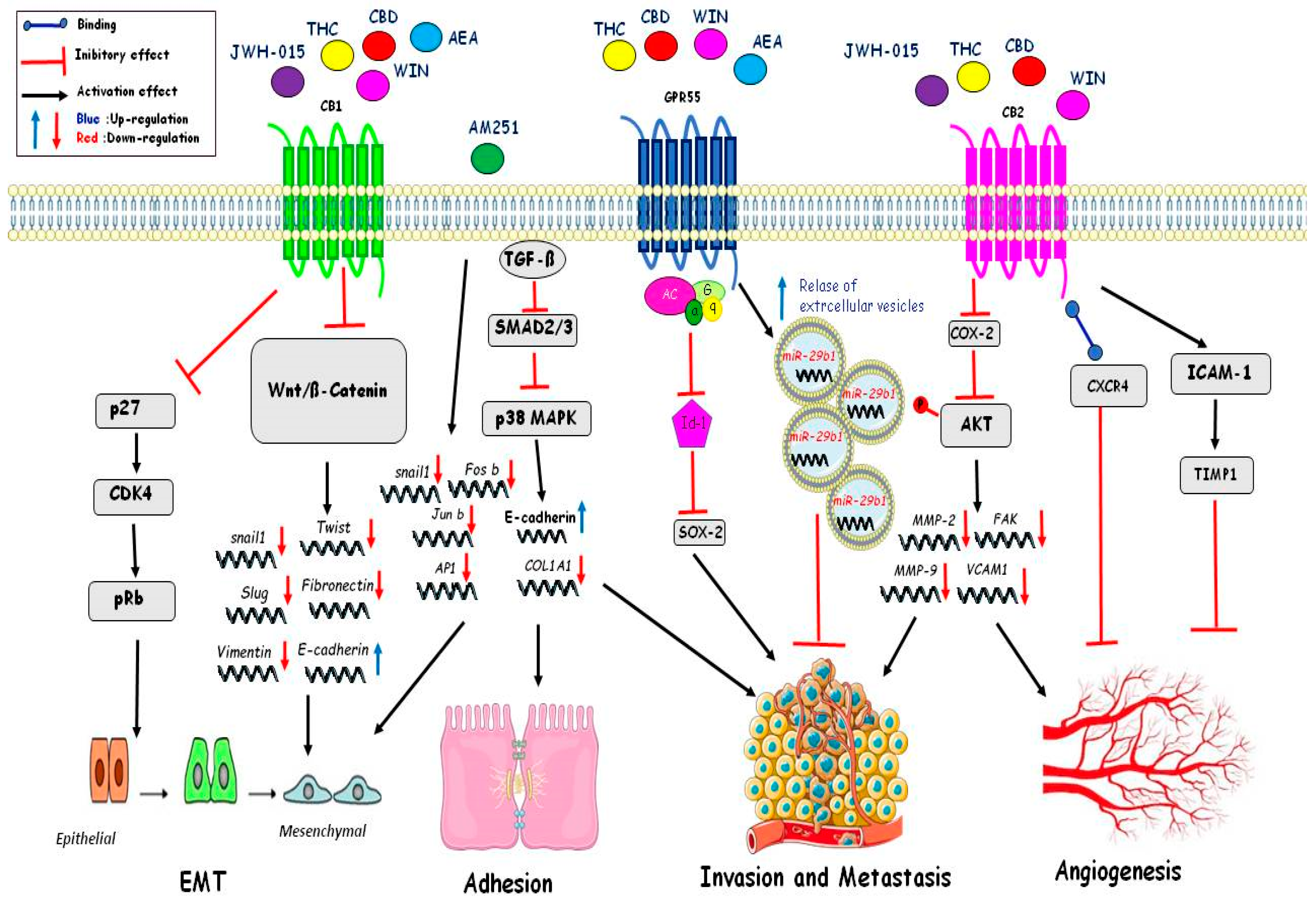
4. Mechanism of Action of the Cannabinoids on Cancer Progression
4.1. Cell Cycle
4.2. Apoptosis
4.3. Autophagy
4.4. Migration
4.5. Angiogenesis
4.6. Epithelial to Mesenchymal Transition (EMT)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-AG | 2-Arachidonoylglycerol |
| 2-AGE | 2-Arachidonyl glyceryl ether |
| AA-5HT | Arachidonoyl serotonin |
| ACEA | Arachidonyl-2’-chloroethylamide |
| ACF | Aberrant Crypt Foci |
| AEA | N-arachidonoylethanolamine |
| AI | Alkylindole synthetic |
| AM251 | Inverse agonist at the CB1R |
| AM404 | N-arachidonoylaminophenol |
| AMPK 5’ | AMP-activated protein kinase |
| ARA-S | N-arachidonoyl serine |
| ATM | Ataxia–telangiectasia mutated |
| CaCMKKβ | Ca2+/Calmoduline-dependent kinase kinase β |
| CB1R | Cannabinoid1 receptor |
| CB2R | Cannabinoid2 receptor |
| CBC | Cannabichromene |
| CBD | Cannabidiol |
| CBDA | Cannabidiolic acid |
| CBDV | Cannabidivarin |
| CBG | Cannabigerol |
| CBN | Cannabinol |
| Cdk | Cyclin-dependent kinases |
| CFZ | Carfilzomib |
| COL1A1 | Collagen 1A1 |
| COX-2 | Cyclooxygenase-2 |
| CP 55,940 | Synthetic cannadinoid |
| CRC | Colorectal cancer cells |
| DAGL | diacylglycerol lipase |
| DBT | Delayed brain tumor |
| EC | Endometrial cancer |
| ECS | Endocannabinoid system |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FAAH | Hydrolase fatty acid amide hydrolase |
| FAK | Focal adhesion kinase |
| FBXL5 | F-box/LRR-repeat protein 5 |
| FGF-2 | Fibroblast growth factors |
| GC | Gastric cancer |
| GPR119 | G protein-coupled receptor 119 |
| GPR18 | G protein-coupled receptor 18 |
| GPR55 | G protein-coupled receptor 55 |
| GRP78 | Glucose-regulated protein 78 |
| HR | Herceptin resistant |
| HRAS | Harvey rat sarcoma virus |
| HS | Herceptin sensitive |
| HUVEC | Human umbilical vein endothelial cells |
| ID1 | Inhibitor of DNA binding 1 |
| IL-6 | Interleukin 6 |
| KRAS | Kirsten murine sarcoma virus |
| LPI | Lysophosphatidylinositol |
| LPS | Lipopolysaccharides |
| MAGL | Monoacylglycerol lipase |
| MAPK | Mitogen Activated-Protein Kinase |
| Met-F-AEA | 2-methyl-2′-F-anandamide |
| MICA/B | MHC class I polypeptide–related sequence A/B |
| MM | Multiple myeloma |
| MMP2/9 | Matrix metalloproteinase-2/9 |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NAAA | N-acylethanolamine acid amide hydrolase |
| NADA | N-Arachidonoyl dopamine |
| NAPE-PLD | N-acyl phosphatidylethanolamine-specific phospholipase D |
| Nf-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK-cells | Natural Killer cells |
| NLRP3 | NLR family pyrin domain containing 3 |
| NMSC | Non-melanoma skin cancer |
| NSCLC | Non-small-cell lung cancer |
| O-1602 | Agonist of GPR55 |
| OA | Oleamide |
| OEA | Oleoylethanolamide |
| oGPCR | Orphan G protein-coupled receptor |
| OMDM-2 | Cannabinoid reuptake inhibitor |
| PEA | Palmitoylethanolamide |
| PGE2 | Prostaglandin E2 |
| PKA | Protein kinase A |
| PLC | Phospholipase C |
| PPAR-α | Peroxisome proliferator-activated receptors α |
| PPAR-γ | Peroxisome proliferator-activated receptors γ |
| PRAS40 | Proline-rich Akt substrate of 40 kDa |
| pRb | Phospho-Retinoblastoma |
| ROS | Reactive oxygen species |
| SOCS3 | Suppressor of cytokine signaling 3 |
| SPARC | Secreted protein acidic and cysteine-rich |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TAMS | Tumor-associated macrophages |
| TCF | T Cell Factor |
| THC | Δ⁹-tetrahydrocannabinol |
| TIMP-1 | Metallopeptidase inhibitor 1 |
| TMZ | Temozolomide |
| TRIB3 | Tribbles pseudokinase 3 |
| TRP | Transient receptor potential |
| TRPV1 | Transient receptor potential vanilloid type-1 |
| TSC2 | Tuberous Sclerosis Complex 2 |
| UCM707 | Cannabinoid reuptake inhibitor |
| URB602 | [1,1’-biphenyl]-3-yl-carbamic acid |
| VCAM1 | Vascular cell adhesion molecule 1 |
| VDM-11 | Cannabinoid reuptake inhibitor |
| VEGF | Vascular endothelial growth factor |
| VWF | Von Willebrand factor |
| XIAP | X-linked inhibitor apoptosis |
References
- Ezzili, C.; Otrubova, K.; Boger, D.L. Fatty acid amide signaling molecules. Bioorg. Med. Chem. Lett. 2010, 20, 5959–5968. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1995, 48, 443–450. [Google Scholar] [PubMed]
- De Petrocellis, L.; Di Marzo, V. An introduction to the endocannabinoid system: From the early to the latest concepts. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. Cannabinoid receptors: An update on cell signaling, pathophysiological roles and therapeutic opportunities in neurological, cardiovascular, and inflammatory diseases. Int. J. Mol. Sci. 2020, 21, 7693. [Google Scholar] [CrossRef]
- Bermudez-Silva, F.; Viveros, M.; McPartland, J.; De Fonseca, F.R. The endocannabinoid system, eating behavior and energy homeostasis: The end or a new beginning? Pharmacol. Biochem. Behav. 2010, 95, 375–382. [Google Scholar] [CrossRef]
- Senn, L.; Cannazza, G.; Biagini, G. Receptors and channels possibly mediating the effects of phytocannabinoids on seizures and epilepsy. Pharmaceuticals 2020, 13, 174. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Martín-Sabroso, C.; Torres-Suárez, A.I. Insights into the effects of the endocannabinoid system in cancer: A review. Br. J. Pharmacol. 2018, 175, 2566–2580. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Andradas, C.; Mira, E.; Pérez-Gómez, E.; Cerutti, C.; Moreno-Bueno, G.; Flores, J.M.; García-Real, I.; Palacios, J.; Mañes, S.; et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef]
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 2009, 8, 3117–3129. [Google Scholar] [CrossRef]
- Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The endocannabinoid system: A target for cancer treatment. Int. J. Mol. Sci. 2020, 21, 747. [Google Scholar] [CrossRef] [PubMed]
- Ayakannu, T.; Taylor, A.H.; Willets, J.M.; Konje, J.C. The evolving role of the endocannabinoid system in gynaecological cancer. Hum. Reprod. Update 2015, 21, 517–535. [Google Scholar] [CrossRef] [PubMed]
- Messalli, E.M.; Grauso, F.; Luise, R.; Angelini, A.; Rossiello, R. Cannabinoid receptor type 1 immunoreactivity and disease severity in human epithelial ovarian tumors. Am. J. Obstet. Gynecol. 2014, 211, 234.e1–234.e6. [Google Scholar] [CrossRef] [PubMed]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non–small cell lung cancer growth and metastasis. Cancer Prev. Res. 2011, 4, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, H.; Ning, W.; Backlund, M.G.; Dey, S.K.; Dubois, R.N. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008, 68, 6468–6476. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Afaq, F.; Adhami, V.M.; Mukhtar, H. Cannabinoid receptor as a novel target for the treatment of prostate cancer. Cancer Res. 2005, 65, 1635–1641. [Google Scholar] [CrossRef]
- Guindon, J.; Hohmann, A.G. The endocannabinoid system and cancer: Therapeutic implication. Br. J. Pharmacol. 2011, 163, 1447–1463. [Google Scholar] [CrossRef]
- Pyszniak, M.; Tabarkiewicz, J.; Łuszczki, J.J. Endocannabinoid system as a regulator of tumor cell malignancy–biological pathways and clinical significance. OncoTargets Ther. 2016, 9, 4323–4336. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bari, M.; Battista, N.; Di Rienzo, M.; Finazzi-Agrò, A. Endogenous cannabinoids in neuronal and immune cells: Toxic effects, levels and degradation. Funct. Neurol. 2001, 16, 53–60. [Google Scholar]
- Wu, X.; Han, L.; Zhang, X.; Li, L.; Jiang, C.; Qiu, Y.; Huang, R.; Xie, B.; Lin, Z.; Ren, J.; et al. Alteration of endocannabinoid system in human gliomas. J. Neurochem. 2012, 120, 842–849. [Google Scholar] [CrossRef]
- Petersen, G.; Moesgaard, B.; Schmid, P.C.; Schmid, H.H.O.; Broholm, H.; Kosteljanetz, M.; Hansen, H.S. Endocannabinoid metabolism in human glioblastomas and meningiomas compared to human non-tumour brain tissue. J. Neurochem. 2005, 93, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.H.; Schmid, P.C.; Berdyshev, E.V. Cell signaling by endocannabinoids and their congeners: Questions of selectivity and other challenges. Chem. Phys. Lipids 2002, 121, 111–134. [Google Scholar] [CrossRef]
- Ligresti, A.; Bisogno, T.; Matias, I.; De Petrocellis, L.; Cascio, M.G.; Cosenza, V.; D’Argenio, G.; Scaglione, G.; Bifulco, M.; Sorrentini, I.; et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology 2003, 125, 677–687. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Benoit, V.M.; Laenkholm, A.V.; Nielsen, O.; Johansen, L.E.; Ditzel, H.J. Identification of genes with altered expression in medullary breast cancer vs. ductal breast cancer and normal breast epithelia. Int. J. Oncol. 2006, 28, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, H.; Li, Y.; Li, L.; Qiu, Y.; Ren, J. Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncol. Rep. 2015, 34, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Ayakannu, T.; Taylor, A.H.; Bari, M.; Mastrangelo, N.; Maccarrone, M.; Konje, J.C. Expression and function of the endocannabinoid modulating enzymes fatty acid amide hydrolase and n-acylphosphatidylethanolamine-specific phospholipase D in endometrial carcinoma. Front. Oncol. 2019, 9, 1363–1376. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, B.; Seviour, E.G.; Tao, K.-X.; Liu, X.-H.; Ling, Y.; Chen, J.-Y.; Wang, G.-B. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 2011, 307, 6–17. [Google Scholar] [CrossRef]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef]
- Zhu, W.; Zhao, Y.; Zhou, J.; Wang, X.; Pan, Q.; Zhang, N.; Wang, L.; Wang, M.; Zhan, D.; Liu, Z.; et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-κB-mediated epithelial-mesenchymal transition. J. Hematol. Oncol. 2016, 9, 1–13. [Google Scholar] [CrossRef]
- Pagotto, U.; Marsicano, G.; Fezza, F.; Theodoropoulou, M.; Grübler, Y.; Stalla, J.; Arzberger, T.; Milone, A.; Losa, M.; Di Marzo, V.; et al. Normal human pituitary gland and pituitary adenomas express cannabinoid receptor Type 1 and synthesize endogenous cannabinoids: First evidence for a direct role of cannabinoids on hormone modulation at the human pituitary level. J. Clin. Endocrinol. Metab. 2001, 86, 2687–2696. [Google Scholar] [CrossRef][Green Version]
- Endsley, M.P.; Thill, R.; Choudhry, I.; Williams, C.L.; Kajdacsy-Balla, A.; Campbell, W.B.; Nithipatikom, K. Expression and function of fatty acid amide hydrolase in prostate cancer. Int. J. Cancer 2008, 123, 1318–1326. [Google Scholar] [CrossRef]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef]
- Michalski, C.W.; Oti, F.E.; Erkan, M.; Sauliunaite, D.; Bergmann, F.; Pacher, P.; Batkai, S.; Müller, M.W.; Giese, N.A.; Friess, H.; et al. Cannabinoids in pancreatic cancer: Correlation with survival and pain. Int. J. Cancer 2007, 122, 742–750. [Google Scholar] [CrossRef]
- Guida, M.; Ligresti, A.; De Filippis, D.; D’Amico, A.; Petrosino, S.; Cipriano, M.; Bifulco, G.; Simonetti, S.; Orlando, P.; Insabato, L.; et al. The Levels of the Endocannabinoid Receptor CB2 and Its Ligand 2-Arachidonoylglycerol Are Elevated in Endometrial Carcinoma. Endocrinology 2010, 151, 921–928. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Isbell, M.A.; Endsley, M.P.; Woodliff, J.E.; Campbell, W.B. Anti-proliferative effect of a putative endocannabinoid, 2-arachidonylglyceryl ether in prostate carcinoma cells. Prostaglandins Lipid Mediat. 2011, 94, 34–43. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Endsley, M.P.; Isbell, M.A.; Falck, J.R.; Iwamoto, Y.; Hillard, C.J.; Campbell, W.B. 2-Arachidonoylglycerol: A novel inhibitor of androgen-independent prostate cancer cell invasion. Cancer Res. 2004, 64, 8826–8830. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Melck, D.; Bobrov, M.Y.; Gretskaya, N.M.; Bezuglov, V.V.; De Petrocellis, L.; Di Marzo, V. N-acyl-dopamines: Novel synthetic CB(1) cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem. J. 2000, 351, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Akimov, M.G.; Gretskaya, N.M.; Zinchenko, G.N.; Bezuglov, V.V. Cytotoxicity of endogenous lipids n-acyl dopamines and their possible metabolic derivatives for human cancer cell lines of different histological origin. Anticancer. Res. 2015, 35, 2657–2661. [Google Scholar] [PubMed]
- Davies, J.W.; Hainsworth, A.H.; Guerin, C.J.; Lambert, D.G. Pharmacology of capsaicin-, anandamide-, and N-arachidonoyl-dopamine-evoked cell death in a homogeneous transient receptor potential vanilloid subtype 1 receptor population. Br. J. Anaesth. 2010, 104, 596–602. [Google Scholar] [CrossRef]
- Wu, M.; Huang, J.; Zhang, J.; Benes, C.; Jiao, B.; Ren, R. N-Arachidonoyl dopamine inhibits NRAS neoplastic transformation by suppressing its plasma membrane translocation. Mol. Cancer Ther. 2016, 16, 57–67. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, structure, and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Maccarrone, M. Missing pieces to the endocannabinoid puzzle. Trends Mol. Med. 2020, 26, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Tomko, A.M.; Whynot, E.G.; Ellis, L.D.; Dupré, D.J. Anti-cancer potential of cannabinoids, terpenes, and flavonoids present in Cannabis. Cancers 2020, 12, 1985. [Google Scholar] [CrossRef]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef]
- Maccarrone, M.; Maldonado, R.; Casas, M.; Henze, T.; Centonze, D. Cannabinoids therapeutic use: What is our current understanding following the introduction of THC, THC:CBD oromucosal spray and others? Expert Rev. Clin. Pharmacol. 2017, 10, 443–455. [Google Scholar] [CrossRef]
- Portenoy, R.K.; Ganae-Motan, E.D.; Allende, S.; Yanagihara, R.; Shaiova, L.; Weinstein, S.; McQuade, R.; E Wright, S.; Fallon, M.T. Nabiximols for opioid-treated cancer patients with poorly-controlled chronic pain: A randomized, placebo-controlled, graded-dose Trial. J. Pain 2012, 13, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; A Ross, R.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef]
- Seltzer, E.S.; Watters, A.K.; MacKenzie, J.D.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a promising anti-cancer drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- López-Valero, I.; Saiz-Ladera, C.; Torres, S.; Hernández-Tiedra, S.; García-Taboada, E.; Rodríguez-Fornés, F.; Barba, M.; Dávila, D.; Salvador-Tormo, N.; Guzmán, M.; et al. Targeting glioma initiating cells with a combined therapy of cannabinoids and temozolomide. Biochem. Pharmacol. 2018, 157, 266–274. [Google Scholar] [CrossRef]
- Orrego-González, E.; Londoño-Tobón, L.; Ardila-González, J.; Polania-Tovar, D.; Valencia-Cárdenas, A.; Meerbeke, A.V.-V. Cannabinoid effects on experimental colorectal cancer models reduce aberrant crypt foci (ACF) and tumor volume: A systematic review. Evid.-Based Complement. Altern. Med. 2020, 2020, 2371527. [Google Scholar] [CrossRef]
- Takeda, S.; Okajima, S.; Miyoshi, H.; Yoshida, K.; Okamoto, Y.; Okada, T.; Amamoto, T.; Watanabe, K.; Omiecinski, C.J.; Aramaki, H. Cannabidiolic acid, a major cannabinoid in fiber-type cannabis, is an inhibitor of MDA-MB-231 breast cancer cell migration. Toxicol. Lett. 2012, 214, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Jinks, N.; Babaei-Jadidi, R.; Kashfi, H.; Castellanos-Uribe, M.; May, S.T.; Mukherjee, A.; Nateri, A.S. Repurposing antibacterial AM404 as a potential anticancer drug for targeting colorectal cancer stem-like cells. Cancers 2019, 12, 106. [Google Scholar] [CrossRef]
- Bifulco, M.; Laezza, C.; Valenti, M.; Ligresti, A.; Portella, G.; Di Marzo, V. A new strategy to block tumor growth by inhibiting endocannabinoid inactivation. FASEB J. 2004, 18, 1606–1608. [Google Scholar] [CrossRef]
- Ammar, R.; Ulrich-Merzenich, G. Curcumin synergizes with the endocannabinoid reuptake inhibitor OMDM-2 in human MCF-7 breast cancer and U-87 glioblastoma cells. Synergy 2017, 5, 7–14. [Google Scholar] [CrossRef]
- Winkler, K.; Ramer, R.; Dithmer, S.; Ivanov, I.; Merkord, J.; Hinz, B. Fatty acid amide hydrolase inhibitors confer anti-invasive and antimetastatic effects on lung cancer cells. Oncotarget 2016, 7, 15047–15064. [Google Scholar] [CrossRef] [PubMed]
- Hamtiaux, L.; Masquelier, J.; Muccioli, G.G.; Bouzin, C.; Feron, O.; Gallez, B.; Lambert, D.M. The association of N-palmitoylethanolamine with the FAAH inhibitor URB597 impairs melanoma growth through a supra-additive action. BMC Cancer 2012, 12, 92. [Google Scholar] [CrossRef]
- Ravi, J.; Sneh, A.; Shilo, K.; Nasser, M.W.; Ganju, R.K. FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget 2014, 5, 2475–2486. [Google Scholar] [CrossRef] [PubMed]
- Hamtiaux, L.; Hansoulle, L.; Dauguet, N.; Muccioli, G.G.; Gallez, B.; Lambert, D.M. Increasing antiproliferative properties of endocannabinoids in N1E-115 neuroblastoma cells through inhibition of their metabolism. PLoS ONE 2011, 6, e26823. [Google Scholar] [CrossRef] [PubMed]
- Proto, M.C.; Gazzerro, P.; Di Croce, L.; Santoro, A.; Malfitano, A.M.; Laezza, C.; Pisanti, S.; Bifulco, M. Interaction of endocannabinoid system and steroid hormones in the control of colon cancer cell growth. J. Cell. Physiol. 2011, 227, 250–258. [Google Scholar] [CrossRef]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef]
- Ma, M.; Bai, J.; Ling, Y.; Chang, W.; Xie, G.; Li, R.; Wang, G.; Tao, K. Monoacylglycerol lipase inhibitor JZL184 regulates apoptosis and migration of colorectal cancer cells. Mol. Med. Rep. 2016, 13, 2850–2856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Z.; Lian, Z.; Liao, R.; Chen, Y.; Qin, Y.; Wang, J.; Jiang, Q.; Wang, X.; Gong, J. Monoacylglycerol lipase: A novel potential therapeutic target and prognostic indicator for hepatocellular carcinoma. Sci. Rep. 2016, 6, 35784. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Kim, S.S.; Choi, E.; Oh, Y.T.; Lin, W.; Kim, T.-H.; Sa, J.K.; Hong, J.H.; Park, S.H.; Kwon, H.J.; et al. ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat. Commun. 2020, 11, 2978. [Google Scholar] [CrossRef]
- Marino, S.; de Ridder, D.; Bishop, R.T.; Renema, N.; Ponzetti, M.; Sophocleous, A.; Capulli, M.; Aljeffery, A.; Carrasco, G.; Gens, M.D.; et al. Paradoxical effects of JZL184, an inhibitor of monoacylglycerol lipase, on bone remodelling in healthy and cancer-bearing mice. EBioMedicine 2019, 44, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Roberto, D.; Klotz, L.H.; Venkateswaran, V. Cannabinoid WIN 55,212-2 induces cell cycle arrest and apoptosis, and inhibits proliferation, migration, invasion, and tumor growth in prostate cancer in a cannabinoid-receptor 2 dependent manner. Prostate 2019, 79, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Pietrovito, L.; Iozzo, M.; Bacci, M.; Giannoni, E.; Chiarugi, P. Treatment with Cannabinoids as a Promising Approach for Impairing Fibroblast Activation and Prostate Cancer Progression. Int. J. Mol. Sci. 2020, 25, 787. [Google Scholar] [CrossRef]
- Xu, D.; Wang, J.; Zhou, Z.; He, Z.; Zhao, Q. Cannabinoid WIN55, 212–2 induces cell cycle arrest and inhibits the proliferation and migration of human BEL7402 hepatocellular carcinoma cells. Mol. Med. Rep. 2015, 12, 7963–7970. [Google Scholar] [CrossRef]
- Ortega, A.; Rangel-López, E.; Hidalgo-Miranda, A.; Morales, A.; Ruiz-García, E.; Meneses-García, A.; Herrera-Gómez, A.; Aguilar-Ponce, J.; González-Herrera, I.; Guevara-Salazar, P.; et al. On the effects of CP 55-940 and other cannabinoid receptor agonists in C6 and U373 cell lines. Toxicol. In Vitro 2015, 29, 1941–1951. [Google Scholar] [CrossRef]
- Soto-Mercado, V.; Mendivil-Perez, M.; Jimenez-Del-Rio, M.; E Fox, J.; Velez-Pardo, C. Cannabinoid CP55940 selectively induces apoptosis in Jurkat cells and in ex vivo T-cell acute lymphoblastic leukemia through H2O2 signaling mechanism. Leuk. Res. 2020, 95, 106389. [Google Scholar] [CrossRef]
- Gustafsson, S.B.; Wallenius, A.; Zackrisson, H.; Popova, D.; Forshell, L.P.; Jacobsson, S.O.P. Effects of cannabinoids and related fatty acids upon the viability of P19 embryonal carcinoma cells. Arch. Toxicol. 2013, 87, 1939–1951. [Google Scholar] [CrossRef][Green Version]
- De Petrocellis, L.; Melck, D.; Palmisano, A.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef] [PubMed]
- Eichele, K.; Ramer, R.; Hinz, B. R(+)-methanandamide-induced apoptosis of human cervical carcinoma cells involves a cyclooxygenase-2-dependent pathway. Pharm. Res. 2008, 26, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Oleaherrero, N.; Vara, D.; Malagariecazenave, S.; Díaz-Laviada, I. Inhibition of human tumour prostate PC-3 cell growth by cannabinoids R(+)-methanandamide and JWH-015: Involvement of CB2. Br. J. Cancer 2009, 101, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, F.; Ostad, S.N.; Aliebrahimi, S.; Daman, Z. Anti-invasion effects of cannabinoids agonist and antagonist on human breast cancer stem cells. Iran. J. Pharm. Res. 2017, 16, 1479–1486. [Google Scholar]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Pozza, E.D.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Jagerovic, N. Antitumor cannabinoid chemotypes: Structural insights. Front. Pharmacol. 2019, 10, 621–638. [Google Scholar] [CrossRef]
- Elbaz, M.; Ahirwar, D.; Ravi, J.; Nasser, M.W.; Ganju, R.K. Novel role of cannabinoid receptor 2 in inhibiting EGF/EGFR and IGF-I/IGF-IR pathways in breast cancer. Oncotarget 2017, 8, 29668–29678. [Google Scholar] [CrossRef] [PubMed]
- Ravi, J.; Elbaz, M.; Wani, N.A.; Nasser, M.W.; Ganju, R.K. Cannabinoid receptor-2 agonist inhibits macrophage induced EMT in non-small cell lung cancer by downregulation of EGFR pathway. Mol. Carcinog. 2016, 55, 2063–2076. [Google Scholar] [CrossRef]
- Rosengren, R.J.; Cridge, B.J. Critical appraisal of the potential use of cannabinoids in cancer management. Cancer Manag. Res. 2013, 5, 301–313. [Google Scholar] [CrossRef]
- Haskó, J.; Fazakas, C.; Molnar, J.; Nyúl-Tóth, Á.; Herman, H.; Hermenean, A.; Wilhelm, I.; Persidsky, Y.; Krizbai, I.A. CB2 receptor activation inhibits melanoma cell transmigration through the blood-brain barrier. Int. J. Mol. Sci. 2014, 15, 8063–8074. [Google Scholar] [CrossRef]
- Staiano, R.I.; Loffredo, S.; Borriello, F.; Iannotti, F.A.; Piscitelli, F.; Orlando, P.; Secondo, A.; Granata, F.; Lepore, M.T.; Fiorelli, A.; et al. Human lung-resident macrophages express CB1 and CB2 receptors whose activation inhibits the release of angiogenic and lymphangiogenic factors. J. Leukoc. Biol. 2016, 99, 531–540. [Google Scholar] [CrossRef]
- Santoro, A.; Pisanti, S.; Grimaldi, C.; Izzo, A.A.; Borrelli, F.; Proto, M.C.; Malfitano, A.M.; Gazzerro, P.; Laezza, C.; Bifulco, M. Rimonabant inhibits human colon cancer cell growth and reduces the formation of precancerous lesions in the mouse colon. Int. J. Cancer 2009, 125, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Ciaglia, E.; Laezza, C.; Abate, M.; Pisanti, S.; Ranieri, R.; D’Alessandro, A.; Picardi, P.; Gazzerro, P.; Bifulco, M. Recognition by natural killer cells of N6-isopentenyladenosine-treated human glioma cell lines. Int. J. Cancer 2018, 142, 176–190. [Google Scholar] [CrossRef]
- Sarnataro, D.; Grimaldi, C.; Pisanti, S.; Gazzerro, P.; Laezza, C.; Zurzolo, C.; Bifulco, M. Plasma membrane and lysosomal localization of CB1 cannabinoid receptor are dependent on lipid rafts and regulated by anandamide in human breast cancer cells. FEBS Lett. 2005, 579, 6343–6349. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Y.; Zou, Y.; Zhu, L.; Dong, B.; Huang, J.; Chen, Y.; Xue, W.; Huang, Y.; Kong, W.; et al. Overexpression of cannabinoid receptor 1 promotes renal cell carcinoma progression. Tumor Biol. 2016, 37, 16237–16247. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, V.; Evangelista, D.; Oddi, S.; Florenzano, F.; Chiurchiù, V.; Avigliano, L.; Catani, M.V.; Maccarrone, M. Regulation of inflammation and proliferation of human bladder carcinoma cells by type-1 and type-2 cannabinoid receptors. Life Sci. 2015, 138, 41–51. [Google Scholar] [CrossRef]
- An, D.; Peigneur, S.; Hendrickx, L.A.; Tytgat, J. Targeting cannabinoid receptors: Current status and prospects of natural products. Int. J. Mol. Sci. 2020, 21, 5064. [Google Scholar] [CrossRef]
- Hanlon, K.E.; Lozano-Ondoua, A.N.; Umaretiya, P.J.; Symons-Liguori, A.M.; Chandramouli, A.; Moy, J.K.; Kwass, W.K.; Mantyh, P.W.; Nelson, M.A.; Vanderah, T.W. Modulation of breast cancer cell viability by a cannabinoid receptor 2 agonist, JWH-015, is calcium dependent. Breast Cancer Targets Ther. 2016, 8, 59–71. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Y.; Zhu, L.; Zou, Y.; Kong, W.; Dong, B.; Huang, J.; Chen, Y.; Xue, W.; Huang, Y.; et al. Cannabinoid receptor 2 as a novel target for promotion of renal cell carcinoma prognosis and progression. J. Cancer Res. Clin. Oncol. 2017, 144, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qin, Y.; Pan, Z.; Li, M.; Liu, X.; Chen, X.; Qu, G.; Zhou, L.; Xu, M.; Zheng, Q.; et al. Cannabidiol induces cell cycle arrest and cell apoptosis in human gastric cancer SGC-7901 cells. Biomolecules 2019, 9, 302. [Google Scholar] [CrossRef]
- Kisková, T.; Mungenast, F.; Suváková, M.; Jäger, W.; Thalhammer, T. Future aspects for cannabinoids in breast cancer therapy. Int. J. Mol. Sci. 2019, 20, 1673. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Offidani, M.; Amantini, C.; Gentili, S.; Soriani, A.; Cardinali, C.; Leoni, P.; Santoni, G. Cannabinoids synergize with carfilzomib, reducing multiple myeloma cells viability and migration. Oncotarget 2016, 7, 77543–77557. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Delta9-Tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arencibia, M.; Molina-Holgado, E.; Molina-Holgado, F. Effect of endocannabinoid signalling on cell fate: Life, death, differentiation and proliferation of brain cells. Br. J. Pharmacol. 2019, 176, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Jo, M.J.; Yun, H.K.; Kim, D.Y.; Kim, B.R.; Kim, J.L.; Park, S.H.; Na, Y.J.; Jeong, Y.A.; Kim, B.G.; et al. Cannabidiol promotes apoptosis via regulation of XIAP/Smac in gastric cancer. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Schoeman, R.; Beukes, N.; Frost, C. Cannabinoid combination induces cytoplasmic vacuolation in MCF-7 breast cancer cells. Molecules 2020, 25, 4682. [Google Scholar] [CrossRef]
- Maccarrone, M.; Finazzi-Agró, A. The endocannabinoid system, anandamide and the regulation of mammalian cell apoptosis. Cell Death Differ. 2003, 10, 946–955. [Google Scholar] [CrossRef]
- Huang, L.; Ramirez, J.C.; Frampton, G.A.; Golden, L.E.; Quinn, M.A.; Pae, H.Y.; Horvat, D.; Liang, L.-J.; DeMorrow, S. Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab. Investig. 2011, 91, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Soliman, E.; Van Dross, R. Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: Receptor-independent endocannabinoid signaling. Mol. Carcinog. 2016, 55, 1807–1821. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Vecera, L.; Gabrhelik, T.; Prasil, P.; Stourac, P. The role of cannabinoids in the treatment of cancer. Bratisl. Lek. Listy 2020, 121, 79–95. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Cannabinoid signaling in glioma cells. Adv. Exp. Med. Biol. 2020, 1202, 223–241. [Google Scholar] [CrossRef]
- Notaro, A.; Emanuele, S.; Geraci, F.; D’Anneo, A.; Lauricella, M.; Calvaruso, G.; Giuliano, M. WIN55,212-2-Induced expression of Mir-29b1 favours the suppression of osteosarcoma cell migration in a SPARC-independent manner. Int. J. Mol. Sci. 2019, 20, 5235. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.; Xu, C.; Hamel, E.; Wager-Miller, J.B.; Woodruff, G.; Miller, A.; Sanford, C.; Mackie, K.; Stella, N. Novel indole-based compounds that differentiate alkylindole-sensitive receptors from cannabinoid receptors and microtubules: Characterization of their activity on glioma cell migration. Pharmacol. Res. 2017, 115, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Hinz, B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J. Natl. Cancer Inst. 2008, 100, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Carracedo, A.; Salazar, M.; Lorente, M.; Egia, A.; González-Feria, L.; Haro, A.; Velasco, G.; Guzmán, M. Down-regulation of tissue inhibitor of metalloproteinases-1 in gliomas: A new marker of cannabinoid antitumoral activity? Neuropharmacology 2008, 54, 235–243. [Google Scholar] [CrossRef]
- Grimaldi, C.; Pisanti, S.; Laezza, C.; Malfitano, A.M.; Santoro, A.; Vitale, M.; Caruso, M.G.; Notarnicola, M.; Iacuzzo, I.; Portella, G.; et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp. Cell Res. 2006, 312, 363–373. [Google Scholar] [CrossRef]
- Preet, A.; Ganju, R.K.; Groopman, J.E. Delta9-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene 2007, 27, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Andersen, L.; Hasenöhrl, C.; Feuersinger, D.; Stancic, A.; Fauland, A.; Magnes, C.; El-Heliebi, A.; Lax, S.; Uranitsch, S.; et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis. Br. J. Pharmacol. 2015, 173, 142–154. [Google Scholar] [CrossRef]
- Bifulco, M.; Laezza, C.; Gazzerro, P.; Pentimalli, F. Endocannabinoids as emerging suppressors of angiogenesis and tumor invasion. Oncol. Rep. 2007, 17, 813–816. [Google Scholar] [CrossRef]
- Ramer, R.; Bublitz, K.; Freimuth, N.; Merkord, J.; Rohde, H.; Haustein, M.; Borchert, P.; Schmuhl, E.; Linnebacher, M.; Hinz, B. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule-1. FASEB J. 2011, 26, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Kogan, N.M.; Blázquez, C.; Álvarez, L.; Gallily, R.; Schlesinger, M.; Guzmán, M.; Mechoulam, R. A cannabinoid quinone inhibits angiogenesis by targeting vascular endothelial cells. Mol. Pharmacol. 2006, 70, 51–59. [Google Scholar] [CrossRef]
- West, D.C.; Thompson, W.D.; Sells, P.G.; Burbridge, M.F. Angiogenesis assays using chick chorioallantoic membrane. Methods Mol. Med. 2003, 46, 107–129. [Google Scholar] [CrossRef]
- Zhang, X.; Maor, Y.; Wang, J.F.; Kunos, G.; Groopman, J.E. Endocannabinoid-like N-arachidonoyl serine is a novel pro-angiogenic mediator. Br. J. Pharmacol. 2010, 160, 1583–1594. [Google Scholar] [CrossRef]
- Mosca, L.; Vitiello, F.; Coppola, A.; Borzacchiello, L.; Ilisso, C.P.; Pagano, M.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. Therapeutic potential of the natural compound S-adenosylmethionine as a chemoprotective synergistic agent in breast, and head and neck cancer treatment: Current status of research. Int. J. Mol. Sci. 2020, 21, 8547. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Huang, L.; Zhang, B.; Wu, C.; Cui, J.; Wang, Z. WIN 55,212-2 inhibits the epithelial mesenchymal transition of gastric cancer cells via COX-2 signals. Cell. Physiol. Biochem. 2016, 39, 2149–2157. [Google Scholar] [CrossRef]
- Milian, L.; Mata, M.; Alcacer, J.; Oliver, M.; Sancho-Tello, M.; De Llano, J.J.M.; Camps, C.; Galbis, J.; Carretero, J.; Carda, C. Cannabinoid receptor expression in non-small cell lung cancer. Effectiveness of tetrahydrocannabinol and cannabidiol inhibiting cell proliferation and epithelial-mesenchymal transition in vitro. PLoS ONE 2020, 15, e0228909. [Google Scholar] [CrossRef]
- Yoshinaga, T.; Uwabe, K.; Naito, S.; Higashino, K.; Nakano, T.; Numata, Y.; Kihara, A. AM251 suppresses epithelial-mesenchymal transition of renal tubular epithelial cells. PLoS ONE 2016, 11, e0167848. [Google Scholar] [CrossRef]
- Laezza, C.; D’Alessandro, A.; Paladino, S.; Malfitano, A.M.; Proto, M.C.; Gazzerro, P.; Pisanti, S.; Santoro, A.; Ciaglia, E.; Bifulco, M. Anandamide inhibits the Wnt/β-catenin signalling pathway in human breast cancer MDA MB 231 cells. Eur. J. Cancer 2012, 48, 3112–3122. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mediators | Effects | References | |
|---|---|---|---|
| Cell Cycle | WIN 55,212-2 CBD THC AEA | Inhibition of Cdk4/CycD Inhibition of Cdk2/CycE Downregulation of Cdc2 Inhibition of cAMP Inhibition of AKT pathway | [65] [87] [88] [90] [91] |
| Apoptosis | CBD C6 THC AEA metAEA | Inhibition of XIAP Downregulation Bcl-2 Upregulation Noxa Enhance ROS generation Upregulation of GRP78 Inhibition of ERK/AKT signalling Accumulation of Ceramide Activation of Raf1/ERK Downregulation AKT pathway Increase of Ca2+ influx Activation of Fas/FasL Activation of PPARγ | [92] [87] [92] [93] [94] [93] [95] [95] [91] [95] [96] [72] |
| Autophagy | THC-CBD THC AEA | Activation of CaCMKKβ Inactivation of mTORC1 Activation of LC3-II Inhibition of mTORC1 Downregulation SOCS3 | [91] [91] [89] [100] [100] |
| MEDIATORS | EFFECTS | REF | |
|---|---|---|---|
| Epithelial Mesenchimal Transition (EMT) | JWH-015 THC CBD WIN AEA | InhibitionWnt/ßcatenina:Downregulation of Snail, Twist, Slug, Vimentin, Fibronectin; Upregulation of E-Cadherin. Inhibition of p27/CDK4/pRb. | [120] [119] |
| AM251 | Downregulation of Snail1, Fos-b,Jun-b, Ap1. | [119] | |
| Adhesion | AM251 | TGFß inhibit Smad2/3-p38MAPK: Downregulation of COL1A1 e Upregulation of E-Cadherin | [119] |
| Invasionand metastasis | THC CBD WIN AEA | Inhibition Id1 and Sox2. Relase of extracellular vesicles with miR-29b1. Inhibition of Cox-2 and of phosphorylation AKT and inhibition of MMP2;FAK,MMP-9,VCAM1 | [101] [104] [106] |
| Angiogenesis | JWH-015 THC CBD WIN | Inhibition of Cox-2 and phosphorylation of AKTand inhibition of MMP2; FAK; MMP-9; VCAM. Binding CXCR4 Activation of ICAM-1 and Timp1 | [90,117] [92] [112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, C.; Navarra, G.; Coppola, L.; Bifulco, M.; Laezza, C. Molecular Mechanism of Cannabinoids in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 3680. https://doi.org/10.3390/ijms22073680
Pagano C, Navarra G, Coppola L, Bifulco M, Laezza C. Molecular Mechanism of Cannabinoids in Cancer Progression. International Journal of Molecular Sciences. 2021; 22(7):3680. https://doi.org/10.3390/ijms22073680
Chicago/Turabian StylePagano, Cristina, Giovanna Navarra, Laura Coppola, Maurizio Bifulco, and Chiara Laezza. 2021. "Molecular Mechanism of Cannabinoids in Cancer Progression" International Journal of Molecular Sciences 22, no. 7: 3680. https://doi.org/10.3390/ijms22073680
APA StylePagano, C., Navarra, G., Coppola, L., Bifulco, M., & Laezza, C. (2021). Molecular Mechanism of Cannabinoids in Cancer Progression. International Journal of Molecular Sciences, 22(7), 3680. https://doi.org/10.3390/ijms22073680